
Single Cell Gene Expression Analysis in a 3D Microtissue Liver Model Reveals Cell Type-Specific Responses to Pro-Fibrotic TGF-β1 Stimulation

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
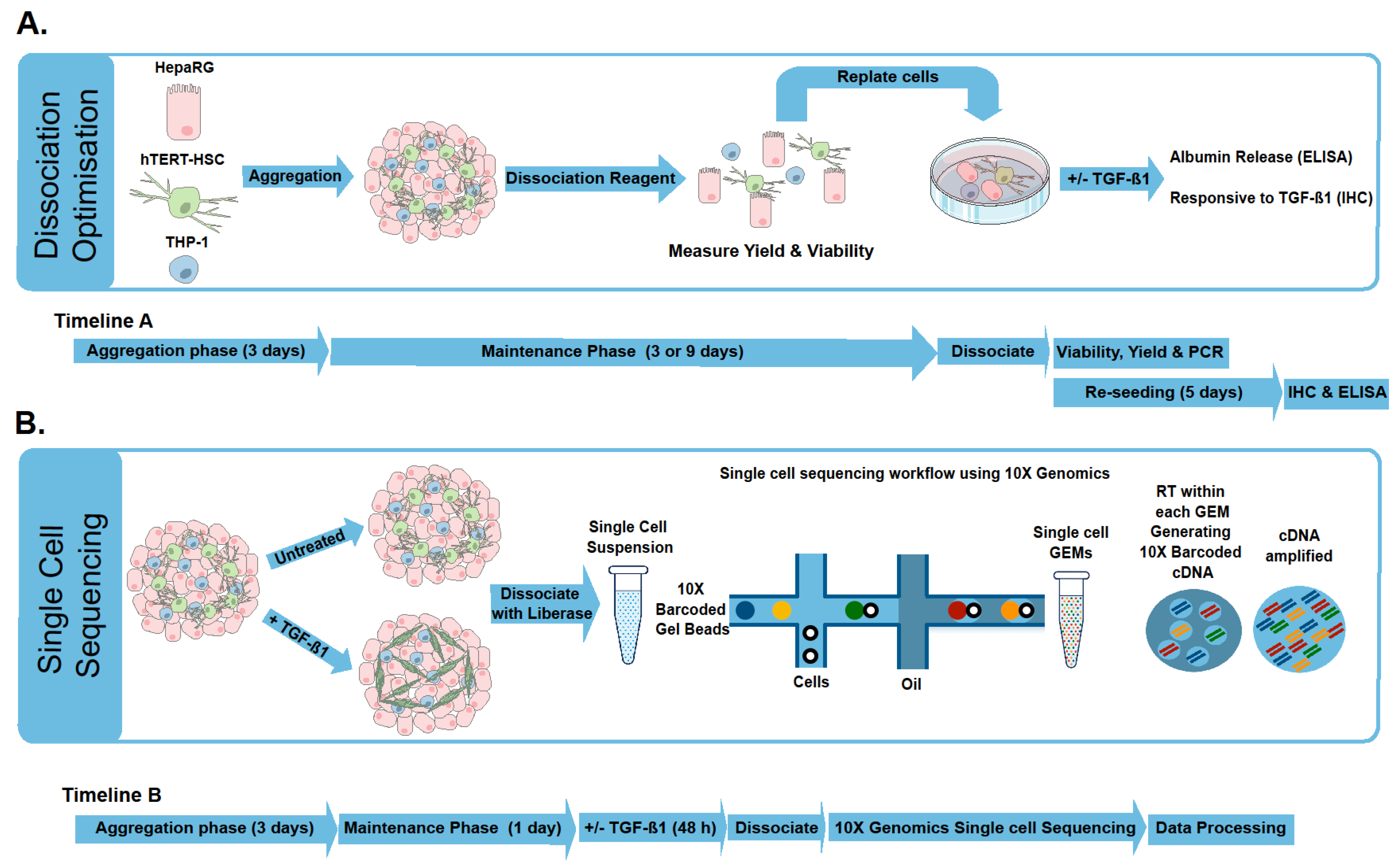
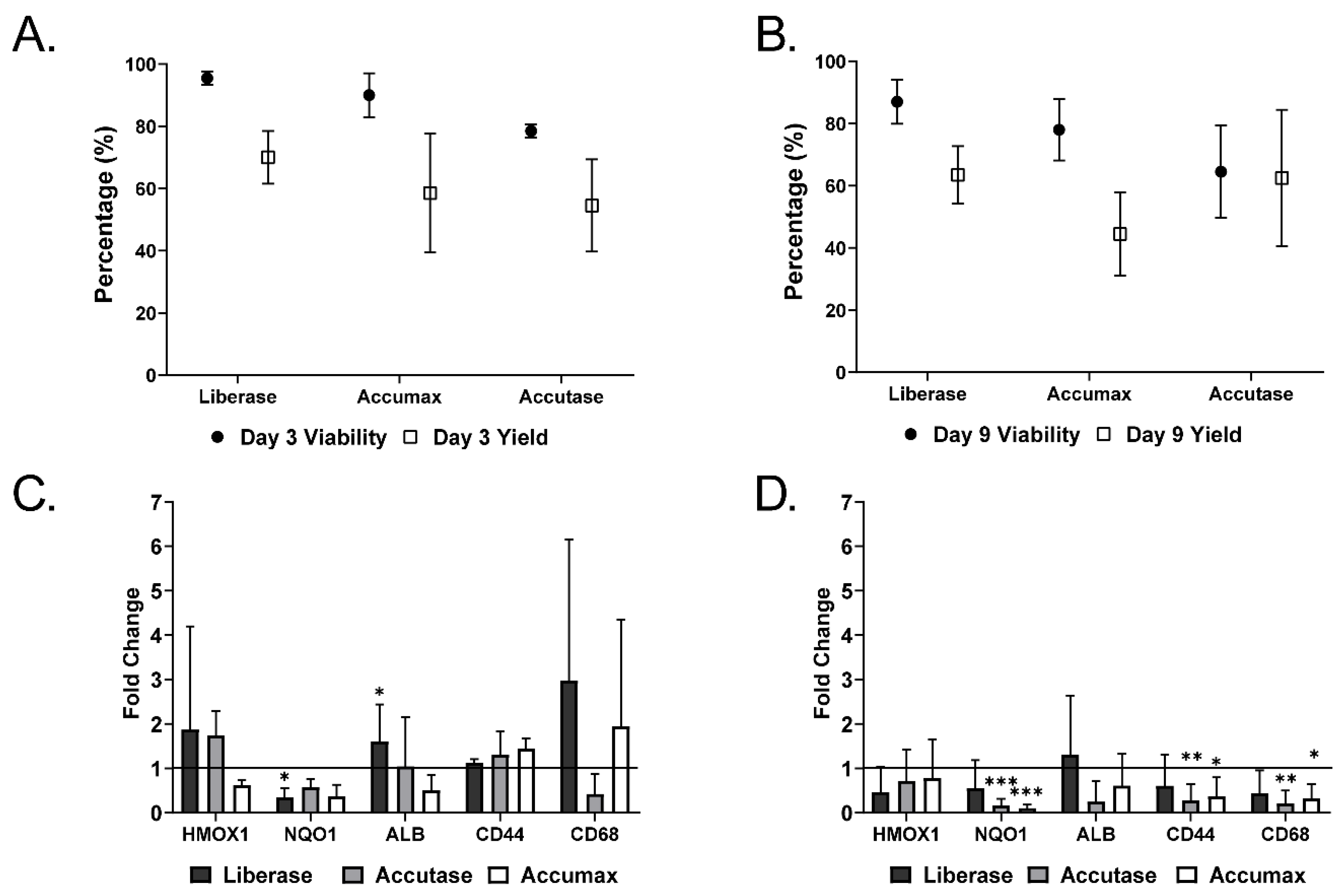
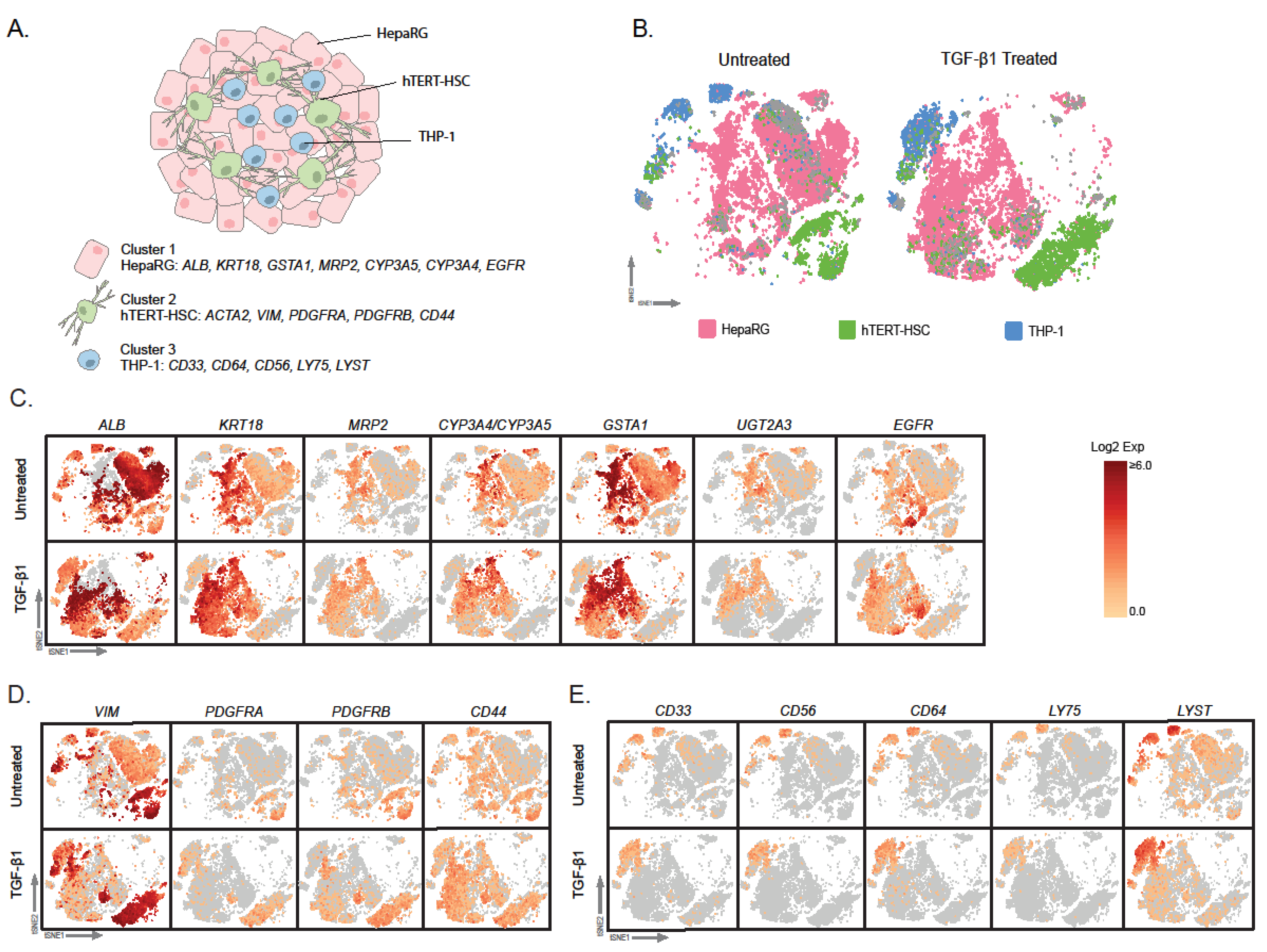
2.1. Microtissue Dissociation
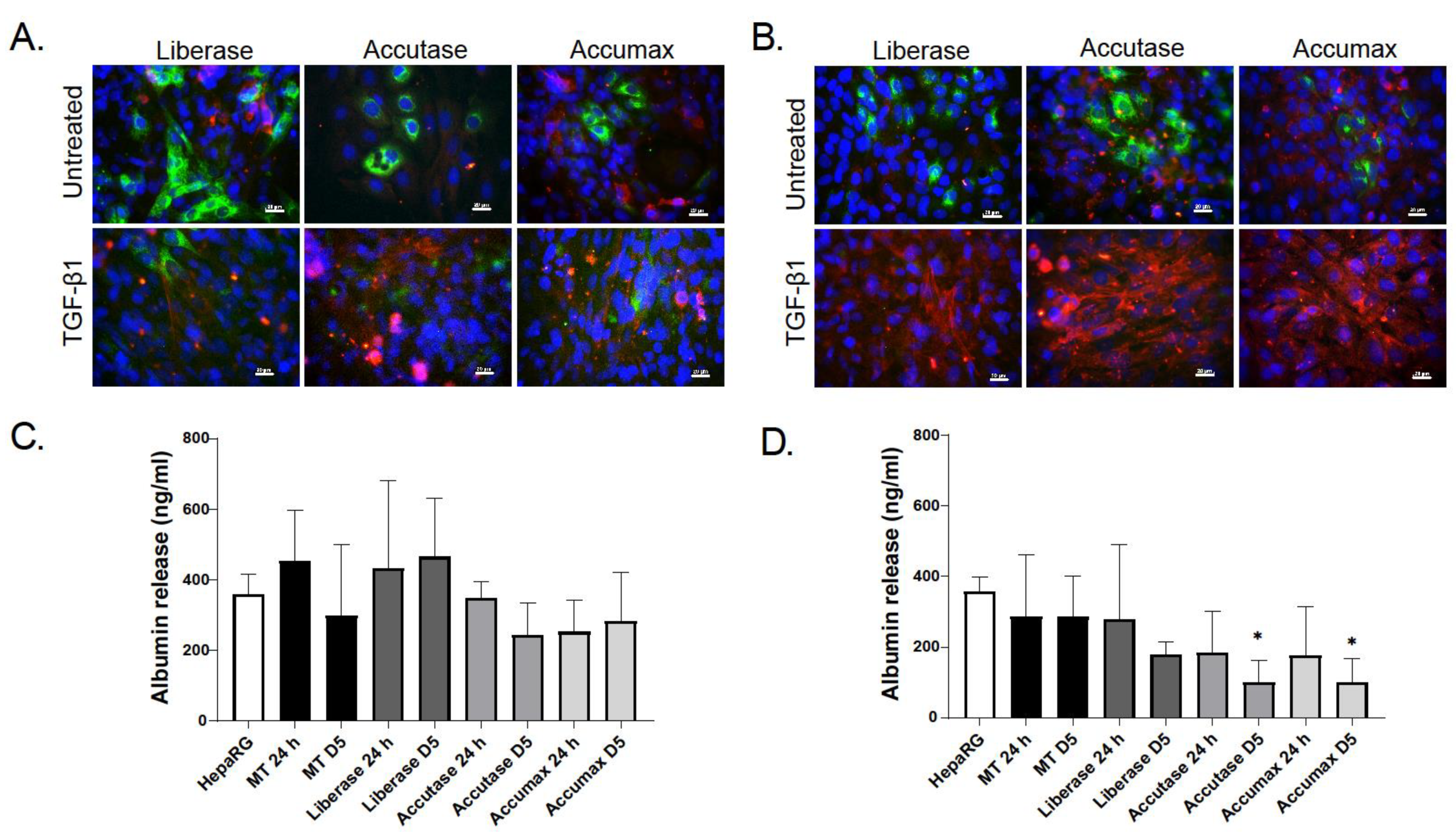
2.2. Characterization of Dissociated Cells
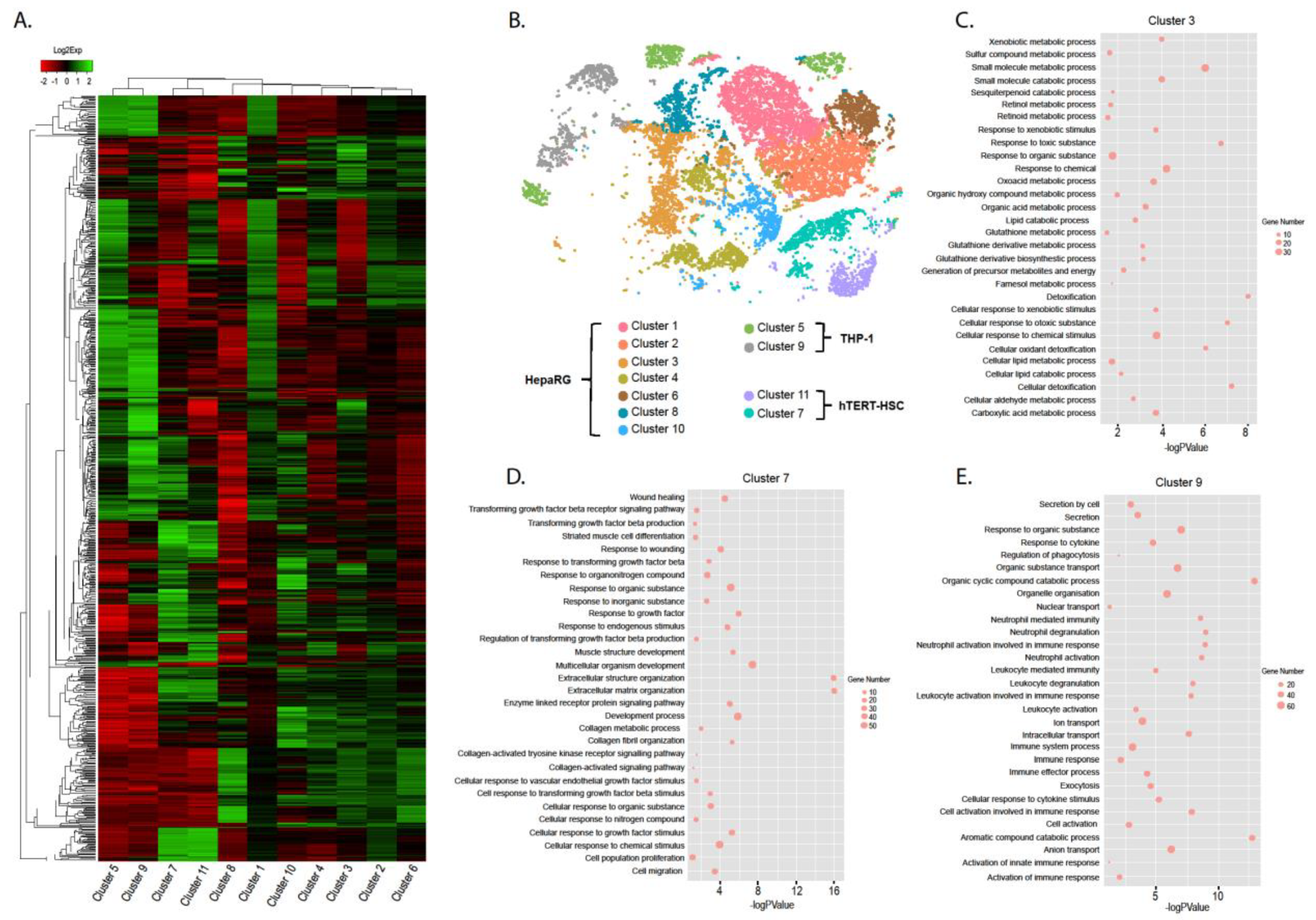
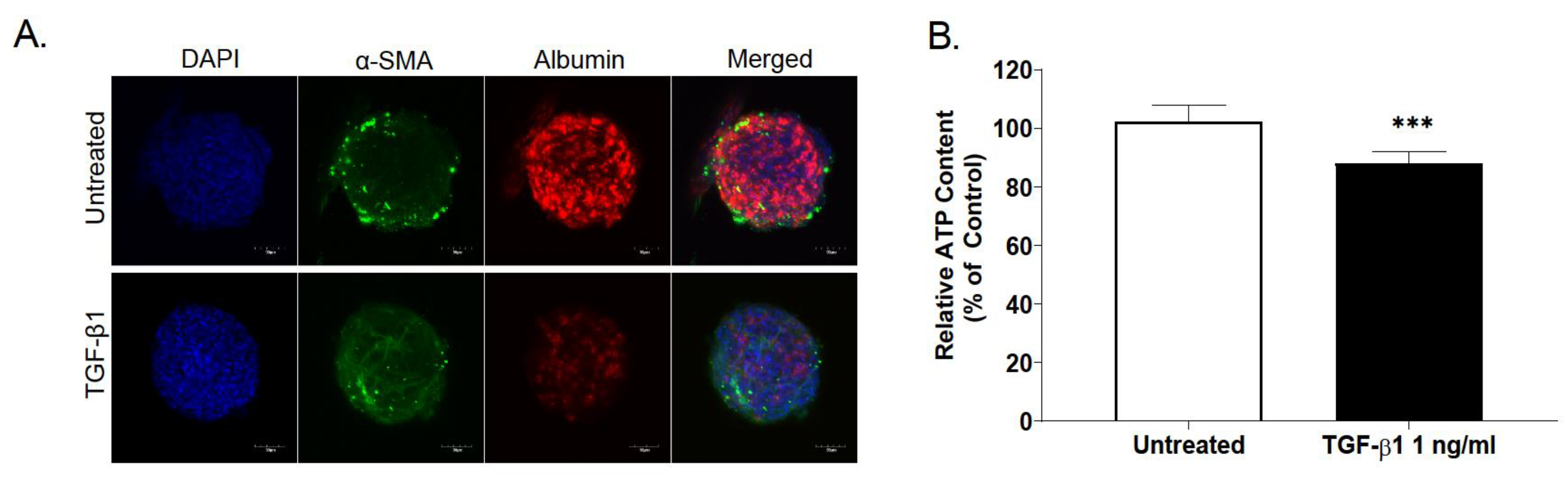
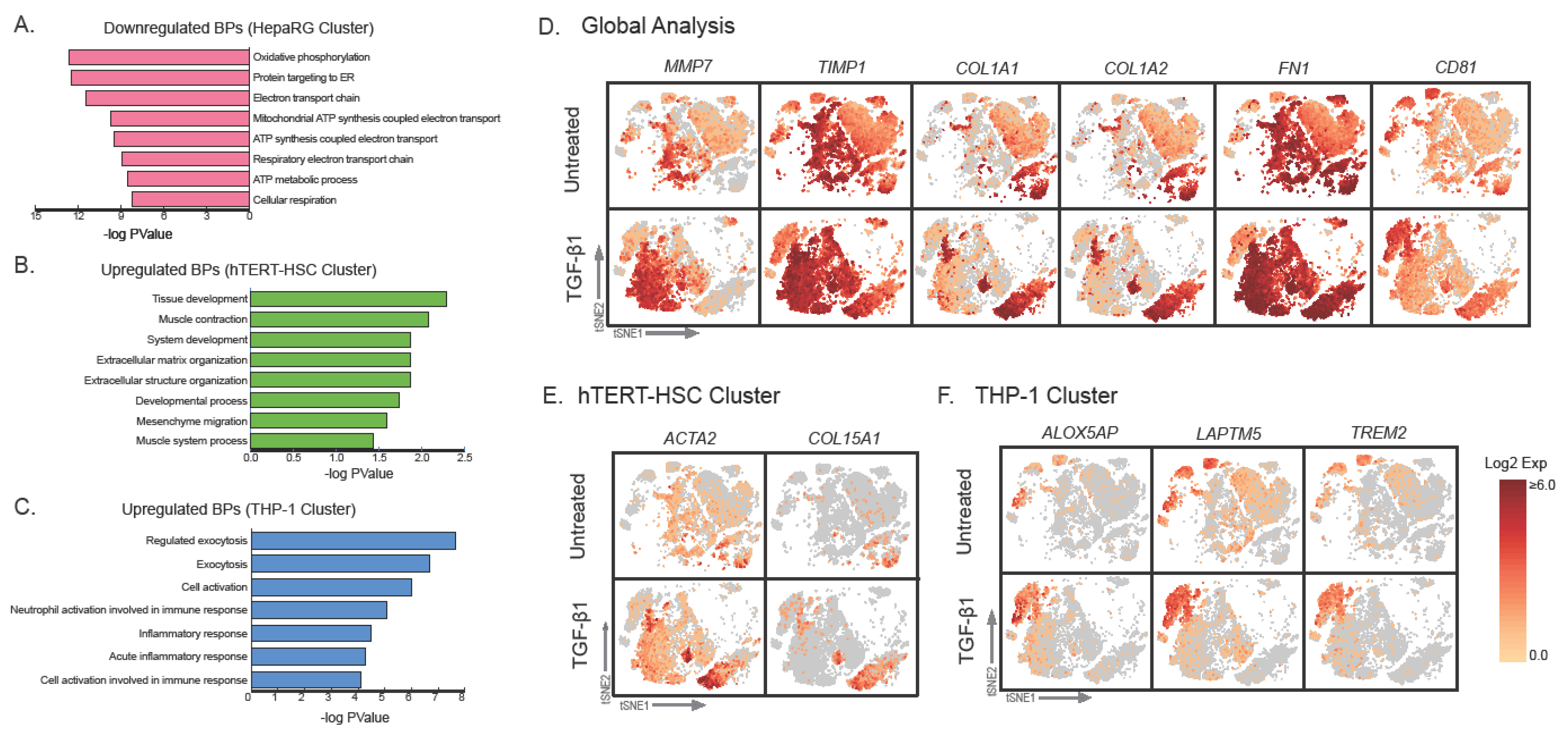
2.3. Effect of TGF-β Treatment on Multicellular MTs
3. Materials and Methods
3.1. Generation of MT
3.2. Viability Assay
3.3. Immunostaining
3.4. Quantitative Real Time Polymerase Chain Reaction
3.5. Albumin Enzyme-Linked Immunosorbent Assay
3.6. Dissociation of the Microtissues
3.7. scRNA-Seq
3.8. Sequencing
3.9. Data Analysis Using Cell Ranger Loupe Browser
3.10. Gene-Based Identification of Specific Cell Clusters
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lauschke, V.M.; Shafagh, R.Z.; Hendriks, D.F.G.; Ingelman-Sundberg, M. 3D Primary Hepatocyte Culture Systems for Analyses of Liver Diseases, Drug Metabolism, and Toxicity: Emerging Culture Paradigms and Applications. Biotechnol. J. 2019, 14, 1800347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, R.; Schrooders, Y.; Hauser, D.; van Herwijnen, M.; Albrecht, W.; ter Braak, B.; Brecklinghaus, T.; Castell, J.V.; Elenschneider, L.; Escher, S.; et al. Comparing in Vitro Human Liver Models to in Vivo Human Liver Using RNA-Seq. Arch. Toxicol. 2021, 95, 573–589. [Google Scholar] [CrossRef] [PubMed]
- Hart, S.N.; Li, Y.; Nakamoto, K.; Subileau, E.; Steen, D.; Zhong, X. A Comparison of Whole Genome Gene Expression Profiles of HepaRG Cells and HepG2 Cells to Primary Human Hepatocytes and Human Liver Tissues. Drug Metab. Dispos. 2010, 38, 988–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunness, P.; Mueller, D.; Shevchenko, V.; Heinzle, E.; Ingelman-Sundberg, M.; Noor, F. 3D Organotypic Cultures of Human HepaRG Cells: A Tool for In Vitro Toxicity Studies. Toxicol. Sci. 2013, 133, 67–78. [Google Scholar] [CrossRef]
- Horvat, T.; Landesmann, B.; Lostia, A.; Vinken, M.; Munn, S.; Whelan, M. Adverse Outcome Pathway Development from Protein Alkylation to Liver Fibrosis. Arch. Toxicol. 2017, 91, 1523–1543. [Google Scholar] [CrossRef] [Green Version]
