
Connectivity Map Analysis of a Single-Cell RNA-Sequencing -Derived Transcriptional Signature of mTOR Signaling


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Overview of scRNA-Seq Connectivity Analysis
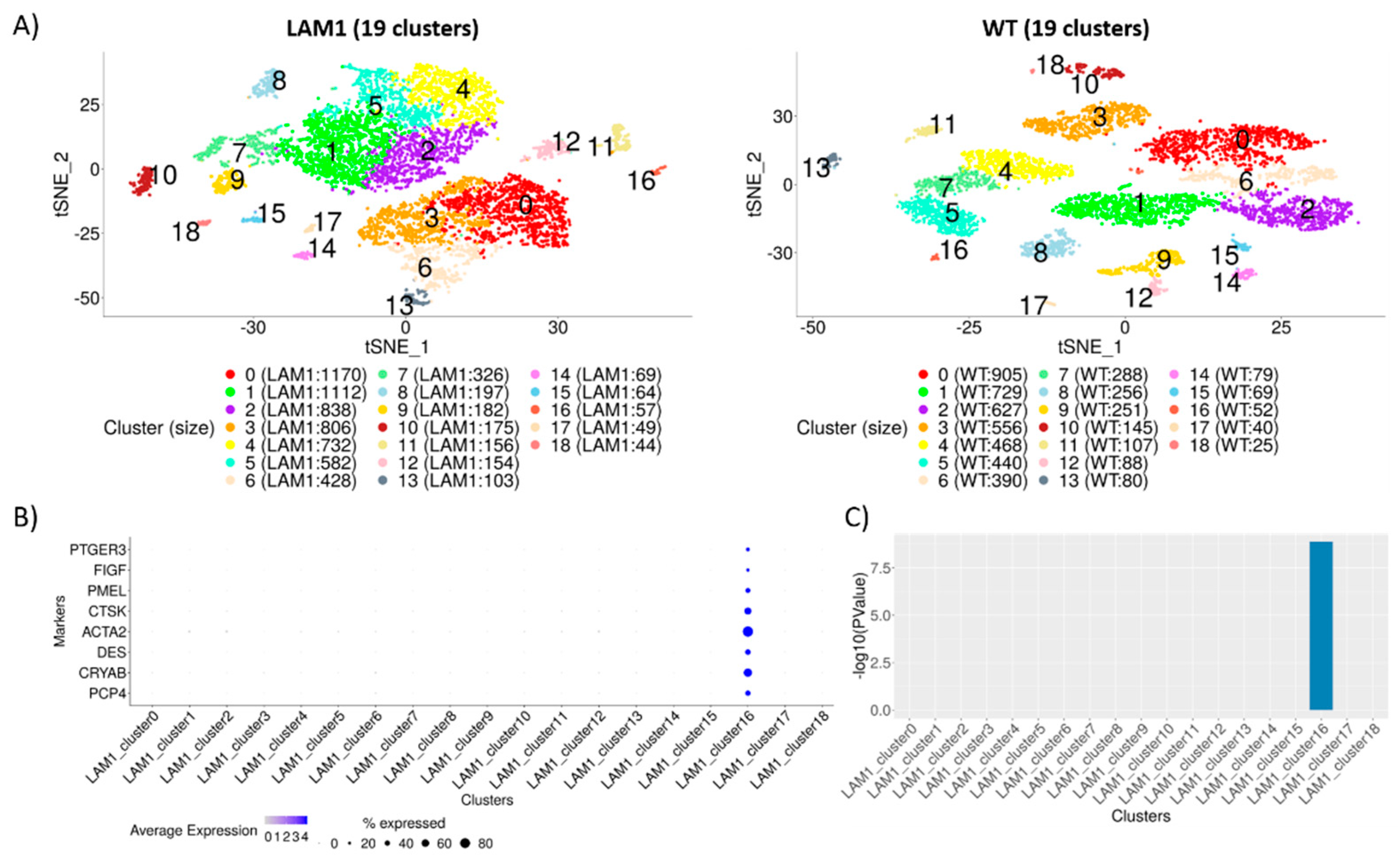
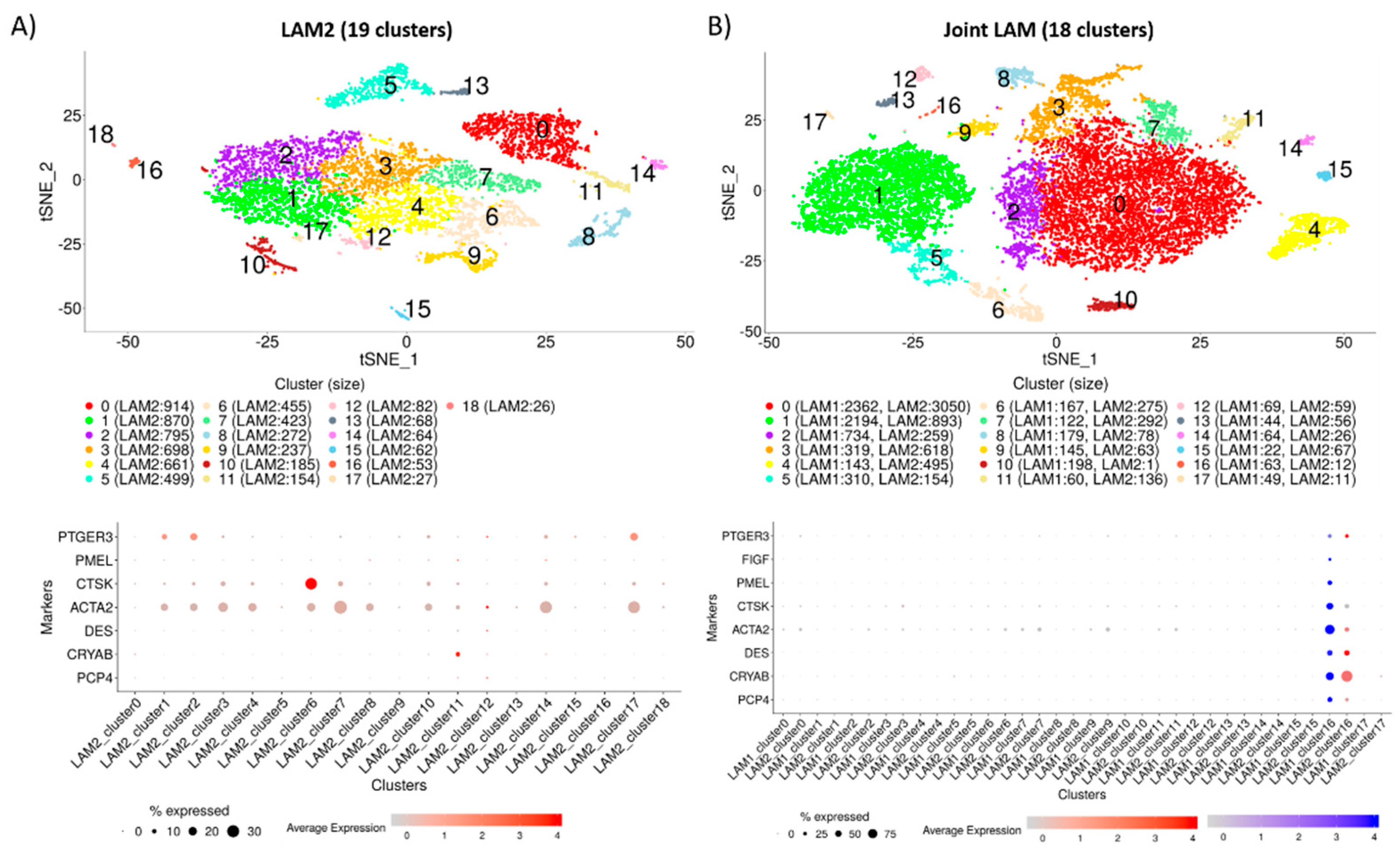
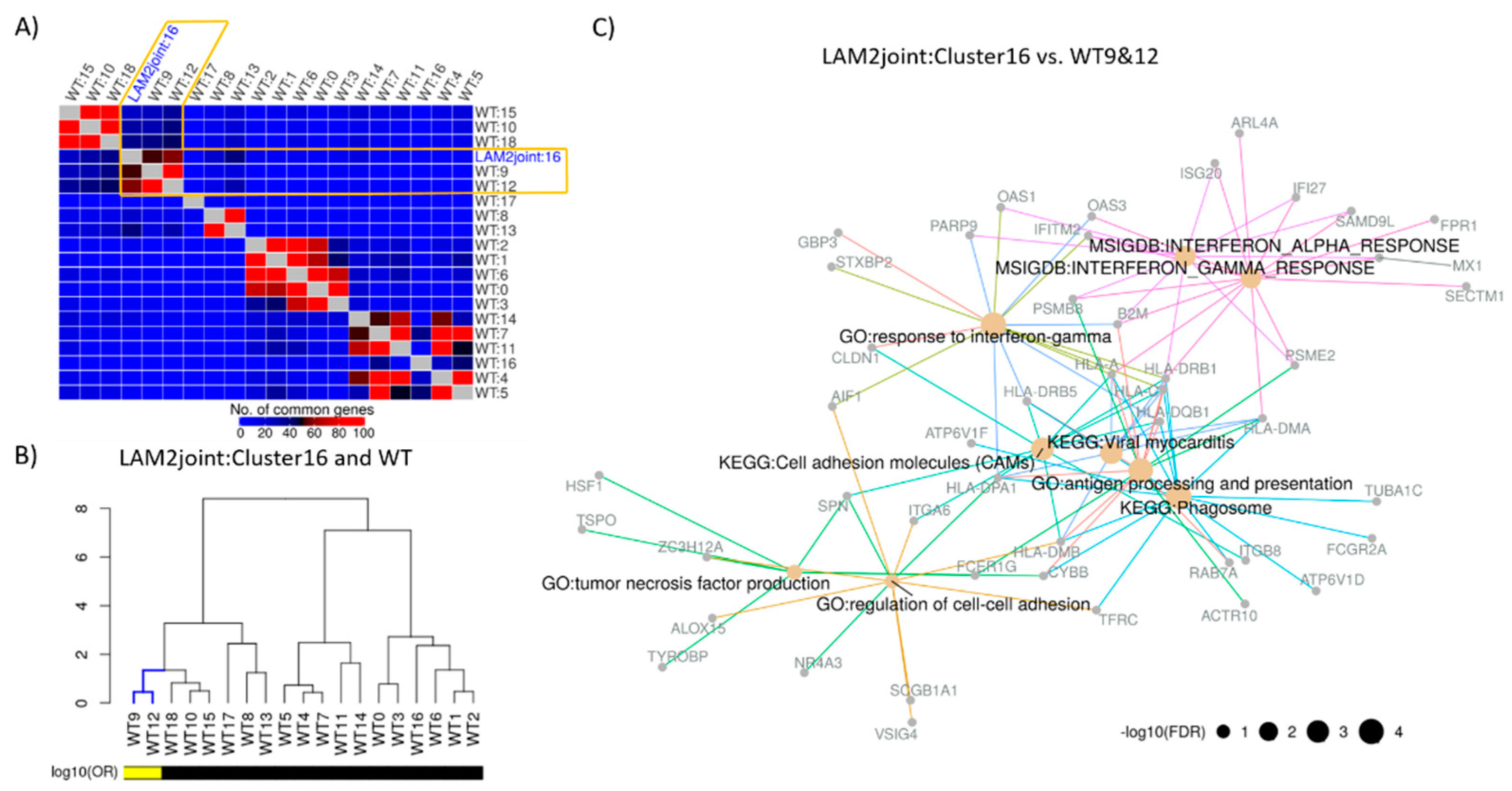
2.2. Identification of Distinct Cell Populations
2.3. Construction of Cluster Annotating Signatures
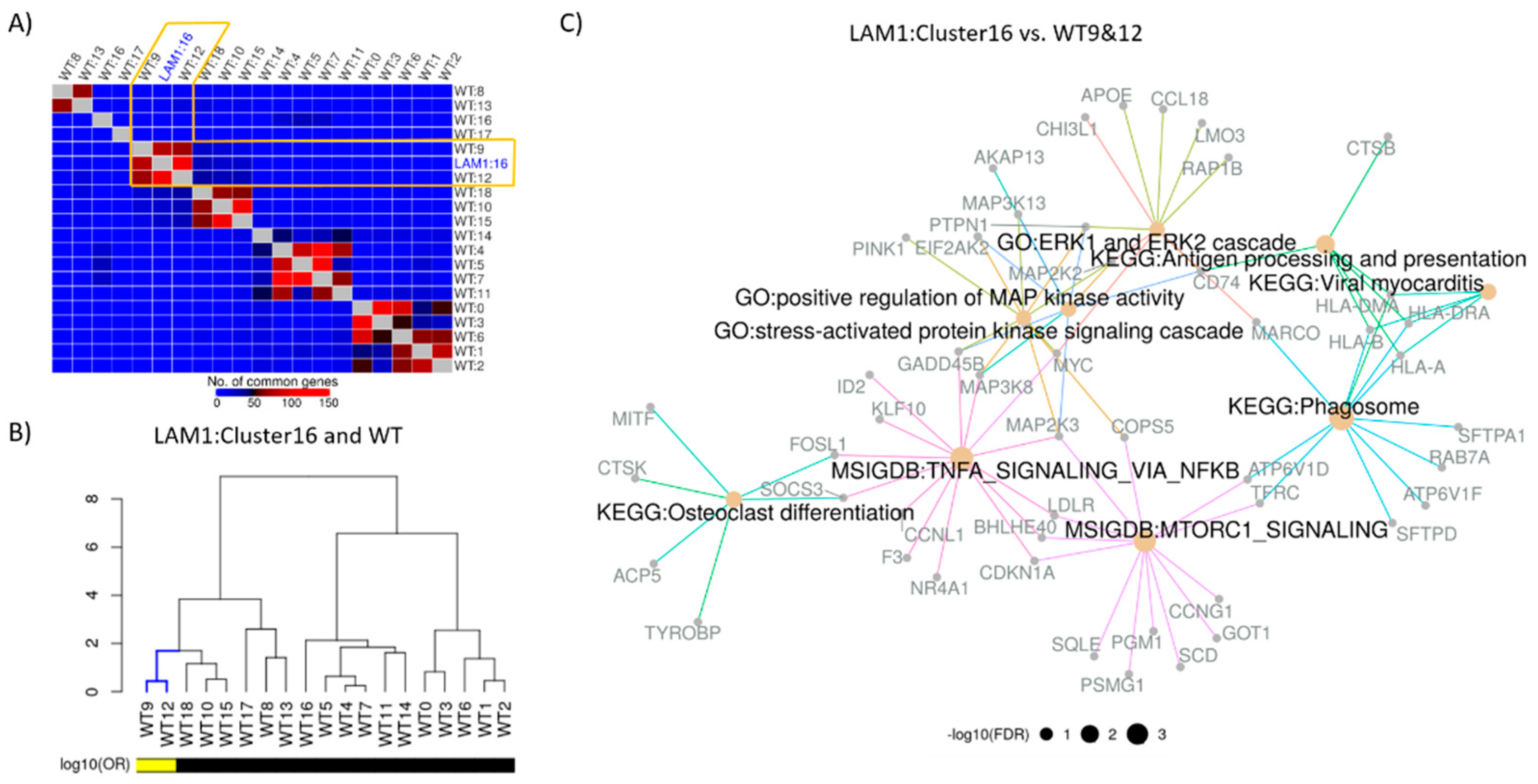
2.4. Construction of Disease Characterizing Signature
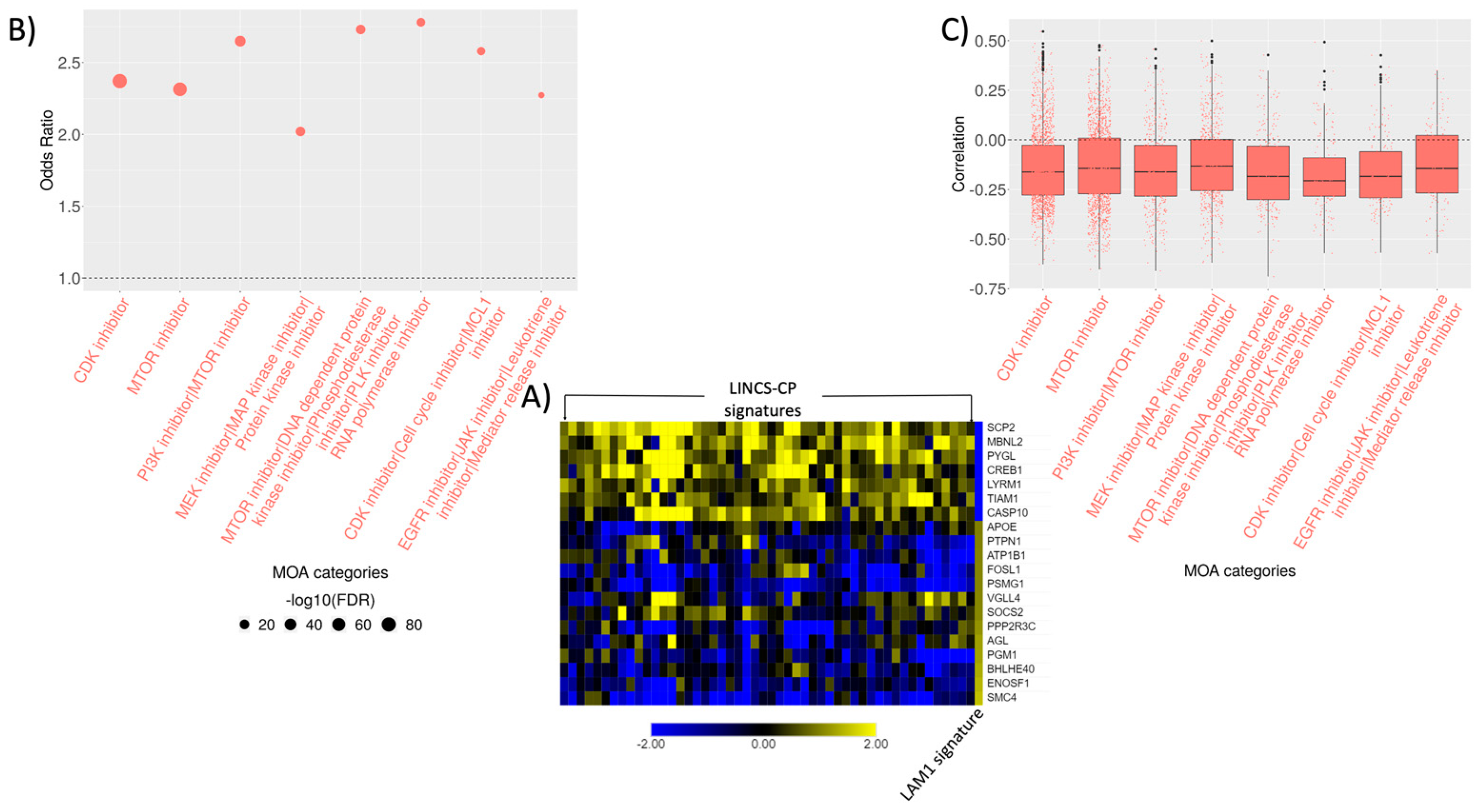
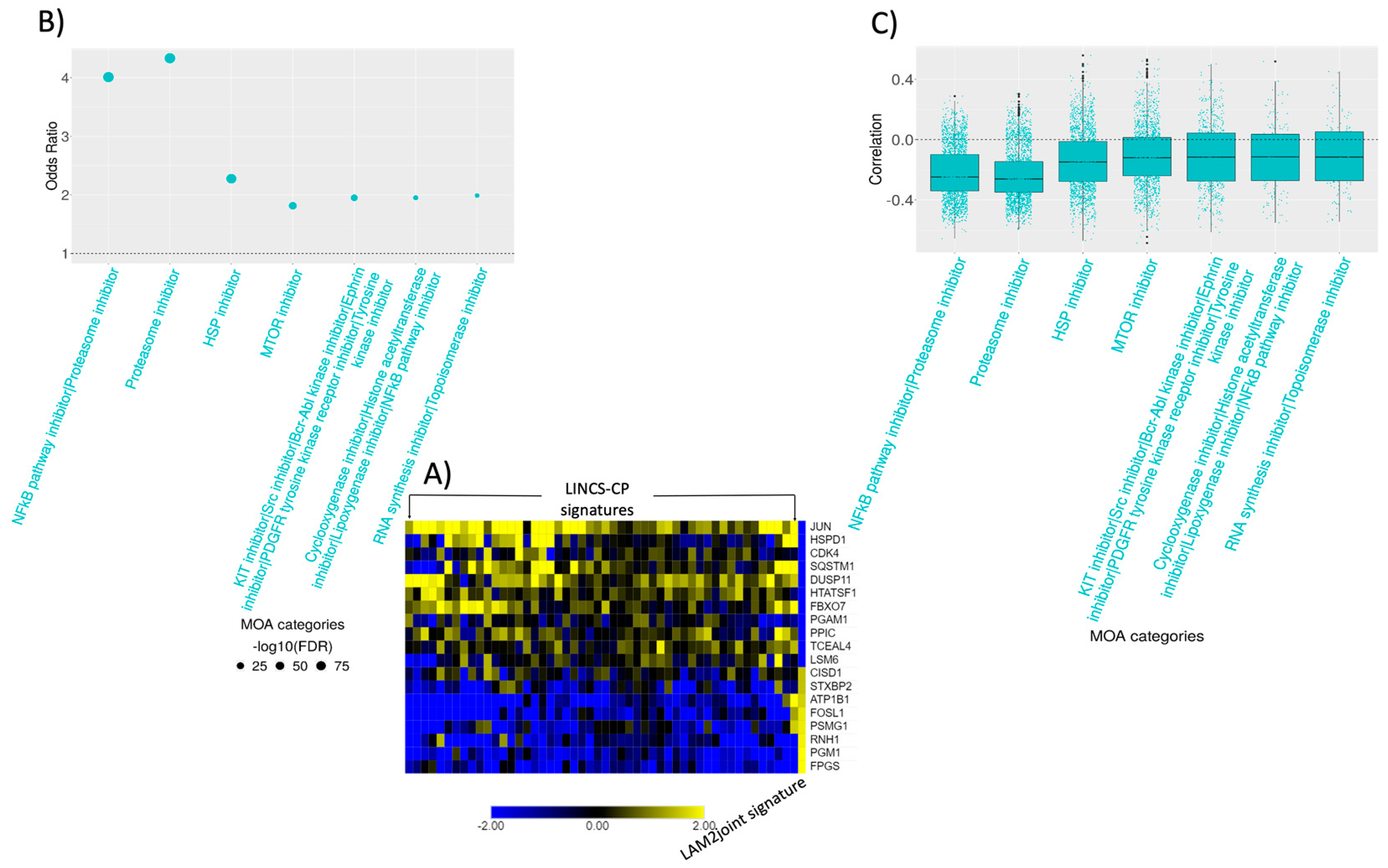
2.5. Connectivity Analysis
2.6. Signature Construction and Connectivity Analysis of Sirolimus Treated LAM
3. Discussion
4. Materials and Methods
4.1. Single-Cell RNA-Seq and LINCS-L1000 Data
4.2. Single-Cell RNA-Seq Data Pre-Processing and Clustering
4.3. Construction of Cluster Annotating and Disease Characterizing Signatures
4.4. Connectivity Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- McCormack, F.X.; Inoue, Y.; Moss, J.; Singer, L.G.; Strange, C.; Nakata, K.; Barker, A.F.; Chapman, J.T.; Brantly, M.L.; Stocks, J.M. Efficacy and safety of sirolimus in lymphangioleiomyomatosis. N. Engl. J. Med. 2011, 364, 1595–1606. [Google Scholar] [CrossRef]
- Bissler, J.J.; McCormack, F.X.; Young, L.R.; Elwing, J.M.; Chuck, G.; Leonard, J.M.; Schmithorst, V.J.; Laor, T.; Brody, A.S.; Bean, J. Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N. Engl. J. Med. 2008, 358, 140–151. [Google Scholar] [CrossRef] [Green Version]
- Taveira-DaSilva, A.M.; Moss, J. Optimizing treatments for lymphangioleiomyomatosis. Expert Rev. Respir. Med. 2012, 6, 267–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sirota, M.; Dudley, J.T.; Kim, J.; Chiang, A.P.; Morgan, A.A.; Sweet-Cordero, A.; Sage, J.; Butte, A.J. Discovery and preclinical validation of drug indications using compendia of public gene expression data. Sci. Transl. Med. 2011, 3, 96ra77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C. Drug Repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef]
