
CHIR99021 Augmented the Function of Late Endothelial Progenitor Cells by Preventing Replicative Senescence

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
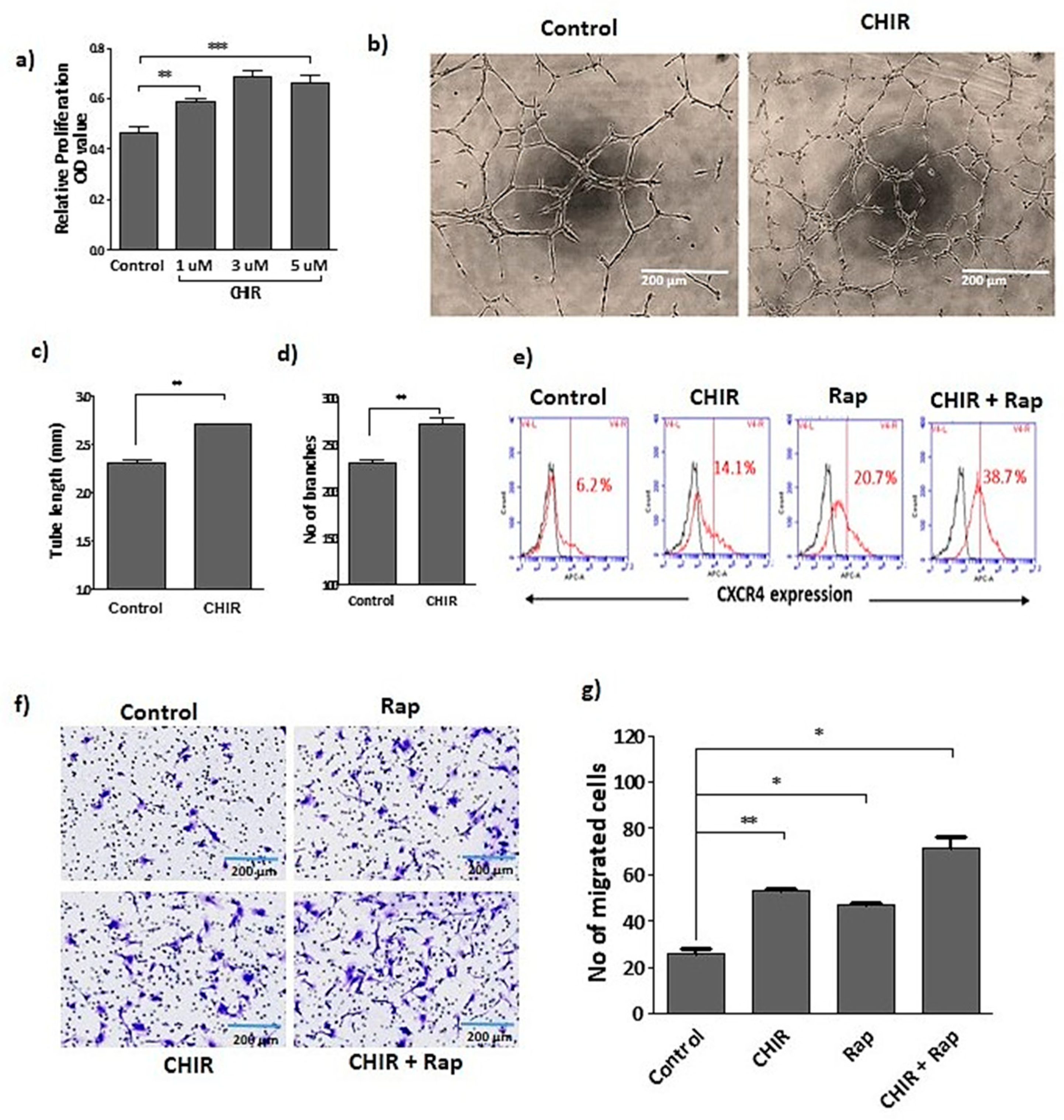
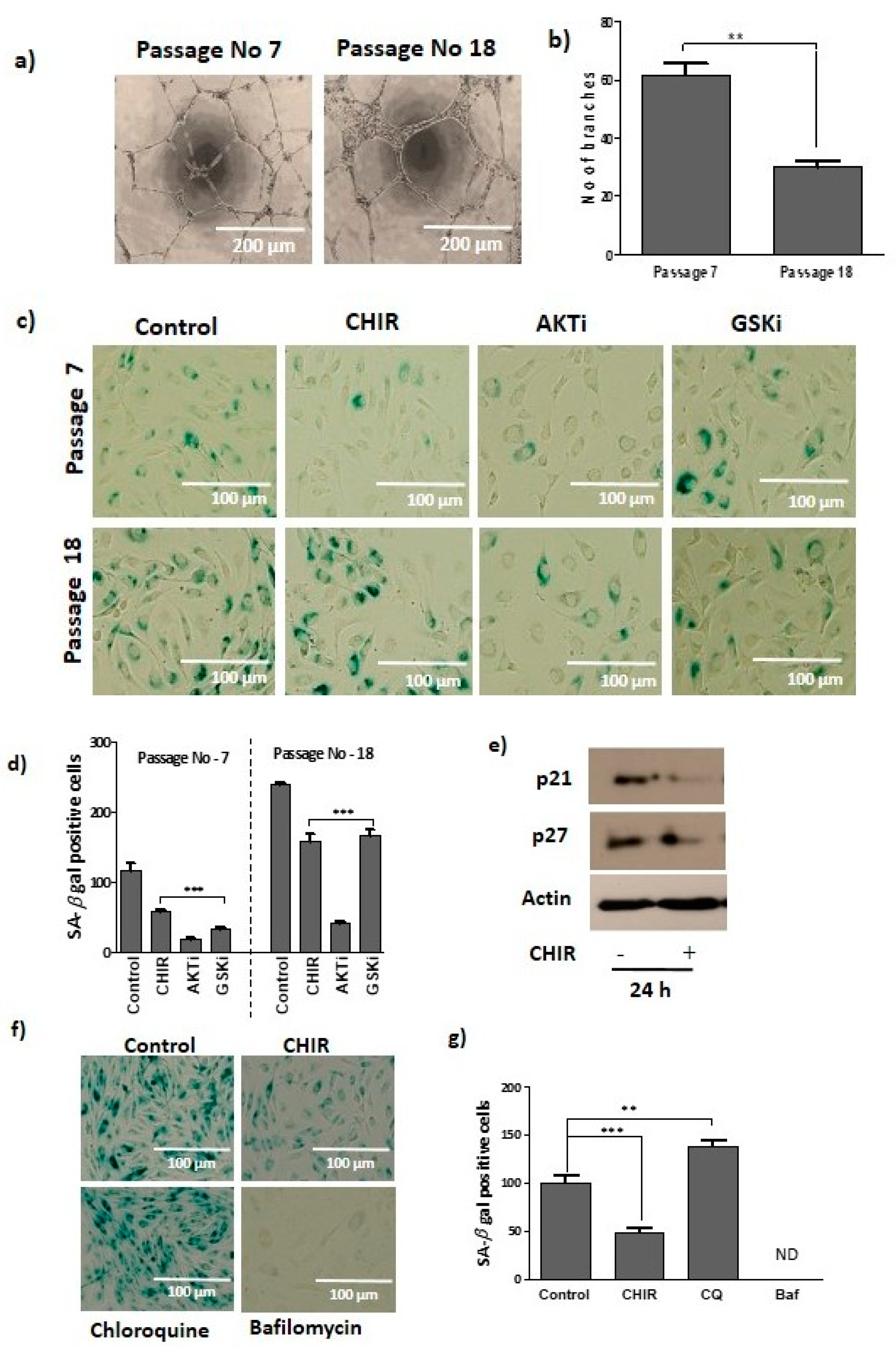
2.1. GSK-3β Inhibition by