- Coll, M.; Perea, L.; Boon, R.; Leite, S.B.; Vallverdú, J.; Mannaerts, I.; Smout, A.; El Taghdouini, A.; Blaya, D.; Rodrigo-Torres, D.; et al. Generation of Hepatic Stellate Cells from Human Pluripotent Stem Cells Enables In Vitro Modeling of Liver Fibrosis. Cell Stem Cell 2018, 23, 101–113.e7. [Google Scholar] [CrossRef] [Green Version]
- Leite, S.B.; Roosens, T.; El Taghdouini, A.; Mannaerts, I.; Smout, A.J.; Najimi, M.; Sokal, E.; Noor, F.; Chesne, C.; van Grunsven, L.A. Novel Human Hepatic Organoid Model Enables Testing of Drug-Induced Liver Fibrosis in Vitro. Biomaterials 2016, 78, 1–10. [Google Scholar] [CrossRef]
- Prestigiacomo, V.; Weston, A.; Messner, S.; Lampart, F.; Suter-Dick, L. Pro-Fibrotic Compounds Induce Stellate Cell Activation, ECM-Remodelling and Nrf2 Activation in a Human 3D-Multicellular Model of Liver Fibrosis. PLoS ONE 2017, 12, e0179995. [Google Scholar] [CrossRef]
- Dewidar, B.; Meyer, C.; Dooley, S.; Meindl-Beinker, N. TGF-β in Hepatic Stellate Cell Activation and Liver Fibrogenesis—Updated. Cells 2019, 8, 1419. [Google Scholar] [CrossRef] [Green Version]
- Saile, B.; Matthes, N.; Knittel, T.; Ramadori, G. Transforming Growth Factor β and Tumor Necrosis Factor α Inhibit Both Apoptosis and Proliferation of Activated Rat Hepatic Stellate Cells. Hepatology 1999, 30, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, K.; Lindhorst, A.; Tron, K.; Ramadori, G.; Saile, B. Decrease of PECAM-1-Gene-Expression Induced by Proinflammatory Cytokines IFN-γ and IFN-α Is Reversed by TGF-β in Sinusoidal Endothelial Cells and Hepatic Mononuclear Phagocytes. BMC Physiol. 2008, 8, 9. [Google Scholar] [CrossRef] [Green Version]
- Messner, C.J.; Mauch, L.; Suter-Dick, L. Bile Salts Regulate CYP7A1 Expression and Elicit a Fibrotic Response and Abnormal Lipid Production in 3D Liver Microtissues. Toxicol. In Vitro 2019, 60, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Messner, C.J.; Zhang, X.; Suter-Dick, L. Assessment of Fibrotic Pathways Induced by Environmental Chemicals Using 3D-Human Liver Microtissue Model. Environ. Res. 2021, 194, 110679. [Google Scholar] [CrossRef] [PubMed]