- Lamb, J.; Crawford, E.D.; Peck, D.; Modell, J.W.; Blat, I.C.; Wrobel, M.J.; Lerner, J.; Brunet, J.-P.; Subramanian, A.; Ross, K.N. The connectivity map: Using gene-expression signatures to connect small molecules, genes, and disease. Science 2006, 313, 1929–1935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Claerhout, S.; Lim, J.Y.; Choi, W.; Park, Y.-Y.; Kim, K.; Kim, S.-B.; Lee, J.-S.; Mills, G.B.; Cho, J.Y. Gene expression signature analysis identifies vorinostat as a candidate therapy for gastric cancer. PLoS ONE 2011, 6. [Google Scholar] [CrossRef]
- Dudley, J.T.; Sirota, M.; Shenoy, M.; Pai, R.K.; Roedder, S.; Chiang, A.P.; Morgan, A.A.; Sarwal, M.M.; Pasricha, P.J.; Butte, A.J. Computational repositioning of the anticonvulsant topiramate for inflammatory bowel disease. Sci. Transl. Med. 2011, 3, 96ra76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, A.; Narayan, R.; Corsello, S.M.; Peck, D.D.; Natoli, T.E.; Lu, X.; Gould, J.; Davis, J.F.; Tubelli, A.A.; Asiedu, J.K. A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell 2017, 171, 1437–1452. [Google Scholar] [CrossRef]
- Keenan, A.B.; Jenkins, S.L.; Jagodnik, K.M.; Koplev, S.; He, E.; Torre, D.; Wang, Z.; Dohlman, A.B.; Silverstein, M.C.; Lachmann, A. The library of integrated network-based cellular signatures NIH program: System-level cataloging of human cells response to perturbations. Cell Syst. 2018, 6, 13–24. [Google Scholar] [CrossRef] [Green Version]
- Guo, M.; Yu, J.J.; Perl, A.K.; Wikenheiser-Brokamp, K.A.; Riccetti, M.; Zhang, E.Y.; Sudha, P.; Adam, M.; Potter, A.; Kopras, E.J. Single cell transcriptomic analysis identifies a unique pulmonary lymphangioleiomyomatosis cell. Am. J. Respir. Crit. Care Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck III, W.M.; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive integration of single-cell data. Cell 2019, 177, 1888–1902. [Google Scholar] [CrossRef]
