
iPSCs: A Preclinical Drug Research Tool for Neurological Disorders

 ,
,  ,
, 
Abstract
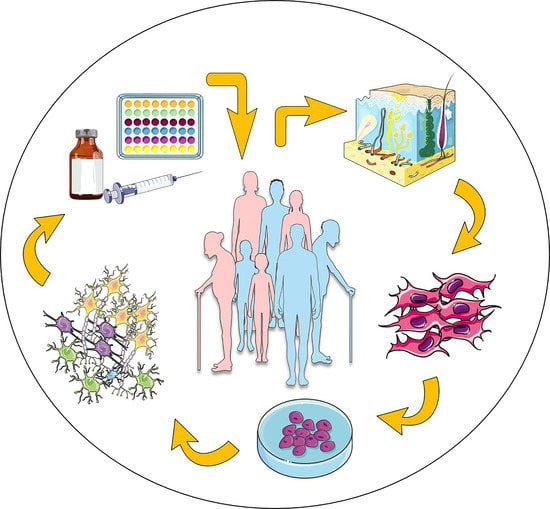
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. iPSC-Based Drug Testing for Alzheimer Disease
3. iPSC-Based Drug Testing for Amyotrophic Lateral Sclerosis
4. iPSC-Based Drug Testing for Parkinson’s Disease
5. iPSC-Based Drug Testing for Fragile X Syndrome
6. Conclusions: Challenges and Perspectives for iPSC Use in Drug Screening
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mager, D.E.; Woo, S.; Jusko, W.J. Scaling pharmacodynamics from in vitro and preclinical animal studies to humans. Drug Metab. Pharmacokinet. 2009, 24, 16–24. [Google Scholar] [CrossRef] [Green Version]
- Zang, R.; Li, D.; Tang, I.-C.; Wang, J.; Yang, S.-T. Cell-based assays in high-throughput screening for drug discovery. Int. J. Biotechnol. Wellness Ind. 2012, 1, 31–51. [Google Scholar]
- Ransohoff, R.M. All (animal) models (of neurodegeneration) are wrong. Are they also useful? J. Exp. Med. 2018, 215, 2955–2958. [Google Scholar] [CrossRef] [Green Version]
- Elitt, M.S.; Barbar, L.; Tesar, P.J. Drug screening for human genetic diseases using iPSC models. Hum. Mol. Genet. 2018, 27, R89–R98. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Seki, T.; Fukuda, K. Methods of induced pluripotent stem cells for clinical application. World J. Stem Cells 2015, 7, 116–125. [Google Scholar] [CrossRef]
- Liu, G.; David, B.T.; Trawczynski, M.; Fessler, R.G. Advances in pluripotent stem cells: History, mechanisms, technologies, and applications. Stem Cell Rev. Rep. 2020, 16, 3–32. [Google Scholar] [CrossRef] [Green Version]
- Ferri, C.P.; Prince, M.; Brayne, C.; Brodaty, H.; Fratiglioni, L.; Ganguli, M.; Hall, K.; Hasegawa, K.; Hendrie, H.; Huang, Y. Global prevalence of dementia: A Delphi consensus study. Lancet 2005, 366, 2112–2117. [Google Scholar] [CrossRef]
- Hyman, B.T.; Van Hoesen, G.W.; Damasio, A.R.; Barnes, C.L. Alzheimer’s disease: Cell-specific pathology isolates the hippocampal formation. Science 1984, 225, 1168–1170. [Google Scholar] [CrossRef] [PubMed]
- Tanzi, R.E. The genetics of Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006296. [Google Scholar] [CrossRef] [PubMed]
- Lanoiselée, H.-M.; Nicolas, G.; Wallon, D.; Rovelet-Lecrux, A.; Lacour, M.; Rousseau, S.; Richard, A.-C.; Pasquier, F.; Rollin-Sillaire, A.; Martinaud, O. APP, PSEN1, and PSEN2 mutations in early-onset Alzheimer disease: A genetic screening study of familial and sporadic cases. PLoS Med. 2017, 14, e1002270. [Google Scholar] [CrossRef] [Green Version]
- Bekris, L.M.; Yu, C.-E.; Bird, T.D.; Tsuang, D.W. Genetics of Alzheimer disease. J. Geriatr. Psychiatry Neurol. 2010, 23, 213–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satoh, K.; Abe-Dohmae, S.; Yokoyama, S.; St George-Hyslop, P.; Fraser, P.E. ATP-binding cassette transporter A7 (ABCA7) loss of function alters Alzheimer amyloid processing. J. Biol. Chem. 2015, 290, 24152–24165. [Google Scholar] [CrossRef] [Green Version]