- MacParland, S.A.; Liu, J.C.; Ma, X.-Z.; Innes, B.T.; Bartczak, A.M.; Gage, B.K.; Manuel, J.; Khuu, N.; Echeverri, J.; Linares, I.; et al. Single Cell RNA Sequencing of Human Liver Reveals Distinct Intrahepatic Macrophage Populations. Nat. Commun. 2018, 9, 4383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krenkel, O.; Hundertmark, J.; Ritz, T.P.; Weiskirchen, R.; Tacke, F. Single Cell RNA Sequencing Identifies Subsets of Hepatic Stellate Cells and Myofibroblasts in Liver Fibrosis. Cells 2019, 8, 503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waise, S.; Parker, R.; Rose-Zerilli, M.J.J.; Layfield, D.M.; Wood, O.; West, J.; Ottensmeier, C.H.; Thomas, G.J.; Hanley, C.J. An Optimised Tissue Disaggregation and Data Processing Pipeline for Characterising Fibroblast Phenotypes Using Single-Cell RNA Sequencing. Sci. Rep. 2019, 9, 9580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vos, J.A.; Simurdak, J.H.; Davis, B.J.; Myers, J.B.; Brissette, M.D. Vortex Disaggregation for Flow Cytometry Allows Direct Histologic Correlation: A Novel Approach for Small Biopsies and Inaspirable Bone Marrows. Cytom. B Clin. Cytom. 2003, 52, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Brockhoff, G.; Fleischmann, S.; Meier, A.; Wachs, F.-P.; Hofstaedter, F.; Knuechel, R. Use of a Mechanical Dissociation Device to Improve Standardization of Flow Cytometric Cytokeratin DNA Measurements of Colon Carcinomas. Cytometry 1999, 38, 184–191. [Google Scholar] [CrossRef]
- Collins, S.D.; Yuen, G.; Tu, T.; Budzinska, M.A.; Spring, K.; Bryant, K.; Shackel, N.A. In Vitro Models of the Liver: Disease Modeling, Drug Discovery and Clinical Applications. In Hepatocellular Carcinoma; Tirnitz-Parker, J.E.E., Ed.; Codon Publications: Brisbane, Australia, 2019; Volume 47, ISBN 978-0-9944381-8-8. [Google Scholar]
- Deng, Z.; Wang, X.; Liu, Y.; Tian, X.; Deng, S.; Sun, Y.; Wang, S.; Zheng, D.; Cui, Z.; Pan, Y.; et al. Single-Cell RNA Sequencing Confirms IgG Transcription and Limited Diversity of V H DJ H Rearrangements in Proximal Tubular Epithelial Cells. Sci. Rep. 2020, 10, 19657. [Google Scholar] [CrossRef]