- Han, X.; Zhou, Z.; Fei, L.; Sun, H.; Wang, R.; Chen, Y.; Chen, H.; Wang, J.; Tang, H.; Ge, W. Construction of a human cell landscape at single-cell level. Nature 2020, 581, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lan, Y.; Xu, J.; Quan, F.; Zhao, E.; Deng, C.; Luo, T.; Xu, L.; Liao, G.; Yan, M. CellMarker: A manually curated resource of cell markers in human and mouse. Nucleic Acids Res. 2019, 47, D721–D728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franzén, O.; Gan, L.-M.; Björkegren, J.L.M. PanglaoDB: A web server for exploration of mouse and human single-cell RNA sequencing data. Database 2019, 2019, baz046. [Google Scholar] [CrossRef] [Green Version]
- Su, A.I.; Wiltshire, T.; Batalov, S.; Lapp, H.; Ching, K.A.; Block, D.; Zhang, J.; Soden, R.; Hayakawa, M.; Kreiman, G. A Gene Atlas of the Mouse and Human Protein-Encoding Transcriptomes. Proc. Natl. Acad. Sci. USA 2004, 101, 6062–6067. [Google Scholar] [CrossRef] [Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The molecular signatures database hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. ClusterProfiler: An R package for comparing biological themes among gene clusters. Omi. A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Besson, A.; Dowdy, S.F.; Roberts, J.M. CDK inhibitors: Cell cycle regulators and beyond. Dev. Cell 2008, 14, 159–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malumbres, M.; Barbacid, M. Cell cycle, CDKs and cancer: A changing paradigm. Nat. Rev. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Astrinidis, A.; Howard, S.; Henske, E.P. Estradiol and tamoxifen stimulate LAM-associated angiomyolipoma cell growth and activate both genomic and nongenomic signaling pathways. Am. J. Physiol. Cell. Mol. Physiol. 2004, 286, L694–L700. [Google Scholar] [CrossRef] [PubMed]