CHIR99021 Modulates Late EPCs Functional Activities
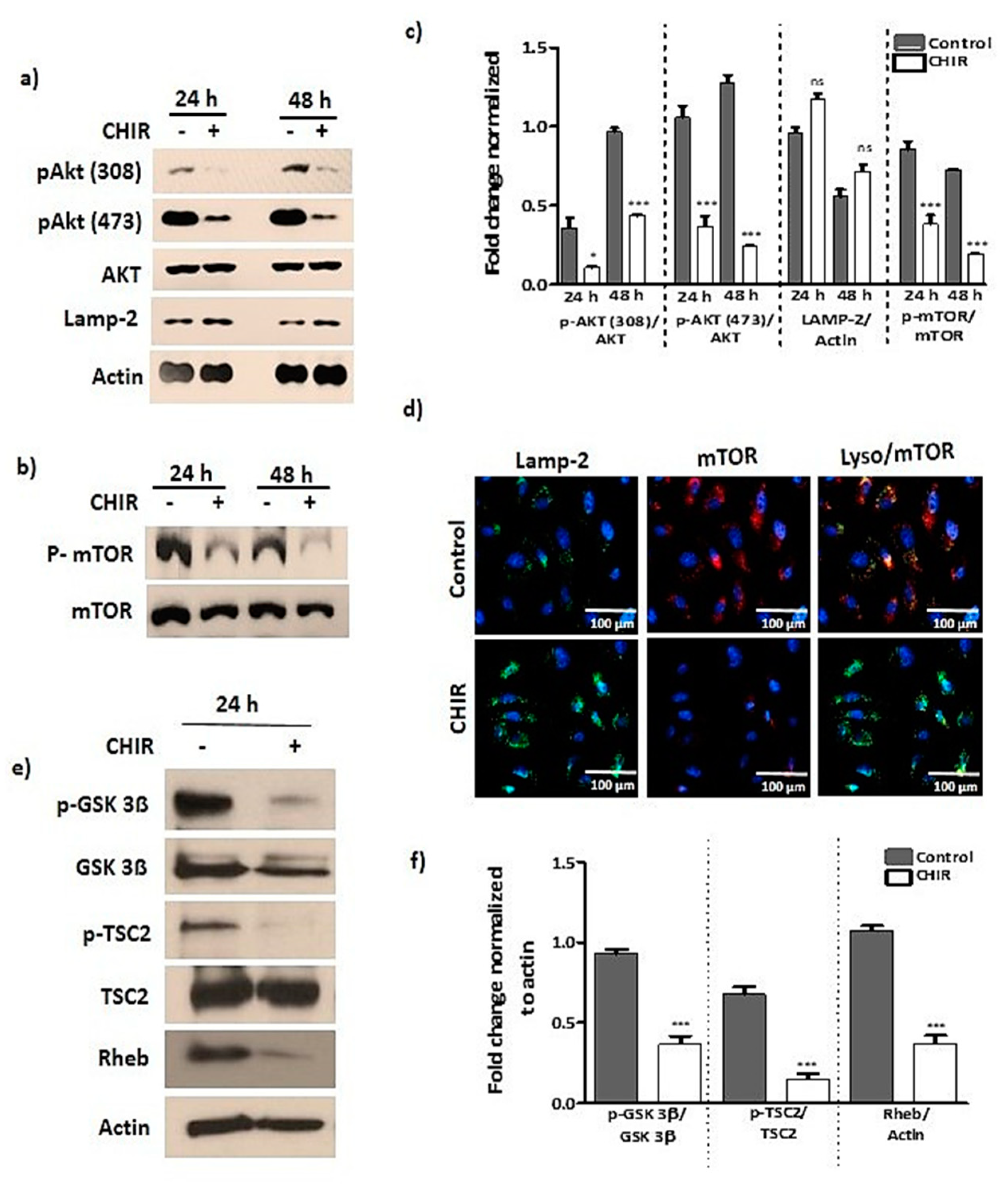
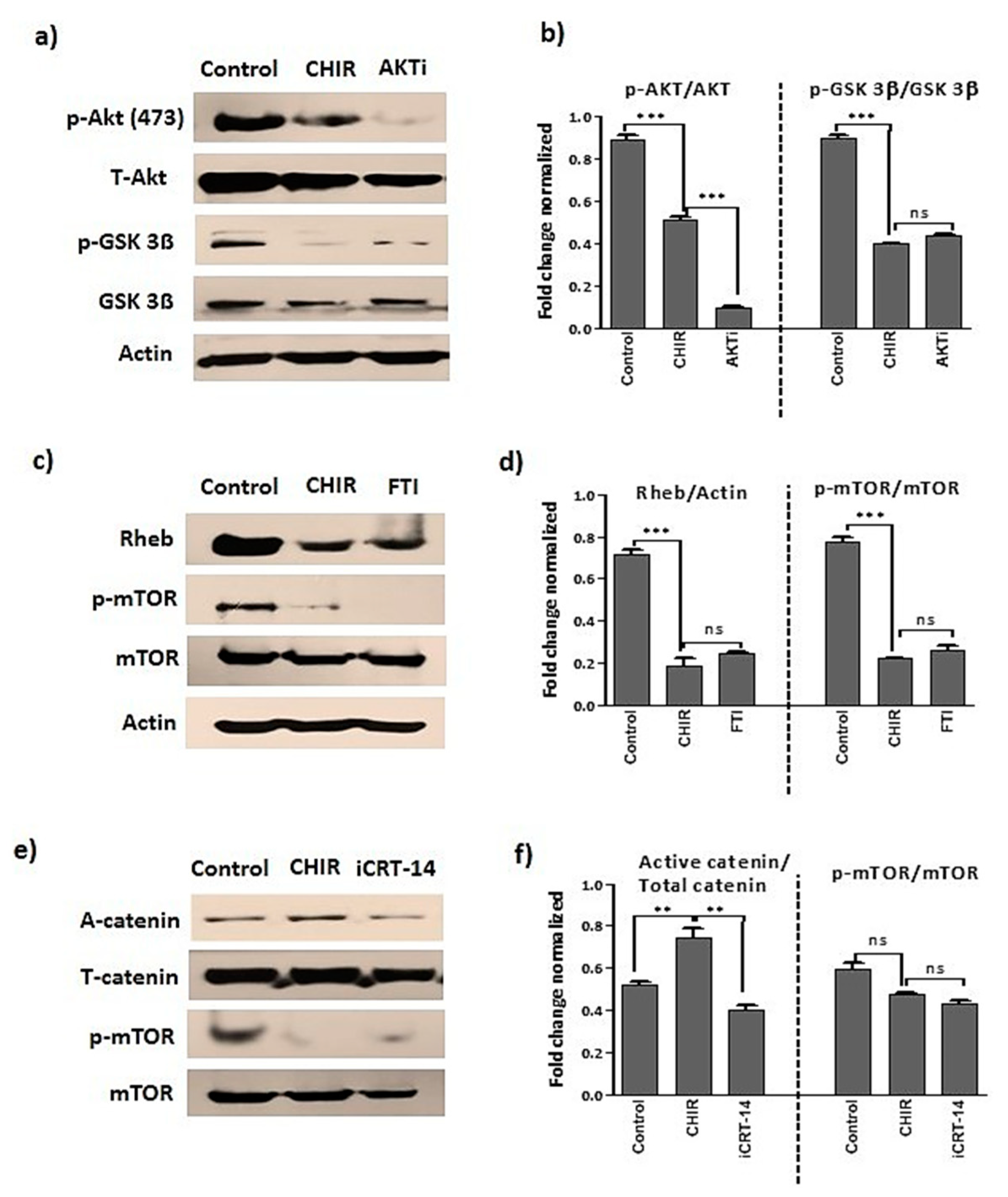