- Ferrall-Fairbanks, M.C.; Ball, M.; Padron, E.; Altrock, P.M. Leveraging Single-Cell RNA Sequencing Experiments to Model Intratumor Heterogeneity. JCO Clin. Cancer Inform. 2018, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, P.A.; Smiljanic, K.; Maso Prévide, R.; Iben, J.R.; Li, T.; Rokic, M.B.; Sherman, A.; Coon, S.L.; Stojilkovic, S.S. Cell Type- and Sex-Dependent Transcriptome Profiles of Rat Anterior Pituitary Cells. Front. Endocrinol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Ravi, M.; Paramesh, V.; Kaviya, S.R.; Anuradha, E.; Solomon, F.D.P. 3D Cell Culture Systems: Advantages and Applications. J. Cell Physiol. 2015, 230, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Casuriaga, R.; Geisinger, A.; López-Carro, B.; Porro, V.; Wettstein, R.; Folle, G.A. Ultra-Fast and Optimized Method for the Preparation of Rodent Testicular Cells for Flow Cytometric Analysis. Biol. Proced Online 2009, 11, 184–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, C.C.; Hendriks, D.F.G.; Moro, S.M.L.; Ellis, E.; Walsh, J.; Renblom, A.; Fredriksson Puigvert, L.; Dankers, A.C.A.; Jacobs, F.; Snoeys, J.; et al. Characterization of Primary Human Hepatocyte Spheroids as a Model System for Drug-Induced Liver Injury, Liver Function and Disease. Sci. Rep. 2016, 6, 25187. [Google Scholar] [CrossRef] [Green Version]
- Gaskell, H.; Sharma, P.; Colley, H.E.; Murdoch, C.; P. Williams, D.; D. Webb, S. Characterization of a Functional C3A Liver Spheroid Model. Toxicol. Res. 2016, 5, 1053–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilz, M.; Kwapiszewska, K.; Kalwarczyk, T.; Bubak, G.; Nowis, D.; Hołyst, R. Transport of Nanoprobes in Multicellular Spheroids. Nanoscale 2020, 12, 19880–19887. [Google Scholar] [CrossRef]
- Pennock, R.; Bray, E.; Pryor, P.; James, S.; McKeegan, P.; Sturmey, R.; Genever, P. Human Cell Dedifferentiation in Mesenchymal Condensates through Controlled Autophagy. Sci. Rep. 2015, 5, 13113. [Google Scholar] [CrossRef] [Green Version]
- Messner, C.J.; Premand, C.; Gaiser, C.; Kluser, T.; Kübler, E.; Suter-Dick, L. Exosomal MicroRNAs Release as a Sensitive Marker for Drug-Induced Liver Injury In Vitro. Appl. In Vitro Toxicol. 2020, 6, 77–89. [Google Scholar] [CrossRef]
- Sun, L.; Yin, H.; Liu, M.; Xu, G.; Zhou, X.; Ge, P.; Yang, H.; Mao, Y. Impaired Albumin Function: A Novel Potential Indicator for Liver Function Damage? Ann. Med. 2019, 51, 333–344. [Google Scholar] [CrossRef]