- Jane, J.Y.; Robb, V.A.; Morrison, T.A.; Ariazi, E.A.; Karbowniczek, M.; Astrinidis, A.; Wang, C.; Hernandez-Cuebas, L.; Seeholzer, L.F.; Nicolas, E. Estrogen promotes the survival and pulmonary metastasis of tuberin-null cells. Proc. Natl. Acad. Sci. USA 2009, 106, 2635–2640. [Google Scholar]
- Seoane, J.; Le, H.-V.; Massagué, J. Myc suppression of the P21 Cip1 cdk inhibitor influences the outcome of the P53 response to DNA damage. Nature 2002, 419, 729–734. [Google Scholar] [CrossRef]
- Gramling, M.W.; Eischen, C.M. Suppression of ras/mapk pathway signaling inhibits myc-induced lymphomagenesis. Cell Death Differ. 2012, 19, 1220–1227. [Google Scholar] [CrossRef] [Green Version]
- Carracedo, A.; Ma, L.; Teruya-Feldstein, J.; Rojo, F.; Salmena, L.; Alimonti, A.; Egia, A.; Sasaki, A.T.; Thomas, G.; Kozma, S.C. Inhibition of MTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J. Clin. Investig. 2008, 118, 3065–3074. [Google Scholar] [CrossRef] [PubMed]
- Mi, R.; Ma, J.; Zhang, D.; Li, L.; Zhang, H. Efficacy of combined inhibition of MTOR and ERK/MAPK pathways in treating a tuberous sclerosis complex cell model. J. Genet. Genomics 2009, 36, 355–361. [Google Scholar] [CrossRef]
- Zhang, Y.; Nicholatos, J.; Dreier, J.R.; Ricoult, S.J.H.; Widenmaier, S.B.; Hotamisligil, G.S.; Kwiatkowski, D.J.; Manning, B.D. Coordinated regulation of protein synthesis and degradation by MTORC1. Nature 2014, 513, 440–443. [Google Scholar] [CrossRef] [Green Version]
- Johnson, C.E.; Dunlop, E.A.; Seifan, S.; McCann, H.D.; Hay, T.; Parfitt, G.J.; Jones, A.T.; Giles, P.J.; Shen, M.H.; Sampson, J.R. Loss of tuberous sclerosis complex 2 sensitizes tumors to nelfinavir-bortezomib therapy to intensify endoplasmic reticulum stress-induced cell death. Oncogene 2018, 37, 5913–5925. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Li, N.; Liu, X.; Zhang, E.Y.; Sun, Y.; Masuda, K.; Li, J.; Sun, J.; Morrison, T.; Li, X. Proapoptotic protein bim attenuates estrogen-enhanced survival in lymphangioleiomyomatosis. JCI Insight 2016, 1, e86629. [Google Scholar] [CrossRef] [Green Version]