- Bettens, K.; Vermeulen, S.; Van Cauwenberghe, C.; Heeman, B.; Asselbergh, B.; Robberecht, C.; Engelborghs, S.; Vandenbulcke, M.; Vandenberghe, R.; De Deyn, P.P. Reduced secreted clusterin as a mechanism for Alzheimer-associated CLU mutations. Mol. Neurodegener. 2015, 10, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, J.-C.; Heath, S.; Even, G.; Campion, D.; Sleegers, K.; Hiltunen, M.; Combarros, O.; Zelenika, D.; Bullido, M.J.; Tavernier, B. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1094–1099. [Google Scholar] [CrossRef]
- Vardarajan, B.N.; Ghani, M.; Kahn, A.; Sheikh, S.; Sato, C.; Barral, S.; Lee, J.H.; Cheng, R.; Reitz, C.; Lantigua, R. Rare coding mutations identified by sequencing of A lzheimer disease genome-wide association studies loci. Ann. Neurol. 2015, 78, 487–498. [Google Scholar] [CrossRef]
- Patel, D.; Mez, J.; Vardarajan, B.N.; Staley, L.; Chung, J.; Zhang, X.; Farrell, J.J.; Rynkiewicz, M.J.; Cannon-Albright, L.A.; Teerlink, C.C. Association of rare coding mutations with Alzheimer disease and other dementias among adults of European ancestry. JAMA Netw. Open 2019, 2, e191350. [Google Scholar] [CrossRef] [Green Version]
- Cuccaro, M.L.; Carney, R.M.; Zhang, Y.; Bohm, C.; Kunkle, B.W.; Vardarajan, B.N.; Whitehead, P.L.; Cukier, H.N.; Mayeux, R.; George-Hyslop, P.S. SORL1 mutations in early-and late-onset Alzheimer disease. Neurol. Genet. 2016, 2, e116. [Google Scholar] [CrossRef] [Green Version]
- Bailey, C.C.; DeVaux, L.B.; Farzan, M. The triggering receptor expressed on myeloid cells 2 binds apolipoprotein E. J. Biol. Chem. 2015, 290, 26033–26042. [Google Scholar] [CrossRef] [Green Version]
- Bellenguez, C.; Grenier-Boley, B.; Lambert, J.-C. Genetics of Alzheimer’s disease: Where we are, and where we are going. Curr. Opin. Neurobiol. 2020, 61, 40–48. [Google Scholar] [CrossRef]
- Ashford, J.W. APOE genotype effects on Alzheimer’s disease onset and epidemiology. J. Mol. Neurosci. 2004, 23, 157–165. [Google Scholar] [CrossRef]
- Corder, E.H.; Saunders, A.M.; Risch, N.J.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Rimmler, J.B.; Locke, P.A.; Conneally, P.M.; Schmader, K.E.; et al. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat. Genet. 1994, 7, 180–184. [Google Scholar] [CrossRef] [PubMed]
- Cusulin, C.; Wells, I.; Badillo, S.; Duran-Pacheco, G.C.; Baumann, K.; Patsch, C. Gamma secretase modulators and BACE inhibitors reduce Aβ production without altering gene expression in Alzheimer’s disease iPSC-derived neurons and mice. Mol. Cell. Neurosci. 2019, 100, 103392. [Google Scholar] [CrossRef]
- Chang, K.-H.; Lee-Chen, G.-J.; Huang, C.-C.; Lin, J.-L.; Chen, Y.-J.; Wei, P.-C.; Lo, Y.-S.; Yao, C.-F.; Kuo, M.-W.; Chen, C.-M. Modeling Alzheimer’s disease by induced pluripotent stem cells carrying APP D678H mutation. Mol. Neurobiol. 2019, 56, 3972–3983. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Leon, J.A.; Caceres-Palomo, L.; Sanchez-Mejias, E.; Mejias-Ortega, M.; Nuñez-Diaz, C.; Fernandez-Valenzuela, J.J.; Sanchez-Varo, R.; Davila, J.C.; Vitorica, J.; Gutierrez, A. Human Pluripotent Stem Cell-Derived Neural Cells as a Relevant Platform for Drug Screening in Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 6867. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-E.; Shin, N.; Kook, M.G.; Kong, D.; Kim, N.G.; Choi, S.W.; Kang, K.-S. Human iNSC-derived brain organoid model of lysosomal storage disorder in Niemann–Pick disease type C. Cell Death Dis. 2020, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Kim, H.J.; Yang, J.; Chae, S.; Lee, W.; Chung, S.; Kim, J.; Choi, H.; Song, H.; Lee, C.K. Acetylation changes tau interactome to degrade tau in Alzheimer’s disease animal and organoid models. Aging Cell 2020, 19, e13081. [Google Scholar] [CrossRef]
- Couratier, P.; Corcia, P.; Lautrette, G.; Nicol, M.; Preux, P.-M.; Marin, B. Epidemiology of amyotrophic lateral sclerosis: A review of literature. Rev. Neurol. 2016, 172, 37–45. [Google Scholar] [CrossRef]
- Traxinger, K.; Kelly, C.; Johnson, B.A.; Lyles, R.H.; Glass, J.D. Prognosis and epidemiology of amyotrophic lateral sclerosis: Analysis of a clinic population, 1997–2011. Neurol. Clin. Pract. 2013, 3, 313–320. [Google Scholar] [CrossRef] [Green Version]
- Gros-Louis, F.; Gaspar, C.; Rouleau, G.A. Genetics of familial and sporadic amyotrophic lateral sclerosis. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2006, 1762, 956–972. [Google Scholar] [CrossRef] [Green Version]
- Da Cruz, S.; Cleveland, D.W. Understanding the role of TDP-43 and FUS/TLS in ALS and beyond. Curr. Opin. Neurobiol. 2011, 21, 904–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS genetics, mechanisms, and therapeutics: Where are we now? Front. Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, L.; Zhang, T.; Lu, K.; Qi, S. The progress in C9orf72 research: ALS/FTD pathogenesis, functions and structure. Small Gtpases 2021, 1–21. [Google Scholar] [CrossRef]
- Boylan, K. Familial amyotrophic lateral sclerosis. Neurol. Clin. 2015, 33, 807–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, S.; Al Khleifat, A.; Al-Chalabi, A. What causes amyotrophic lateral sclerosis? F1000Research 2017, 6, 371. [Google Scholar] [CrossRef] [Green Version]
- Jaiswal, M.K. Riluzole and edaravone: A tale of two amyotrophic lateral sclerosis drugs. Med. Res. Rev. 2019, 39, 733–748. [Google Scholar] [CrossRef]
- Guo, W.; Naujock, M.; Fumagalli, L.; Vandoorne, T.; Baatsen, P.; Boon, R.; Ordovás, L.; Patel, A.; Welters, M.; Vanwelden, T. HDAC6 inhibition reverses axonal transport defects in motor neurons derived from FUS-ALS patients. Nat. Commun. 2017, 8, 1–15. [Google Scholar] [CrossRef]
- Okano, H.; Yasuda, D.; Fujimori, K.; Morimoto, S.; Takahashi, S. Ropinirole, a New ALS Drug Candidate Developed Using iPSCs. Trends Pharmacol. Sci. 2020, 41, 99–109. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, S.; Morimoto, S.; Fukushima, K.; Nakahara, J.; Okano, H. [Ropinirole Hydrochloride for ALS]. Brain Nerve Shinkei Kenkyu No Shinpo 2019, 71, 1279–1288. [Google Scholar] [CrossRef]