- Tsuchida, T.; Friedman, S.L. Mechanisms of Hepatic Stellate Cell Activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef]
- Trefts, E.; Gannon, M.; Wasserman, D.H. The Liver. Curr. Biol. 2017, 27, R1147–R1151. [Google Scholar] [CrossRef] [PubMed]
- Gebhardt, R.; Matz-Soja, M. Liver Zonation: Novel Aspects of Its Regulation and Its Impact on Homeostasis. World J. Gastroenterol. 2014, 20, 8491–8504. [Google Scholar] [CrossRef] [PubMed]
- Distler, M.G.; Palmer, A.A. Role of Glyoxalase 1 (Glo1) and Methylglyoxal (MG) in Behavior: Recent Advances and Mechanistic Insights. Front. Genet. 2012, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Droin, C.; Kholtei, J.E.; Bahar Halpern, K.; Hurni, C.; Rozenberg, M.; Muvkadi, S.; Itzkovitz, S.; Naef, F. Space-Time Logic of Liver Gene Expression at Sub-Lobular Scale. Nat. Metab. 2021, 3, 43–58. [Google Scholar] [CrossRef]
- Ju, C.; Colgan, S.P.; Eltzschig, H.K. Hypoxia-Inducible Factors as Molecular Targets for Liver Diseases. J. Mol. Med. 2016, 94, 613–627. [Google Scholar] [CrossRef] [Green Version]
- VandeKopple, M.J.; Wu, J.; Auer, E.N.; Giaccia, A.J.; Denko, N.C.; Papandreou, I. HILPDA Regulates Lipid Metabolism, Lipid Droplet Abundance, and Response to Microenvironmental Stress in Solid Tumors. Mol. Cancer Res. 2019, 17, 2089–2101. [Google Scholar] [CrossRef] [Green Version]
- Kietzmann, T. Metabolic Zonation of the Liver: The Oxygen Gradient Revisited. Redox. Biol. 2017, 11, 622–630. [Google Scholar] [CrossRef]
- McEnerney, L.; Duncan, K.; Bang, B.-R.; Elmasry, S.; Li, M.; Miki, T.; Ramakrishnan, S.K.; Shah, Y.M.; Saito, T. Dual Modulation of Human Hepatic Zonation via Canonical and Non-Canonical Wnt Pathways. Exp. Mol. Med. 2017, 49, e413. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.; Moreno-Navarrete, J.M.; Wei, X.; Kikukawa, Y.; Tzameli, I.; Prasad, D.; Lee, Y.; Asara, J.M.; Fernandez-Real, J.M.; Maratos-Flier, E.; et al. Nicotinamide N-Methyltransferase Regulates Hepatic Nutrient Metabolism through Sirt1 Protein Stabilization. Nat. Med. 2015, 21, 887–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, H.; Zhang, S.; Sun, X.; Liu, J.; Wu, Y.; Guo, W.; Wang, F.; Ou, X.; Cong, M.; Jin, E.; et al. Distinct Iron Deposition Profiles of Liver Zones in Various Models with Iron Homeostasis Disorders. Adv. Sci. 2018, 5. [Google Scholar] [CrossRef]