- Karin, M. Nuclear factor-ΚB in cancer development and progression. Nature 2006, 441, 431–436. [Google Scholar] [CrossRef]
- Henske, E.P.; McCormack, F.X. Lymphangioleiomyomatosis—A wolf in sheep’s clothing. J. Clin. Investig. 2012, 122, 3807–3816. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S.; Tergaonkar, V.; Rothlin, C.V.; Correa, R.G.; Bottero, V.; Bist, P.; Verma, I.M.; Hunter, T. Essential role of tuberous sclerosis genes TSC1 and TSC2 in NF-ΚB activation and cell survival. Cancer Cell 2006, 10, 215–226. [Google Scholar] [CrossRef] [Green Version]
- Al Mahi, N.; Najafabadi, M.F.; Pilarczyk, M.; Kouril, M.; Medvedovic, M. GREIN: An interactive web platform for re-analyzing GEO RNA-Seq data. Sci. Rep. 2019, 9, 1–9. [Google Scholar]
- Athar, A.; Füllgrabe, A.; George, N.; Iqbal, H.; Huerta, L.; Ali, A.; Snow, C.; Fonseca, N.A.; Petryszak, R.; Papatheodorou, I. ArrayExpress update–from bulk to single-cell expression data. Nucleic Acids Res. 2019, 47, D711–D715. [Google Scholar] [CrossRef] [PubMed]
- Pilarczyk, M.; Najafabadi, M.F.; Kouril, M.; Vasiliauskas, J.; Niu, W.; Shamsaei, B.; Mahi, N.; Zhang, L.; Clark, N.; Ren, Y.; et al. Connecting omics signatures of diseases, drugs, and mechanisms of actions with ILINCS. bioRxiv 2019, 826271. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Monteiro, C.D.; Jagodnik, K.M.; Fernandez, N.F.; Gundersen, G.W.; Rouillard, A.D.; Jenkins, S.L.; Feldmann, A.S.; Hu, K.S.; McDermott, M.G. Extraction and analysis of signatures from the gene expression omnibus by the crowd. Nat. Commun. 2016, 7, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, J.; Adiconis, X.; Simmons, S.K.; Kowalczyk, M.S.; Hession, C.C.; Marjanovic, N.D.; Hughes, T.K.; Wadsworth, M.H.; Burks, T.; Nguyen, L.T. Systematic comparison of single-cell and single-nucleus RNA-sequencing methods. Nat. Biotechnol. 2020, 38, 737–746. [Google Scholar] [CrossRef]