- McNeish, J.; Gardner, J.P.; Wainger, B.J.; Woolf, C.J.; Eggan, K. From dish to bedside: Lessons learned while translating findings from a stem cell model of disease to a clinical trial. Cell Stem Cell 2015, 17, 8–10. [Google Scholar] [CrossRef] [Green Version]
- Ortuño-Costela, M.d.C.; Cerrada, V.; García-López, M.; Gallardo, M.E. The challenge of bringing iPSCs to the patient. Int. J. Mol. Sci. 2019, 20, 6305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pernia, C.; Tobe, B.T.; O’Donnell, R.; Snyder, E.Y. The evolution of stem cells, disease modeling, and drug discovery for neurological disorders. Stem Cells Dev. 2020, 29, 1131–1141. [Google Scholar] [CrossRef] [PubMed]
- Wainger, B.J.; Macklin, E.A.; Vucic, S.; McIlduff, C.E.; Paganoni, S.; Maragakis, N.J.; Bedlack, R.; Goyal, N.A.; Rutkove, S.B.; Lange, D.J.; et al. Effect of Ezogabine on Cortical and Spinal Motor Neuron Excitability in Amyotrophic Lateral Sclerosis: A Randomized Clinical Trial. JAMA Neurol. 2021, 78, 186–196. [Google Scholar] [CrossRef] [PubMed]
- Aronica, E.; Baas, F.; Iyer, A.; ten Asbroek, A.L.; Morello, G.; Cavallaro, S. Molecular classification of amyotrophic lateral sclerosis by unsupervised clustering of gene expression in motor cortex. Neurobiol. Dis. 2015, 74, 359–376. [Google Scholar] [CrossRef] [Green Version]
- Polanco, M.J.; Pennuto, M. Pituitary adenylyl cyclase activating polypeptide (PACAP) signaling and the cell cycle machinery in neurodegenerative diseases. Curr. Pharm. Des. 2018, 24, 3878–3891. [Google Scholar] [CrossRef]
- Dejda, A.; Jolivel, V.; Bourgault, S.; Seaborn, T.; Fournier, A.; Vaudry, H.; Vaudry, D. Inhibitory Effect of PACAP on Caspase Activity in Neuronal Apoptosis: A Better Understanding Towards Therapeutic Applications in Neurodegenerative Diseases. J. Mol. Neurosci. 2008, 36, 26–37. [Google Scholar] [CrossRef]
- Maugeri, G.; D’Amico, A.G.; Rasà, D.M.; Federico, C.; Saccone, S.; Morello, G.; La Cognata, V.; Cavallaro, S.; D’Agata, V. Molecular mechanisms involved in the protective effect of pituitary adenylate cyclase-activating polypeptide in an in vitro model of amyotrophic lateral sclerosis. J. Cell. Physiol. 2019, 234, 5203–5214. [Google Scholar] [CrossRef]
- Yang, R.; Jiang, X.; Ji, R.; Meng, L.; Liu, F.; Chen, X.; Xin, Y. Therapeutic potential of PACAP for neurodegenerative diseases. Cell. Mol. Biol. Lett. 2015, 20, 265–278. [Google Scholar] [CrossRef]
- Reglodi, D.; Kiss, P.; Lubics, A.; Tamas, A. Review on the protective effects of PACAP in models of neurodegenerative diseases in vitro and in vivo. Curr. Pharm. Des. 2011, 17, 962–972. [Google Scholar] [CrossRef]
- Morello, G.; Spampinato, A.G.; Conforti, F.L.; D’Agata, V.; Cavallaro, S. Selection and prioritization of candidate drug targets for amyotrophic lateral sclerosis through a meta-analysis approach. J. Mol. Neurosci. 2017, 61, 563–580. [Google Scholar] [CrossRef] [Green Version]