- Krähenbühl, S.; Stucki, J.; Reichen, J. Reduced Activity of the Electron Transport Chain in Liver Mitochondria Isolated from Rats with Secondary Biliary Cirrhosis. Hepatology 1992, 15, 1160–1166. [Google Scholar] [CrossRef]
- Mejias, M.; Gallego, J.; Naranjo-Suarez, S.; Ramirez, M.; Pell, N.; Manzano, A.; Suñer, C.; Bartrons, R.; Mendez, R.; Fernandez, M. CPEB4 Increases Expression of PFKFB3 to Induce Glycolysis and Activate Mouse and Human Hepatic Stellate Cells, Promoting Liver Fibrosis. Gastroenterology 2020, 159, 273–288. [Google Scholar] [CrossRef]
- Lemoinne, S.; Thabut, D.; Housset, C. Portal Myofibroblasts Connect Angiogenesis and Fibrosis in Liver. Cell Tissue Res. 2016, 365, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Amenta, P.S.; Scivoletti, N.A.; Newman, M.D.; Sciancalepore, J.P.; Li, D.; Myers, J.C. Proteoglycan-Collagen XV in Human Tissues Is Seen Linking Banded Collagen Fibers Subjacent to the Basement Membrane. J. Histochem. Cytochem. 2005, 53, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, P.; Dobie, R.; Wilson-Kanamori, J.R.; Dora, E.F.; Henderson, B.E.P.; Luu, N.T.; Portman, J.R.; Matchett, K.P.; Brice, M.; Marwick, J.A.; et al. Resolving the Fibrotic Niche of Human Liver Cirrhosis at Single-Cell Level. Nature 2019, 575, 512–518. [Google Scholar] [CrossRef]
- Titos, E.; Clària, J.; Planagumà, A.; López-Parra, M.; González-Périz, A.; Gaya, J.; Miquel, R.; Arroyo, V.; Rodés, J. Inhibition of 5-Lipoxygenase-Activating Protein Abrogates Experimental Liver Injury: Role of Kupffer Cells. J. Leukoc. Biol. 2005, 78, 871–878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahara, Y.; Takahashi, M.; Zhang, Q.-W.; Wagatsuma, H.; Mori, M.; Tamori, A.; Shiomi, S.; Nishiguchi, S. Serial Changes in Expression of Functionally Clustered Genes in Progression of Liver Fibrosis in Hepatitis C Patients. World J. Gastroenterol. 2008, 14, 2010–2022. [Google Scholar] [CrossRef] [PubMed]