- Vieth, B.; Parekh, S.; Ziegenhain, C.; Enard, W.; Hellmann, I. A systematic evaluation of single cell RNA-seq analysis pipelines. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blondel, V.D.; Guillaume, J.-L.; Lambiotte, R.; Lefebvre, E. Fast unfolding of communities in large networks. J. Stat. Mech. Theory Exp. 2008, 2008, P10008. [Google Scholar] [CrossRef] [Green Version]
- Levine, J.H.; Simonds, E.F.; Bendall, S.C.; Davis, K.L.; El-ad, D.A.; Tadmor, M.D.; Litvin, O.; Fienberg, H.G.; Jager, A.; Zunder, E.R. Data-driven phenotypic dissection of AML reveals progenitor-like cells that correlate with prognosis. Cell 2015, 162, 184–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahlo, M.; Tian, L.; Lönnstedt, I.; Ng, M.; Hicks, S. Comparison of clustering tools in R for medium-sized 10× genomics single-cell RNA-sequencing. F1000 Res. 2018, 7, 1–26. [Google Scholar]
- Duò, A.; Robinson, M.D.; Soneson, C. A Systematic Performance Evaluation of Clustering Methods for Single-Cell RNA-Seq Data. F1000 Res. 2018, 7, 1141. [Google Scholar] [CrossRef]
- Yang, Y.; Huh, R.; Culpepper, H.W.; Lin, Y.; Love, M.I.; Li, Y. SAFE-clustering: Single-cell aggregated (from ensemble) clustering for single-cell RNA-seq data. Bioinformatics 2019, 35, 1269–1277. [Google Scholar] [CrossRef]
- Finak, G.; McDavid, A.; Yajima, M.; Deng, J.; Gersuk, V.; Shalek, A.K.; Slichter, C.K.; Miller, H.W.; McElrath, M.J.; Prlic, M. MAST: A Flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 2015, 16, 278. [Google Scholar] [CrossRef] [Green Version]
- Saxton, R.A.; Sabatini, D.M. MTOR signaling in growth, metabolism, and disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carsillo, T.; Astrinidis, A.; Henske, E.P. Mutations in the tuberous sclerosis complex gene TSC2 are a cause of sporadic pulmonary lymphangioleiomyomatosis. Proc. Natl. Acad. Sci. USA 2000, 97, 6085–6090. [Google Scholar] [CrossRef] [Green Version]