- Bonaventura, G.; Iemmolo, R.; D’Amico, A.G.; La Cognata, V.; Costanzo, E.; Zappia, M.; D’Agata, V.; Conforti, F.L.; Aronica, E.; Cavallaro, S. PACAP and PAC1R are differentially expressed in motor cortex of amyotrophic lateral sclerosis patients and support survival of iPSC-derived motor neurons. J. Cell. Physiol. 2018, 233, 3343–3351. [Google Scholar] [CrossRef]
- Osaki, T.; Uzel, S.G.; Kamm, R.D. Microphysiological 3D model of amyotrophic lateral sclerosis (ALS) from human iPS-derived muscle cells and optogenetic motor neurons. Sci. Adv. 2018, 4, eaat5847. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.; Kim, J.; Park, H.; Choi, H.; Kim, J. Modelling neurodegenerative diseases with 3D brain organoids. Biol. Rev. 2020, 95, 1497–1509. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.K.; Farrer, M.J. Genetics and genomics of Parkinson’s disease. Genome Med. 2014, 6, 48. [Google Scholar] [CrossRef] [Green Version]
- La Cognata, V.; Morello, G.; D’Agata, V.; Cavallaro, S. Copy number variability in Parkinson’s disease: Assembling the puzzle through a systems biology approach. Hum. Genet. 2017, 136, 13–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Cognata, V.; D’Agata, V.; Cavalcanti, F.; Cavallaro, S. Splicing: Is there an alternative contribution to Parkinson’s disease? Neurogenetics 2015, 16, 245–263. [Google Scholar] [CrossRef] [Green Version]
- Aflaki, E.; Borger, D.K.; Moaven, N.; Stubblefield, B.K.; Rogers, S.A.; Patnaik, S.; Schoenen, F.J.; Westbroek, W.; Zheng, W.; Sullivan, P. A new glucocerebrosidase chaperone reduces α-synuclein and glycolipid levels in iPSC-derived dopaminergic neurons from patients with Gaucher disease and parkinsonism. J. Neurosci. 2016, 36, 7441–7452. [Google Scholar] [CrossRef]
- Mazzulli, J.R.; Zunke, F.; Tsunemi, T.; Toker, N.J.; Jeon, S.; Burbulla, L.F.; Patnaik, S.; Sidransky, E.; Marugan, J.J.; Sue, C.M. Activation of β-glucocerebrosidase reduces pathological α-synuclein and restores lysosomal function in Parkinson’s patient midbrain neurons. J. Neurosci. 2016, 36, 7693–7706. [Google Scholar] [CrossRef]
- Zheng, J.; Jeon, S.; Jiang, W.; Burbulla, L.F.; Ysselstein, D.; Oevel, K.; Krainc, D.; Silverman, R.B. Conversion of Quinazoline Modulators from Inhibitors to Activators of β-Glucocerebrosidase. J. Med. Chem. 2019, 62, 1218–1230. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Jeon, S.; Burbulla, L.F.; Krainc, D. Acid ceramidase inhibition ameliorates α-synuclein accumulation upon loss of GBA1 function. Hum. Mol. Genet. 2018, 27, 1972–1988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burbulla, L.F.; Jeon, S.; Zheng, J.; Song, P.; Silverman, R.B.; Krainc, D. A modulator of wild-type glucocerebrosidase improves pathogenic phenotypes in dopaminergic neuronal models of Parkinson’s disease. Sci. Transl. Med. 2019, 11, eaau6870. [Google Scholar] [CrossRef] [PubMed]
- Burbulla, L.F.; Song, P.; Mazzulli, J.R.; Zampese, E.; Wong, Y.C.; Jeon, S.; Santos, D.P.; Blanz, J.; Obermaier, C.D.; Strojny, C. Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease. Science 2017, 357, 1255–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambasudhan, R.; Ryan, S.D.; Dolatabadi, N.; Chan, S.F.; Zhang, X.; Akhtar, M.W.; Parker, J.; Soldner, F.; Sunico, C.R.; Nagar, S. Isogenic Human iPSC Parkinson’s Model Shows Nitrosative Stress-Induced Dysfunction in MEF2-PGC1α Transcription. Cell 2013, 155, 1351–1364. [Google Scholar]