- Rådmark, O.; Samuelsson, B. 5-Lipoxygenase: Mechanisms of Regulation. J. Lipid Res. 2009, 50, S40–S45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, C.; Feng, W.; Wei, S.; Wang, Y.; Xu, X.; Wei, J.; Ma, Z.; Du, Y.; Guo, J.; He, Y.; et al. Bioinformatics Analysis of Key Differentially Expressed Genes in Nonalcoholic Fatty Liver Disease Mice Models. Gene Expr. 2018, 19, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Baglama, J.; Reichel, L. Augmented Implicitly Restarted Lanczos Bidiagonalization Methods. SIAM J. Sci. Comput. 2005, 27, 19–42. [Google Scholar] [CrossRef] [Green Version]
- Yu, D.; Huber, W.; Vitek, O. Shrinkage Estimation of Dispersion in Negative Binomial Models for RNA-Seq Experiments with Small Sample Size. Bioinformatics 2013, 29, 1275–1282. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; Smyth, G.K. Small-Sample Estimation of Negative Binomial Dispersion, with Applications to SAGE Data. Biostatistics 2008, 9, 321–332. [Google Scholar] [CrossRef] [Green Version]
- Török, G.; Erdei, Z.; Lilienberg, J.; Apáti, Á.; Homolya, L. The Importance of Transporters and Cell Polarization for the Evaluation of Human Stem Cell-Derived Hepatic Cells. PLoS ONE 2020, 15, e0227751. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Hepatic Stellate Cells: Protean, Multifunctional, and Enigmatic Cells of the Liver. Physiol. Rev. 2008, 88, 125–172. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.-Z.; Chen, Q.; Zhang, W.-Y.; Zhang, H.-H.; Ma, Y.; Zhang, S.-Z.; Fang, J.; Yu, C.-H. PDGF Signaling Pathway in Hepatic Fibrosis Pathogenesis and Therapeutics. Mol. Med. Rep. 2017, 16, 7879–7889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forrester, M.A.; Wassall, H.J.; Hall, L.S.; Cao, H.; Wilson, H.M.; Barker, R.N.; Vickers, M.A. Similarities and Differences in Surface Receptor Expression by THP-1 Monocytes and Differentiated Macrophages Polarized Using Seven Different Conditioning Regimens. Cell. Immunol. 2018, 332, 58–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klijn, C.; Durinck, S.; Stawiski, E.W.; Haverty, P.M.; Jiang, Z.; Liu, H.; Degenhardt, J.; Mayba, O.; Gnad, F.; Liu, J.; et al. A Comprehensive Transcriptional Portrait of Human Cancer Cell Lines. Nat. Biotechnol. 2015, 33, 306–312. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein of Interest | Primary Antibody | Secondary Antibody |
|---|---|---|
| Albumin | Monoclonal rabbit anti-albumin (EPR20195) 1:700 dilution (abcam, ab207327) | F(ab’)2-Goat anti-rabbit IgG (H + L) cross-adsorbed secondary antibody, AF488 conjugated, diluted 1:1000 (Invitrogen, A11070) |