- McCormack, F.X.; Travis, W.D.; Colby, T.V.; Henske, E.P.; Moss, J. Lymphangioleiomyomatosis: Calling it what it is: A low-grade, destructive, metastasizing neoplasm. Am. J. Respir. Crit. Care Med. 2012, 186, 1210–1212. [Google Scholar] [CrossRef]
- Stallone, G.; Infante, B.; Prisciandaro, C.; Grandaliano, G. MTOR and Aging: An old fashioned dress. Int. J. Mol. Sci. 2019, 20, 2774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.Y.; Sabatini, D.M. MTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef] [PubMed]
- Robida-Stubbs, S.; Glover-Cutter, K.; Lamming, D.W.; Mizunuma, M.; Narasimhan, S.D.; Neumann-Haefelin, E.; Sabatini, D.M.; Blackwell, T.K. TOR signaling and rapamycin influence longevity by regulating SKN-1/Nrf and DAF-16/FoxO. Cell Metab. 2012, 15, 713–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjedov, I.; Toivonen, J.M.; Kerr, F.; Slack, C.; Jacobson, J.; Foley, A.; Partridge, L. Mechanisms of life span extension by rapamycin in the fruit fly drosophila melanogaster. Cell Metab. 2010, 11, 35–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, D.E.; Strong, R.; Sharp, Z.D.; Nelson, J.F.; Astle, C.M.; Flurkey, K.; Nadon, N.L.; Wilkinson, J.E.; Frenkel, K.; Carter, C.S. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 2009, 460, 392–395. [Google Scholar] [CrossRef] [Green Version]
- Spilman, P.; Podlutskaya, N.; Hart, M.J.; Debnath, J.; Gorostiza, O.; Bredesen, D.; Richardson, A.; Strong, R.; Galvan, V. Inhibition of MTOR by rapamycin abolishes cognitive deficits and reduces amyloid-β levels in a mouse model of alzheimer’s disease. PLoS ONE 2010, 5, e9979. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Jiang, Y. MTOR Inhibitors at a glance. Mol. Cell. Pharmacol. 2015, 7, 15. [Google Scholar]