- Kouroupi, G.; Taoufik, E.; Vlachos, I.S.; Tsioras, K.; Antoniou, N.; Papastefanaki, F.; Chroni-Tzartou, D.; Wrasidlo, W.; Bohl, D.; Stellas, D.; et al. Defective synaptic connectivity and axonal neuropathology in a human iPSC-based model of familial Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, E3679–E3688. [Google Scholar] [CrossRef] [Green Version]
- Cooper, O.; Seo, H.; Andrabi, S.; Guardia-Laguarta, C.; Graziotto, J.; Sundberg, M.; McLean, J.R.; Carrillo-Reid, L.; Xie, Z.; Osborn, T.; et al. Pharmacological Rescue of Mitochondrial Deficits in iPSC-Derived Neural Cells from Patients with Familial Parkinson’s Disease. Sci. Transl. Med. 2012, 4, 141ra190. [Google Scholar] [CrossRef] [Green Version]
- Ke, M.; Chong, C.-M.; Zeng, H.; Huang, M.; Huang, Z.; Zhang, K.; Cen, X.; Lu, J.-H.; Yao, X.; Qin, D.; et al. Azoramide protects iPSC-derived dopaminergic neurons with PLA2G6 D331Y mutation through restoring ER function and CREB signaling. Cell Death Dis. 2020, 11, 130. [Google Scholar] [CrossRef] [Green Version]
- Baker, B.M.; Chen, C.S. Deconstructing the third dimension—how 3D culture microenvironments alter cellular cues. J. Cell Sci. 2012, 125, 3015–3024. [Google Scholar] [CrossRef] [Green Version]
- Bolognin, S.; Fossépré, M.; Qing, X.; Jarazo, J.; Ščančar, J.; Moreno, E.L.; Nickels, S.L.; Wasner, K.; Ouzren, N.; Walter, J.; et al. 3D Cultures of Parkinson’s Disease-Specific Dopaminergic Neurons for High Content Phenotyping and Drug Testing. Adv. Sci. 2019, 6, 1800927. [Google Scholar] [CrossRef] [PubMed]
- Sethna, F.; Moon, C.; Wang, H. From FMRP Function to Potential Therapies for Fragile X Syndrome. Neurochem. Res. 2014, 39, 1016–1031. [Google Scholar] [CrossRef] [Green Version]
- Erickson, C.A.; Davenport, M.H.; Schaefer, T.L.; Wink, L.K.; Pedapati, E.V.; Sweeney, J.A.; Fitzpatrick, S.E.; Brown, W.T.; Budimirovic, D.; Hagerman, R.J.; et al. Fragile X targeted pharmacotherapy: Lessons learned and future directions. J. Neurodev. Disord. 2017, 9, 7. [Google Scholar] [CrossRef]
- Alisch, R.S.; Wang, T.; Chopra, P.; Visootsak, J.; Conneely, K.N.; Warren, S.T. Genome-wide analysis validates aberrant methylation in fragile X syndrome is specific to the FMR1locus. BMC Med. Genet. 2013, 14, 18. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.W.; Ventola, P.; Budimirovic, D.; Berry-Kravis, E.; Visootsak, J. Clinical Development of Targeted Fragile X Syndrome Treatments: An Industry Perspective. Brain Sci. 2018, 8, 214. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Bray, S.M.; Warren, S.T. New perspectives on the biology of fragile X syndrome. Curr. Opin. Genet. Dev. 2012, 22, 256–263. [Google Scholar] [CrossRef] [Green Version]
- Vershkov, D.; Ben-Hur, T.; Benvenisty, N. Chapter 6—Modeling Fragile X Syndrome Using Human Pluripotent Stem Cells. In Fragile X Syndrome; Willemsen, R., Kooy, R.F., Eds.; Academic Press: London, UK, 2017; pp. 103–121. [Google Scholar] [CrossRef]