| α-Smooth muscle actin (α-SMA) | Monoclonal mouse anti-α-SMA, 1:300 (Sigma, A5228) | F(ab’)2-Goat anti-mouse IgG (H + L) cross-adsorbed secondary antibody, AF546 conjugated, diluted 1:1000 (Invitrogen, A11018) |
| Marker of Interest | Abbreviation | Catalogue Number |
|---|---|---|
| Glyceraldehyde 3-phosphate dehydrogenase | GAPDH | Hs02758991_g1 |
| Vimentin | VIM | Hs00958111_m1 |
| CD44 | CD44 | Hs01075861_m1 |
| NAD(P)H dehydrogenase (quinone) 1 | NQO1 | Hs02512143_s1 |
| Heme Oxygenase 1 | HMOX1 | Hs01110250_m1 |
| Albumin | ALB | Hs00609403_m1 |
| CD68 | CD68 | Hs02836816_g1 |
| Reagent | Enzyme Concentration or Quantity | Dissociation Time Day 3 | Dissociation Time Day 9 | Shaken by Hand |
|---|---|---|---|---|
| Liberase | 1.3 U/mL | 40 min | 60 min | 10× every 20 min |
| Accumax | 900 µL | 50 min | 60 min | 10× every 20 min |
| Accutase | 900 µL | 60 min | 60 min | 10× every 20 min |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Messner, C.J.; Babrak, L.; Titolo, G.; Caj, M.; Miho, E.; Suter-Dick, L. Single Cell Gene Expression Analysis in a 3D Microtissue Liver Model Reveals Cell Type-Specific Responses to Pro-Fibrotic TGF-β1 Stimulation. Int. J. Mol. Sci. 2021, 22, 4372. https://doi.org/10.3390/ijms22094372
Messner CJ, Babrak L, Titolo G, Caj M, Miho E, Suter-Dick L. Single Cell Gene Expression Analysis in a 3D Microtissue Liver Model Reveals Cell Type-Specific Responses to Pro-Fibrotic TGF-β1 Stimulation. International Journal of Molecular Sciences. 2021; 22(9):4372. https://doi.org/10.3390/ijms22094372
Chicago/Turabian StyleMessner, Catherine Jane, Lmar Babrak, Gaia Titolo, Michaela Caj, Enkelejda Miho, and Laura Suter-Dick. 2021. "Single Cell Gene Expression Analysis in a 3D Microtissue Liver Model Reveals Cell Type-Specific Responses to Pro-Fibrotic TGF-β1 Stimulation" International Journal of Molecular Sciences 22, no. 9: 4372. https://doi.org/10.3390/ijms22094372
APA StyleMessner, C. J., Babrak, L., Titolo, G., Caj, M., Miho, E., & Suter-Dick, L. (2021). Single Cell Gene Expression Analysis in a 3D Microtissue Liver Model Reveals Cell Type-Specific Responses to Pro-Fibrotic TGF-β1 Stimulation. International Journal of Molecular Sciences, 22(9), 4372. https://doi.org/10.3390/ijms22094372