- Xiao, Y.; Hsiao, T.-H.; Suresh, U.; Chen, H.-I.H.; Wu, X.; Wolf, S.E.; Chen, Y. A novel significance score for gene selection and ranking. Bioinformatics 2014, 30, 801–807. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Freudenberg, J.M.; Joshi, V.K.; Hu, Z.; Medvedovic, M. CLEAN: CLustering enrichment ANalysis. BMC Bioinform. 2009, 10, 234. [Google Scholar] [CrossRef] [Green Version]
- Sartor, M.A.; Leikauf, G.D.; Medvedovic, M. LRpath: A logistic regression approach for identifying enriched biological groups in gene expression data. Bioinformatics 2009, 25, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Kosmidis, I.; Pagui, E.C.K.; Sartori, N. Mean and median bias reduction in generalized linear models. Stat. Comput. 2020, 30, 43–59. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al Mahi, N.; Zhang, E.Y.; Sherman, S.; Yu, J.J.; Medvedovic, M. Connectivity Map Analysis of a Single-Cell RNA-Sequencing -Derived Transcriptional Signature of mTOR Signaling. Int. J. Mol. Sci. 2021, 22, 4371. https://doi.org/10.3390/ijms22094371
Al Mahi N, Zhang EY, Sherman S, Yu JJ, Medvedovic M. Connectivity Map Analysis of a Single-Cell RNA-Sequencing -Derived Transcriptional Signature of mTOR Signaling. International Journal of Molecular Sciences. 2021; 22(9):4371. https://doi.org/10.3390/ijms22094371
Chicago/Turabian StyleAl Mahi, Naim, Erik Y. Zhang, Susan Sherman, Jane J. Yu, and Mario Medvedovic. 2021. "Connectivity Map Analysis of a Single-Cell RNA-Sequencing -Derived Transcriptional Signature of mTOR Signaling" International Journal of Molecular Sciences 22, no. 9: 4371. https://doi.org/10.3390/ijms22094371
APA StyleAl Mahi, N., Zhang, E. Y., Sherman, S., Yu, J. J., & Medvedovic, M. (2021). Connectivity Map Analysis of a Single-Cell RNA-Sequencing -Derived Transcriptional Signature of mTOR Signaling. International Journal of Molecular Sciences, 22(9), 4371. https://doi.org/10.3390/ijms22094371