- Mor-Shaked, H.; Eiges, R. Modeling Fragile X Syndrome Using Human Pluripotent Stem Cells. Genes 2016, 7, 77. [Google Scholar] [CrossRef] [Green Version]
- Bar-Nur, O.; Caspi, I.; Benvenisty, N. Molecular analysis of FMR1 reactivation in fragile-X induced pluripotent stem cells and their neuronal derivatives. J. Mol. Cell Biol. 2012, 4, 180–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vershkov, D.; Fainstein, N.; Suissa, S.; Golan-Lev, T.; Ben-Hur, T.; Benvenisty, N. FMR1 Reactivating Treatments in Fragile X iPSC-Derived Neural Progenitors In Vitro and In Vivo. Cell Rep. 2019, 26, 2531–2539.e4. [Google Scholar] [CrossRef] [Green Version]
- Silva, M.C.; Haggarty, S.J. Human pluripotent stem cell–derived models and drug screening in CNS precision medicine. Ann. N. Y. Acad. Sci. 2020, 1471, 18–56. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Paul, S.; Lim, H.; Dayem, A.A.; Cho, S.-G. Induced pluripotent stem cell research: A revolutionary approach to face the challenges in drug screening. Arch. Pharmacal Res. 2012, 35, 245–260. [Google Scholar] [CrossRef] [PubMed]
- Liang, N.; Trujillo, C.A.; Negraes, P.D.; Muotri, A.R.; Lameu, C.; Ulrich, H. Stem cell contributions to neurological disease modeling and personalized medicine. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2018, 80, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Little, D.; Ketteler, R.; Gissen, P.; Devine, M.J. Using stem cell–derived neurons in drug screening for neurological diseases. Neurobiol. Aging 2019, 78, 130–141. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bonaventura, G.; Iemmolo, R.; Attaguile, G.A.; La Cognata, V.; Pistone, B.S.; Raudino, G.; D’Agata, V.; Cantarella, G.; Barcellona, M.L.; Cavallaro, S. iPSCs: A Preclinical Drug Research Tool for Neurological Disorders. Int. J. Mol. Sci. 2021, 22, 4596. https://doi.org/10.3390/ijms22094596
Bonaventura G, Iemmolo R, Attaguile GA, La Cognata V, Pistone BS, Raudino G, D’Agata V, Cantarella G, Barcellona ML, Cavallaro S. iPSCs: A Preclinical Drug Research Tool for Neurological Disorders. International Journal of Molecular Sciences. 2021; 22(9):4596. https://doi.org/10.3390/ijms22094596
Chicago/Turabian StyleBonaventura, Gabriele, Rosario Iemmolo, Giuseppe Antonino Attaguile, Valentina La Cognata, Brigida Sabrina Pistone, Giuseppe Raudino, Velia D’Agata, Giuseppina Cantarella, Maria Luisa Barcellona, and Sebastiano Cavallaro. 2021. "iPSCs: A Preclinical Drug Research Tool for Neurological Disorders" International Journal of Molecular Sciences 22, no. 9: 4596. https://doi.org/10.3390/ijms22094596
APA StyleBonaventura, G., Iemmolo, R., Attaguile, G. A., La Cognata, V., Pistone, B. S., Raudino, G., D’Agata, V., Cantarella, G., Barcellona, M. L., & Cavallaro, S. (2021). iPSCs: A Preclinical Drug Research Tool for Neurological Disorders. International Journal of Molecular Sciences, 22(9), 4596. https://doi.org/10.3390/ijms22094596