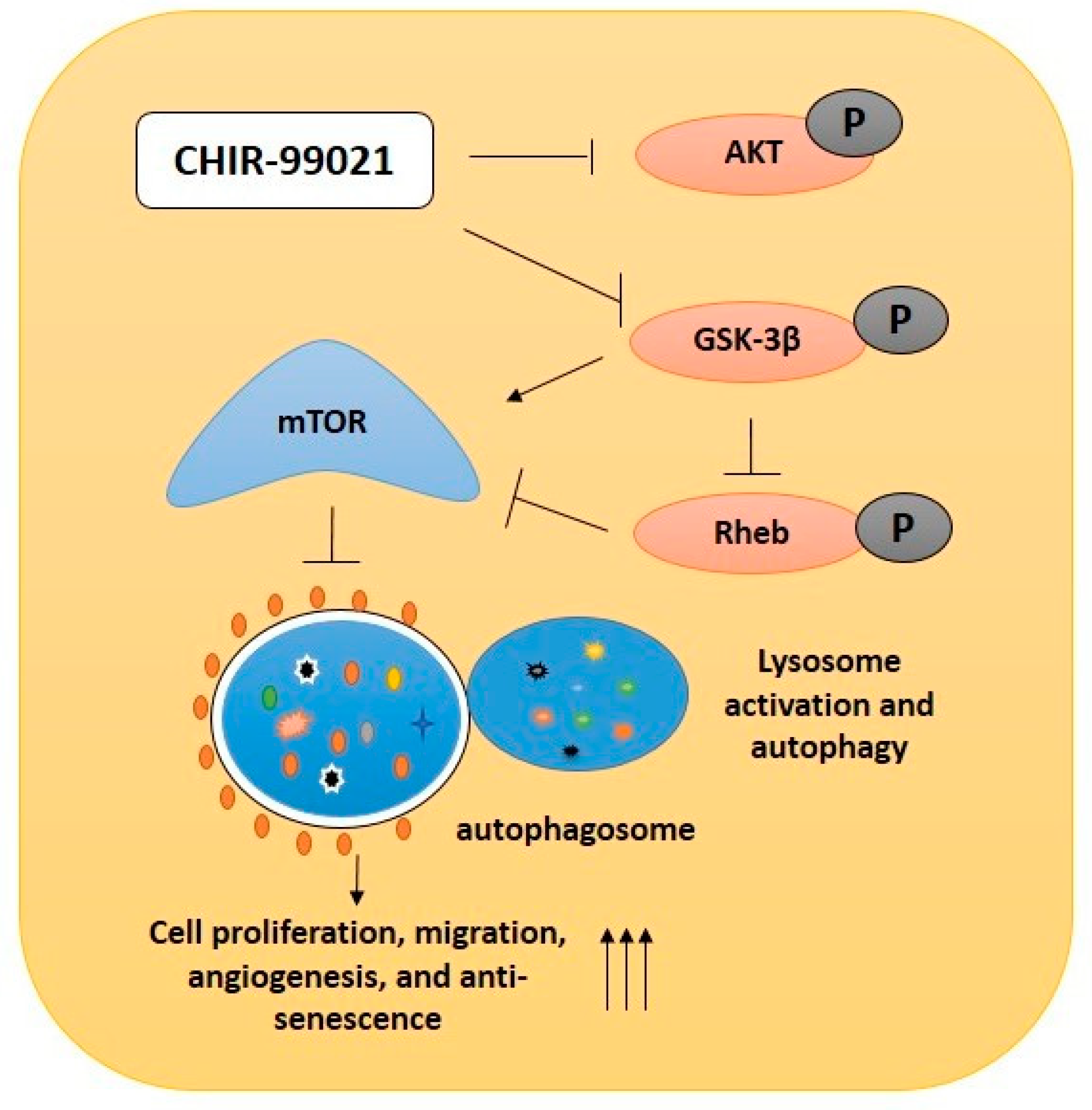
2.2. GSK-3β Inhibition by Using CHIR99021 Deregulates mTOR via Rheb Inhibition
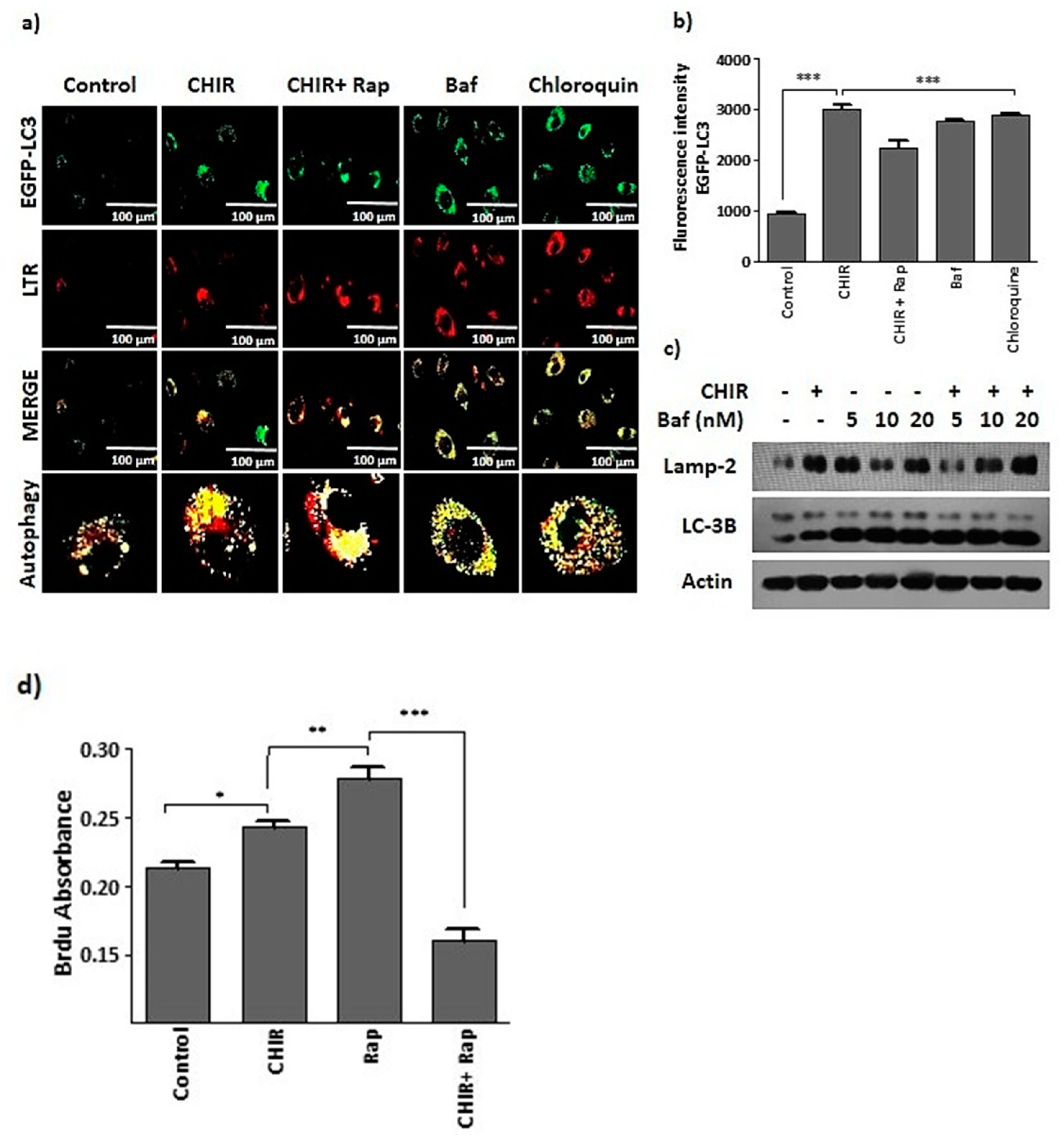
2.3. CHIR99021-Induced GSK-3β Inhibition Enhances Lysosome Activation and Autophagy
2.4. Lysosome Activation and Autophagy by the Way of GSK-3β Inhibition Augments Late EPCs Functional Activity
2.5. Lysosome Activation and Autophagy Prevent Late EPCs Senescence
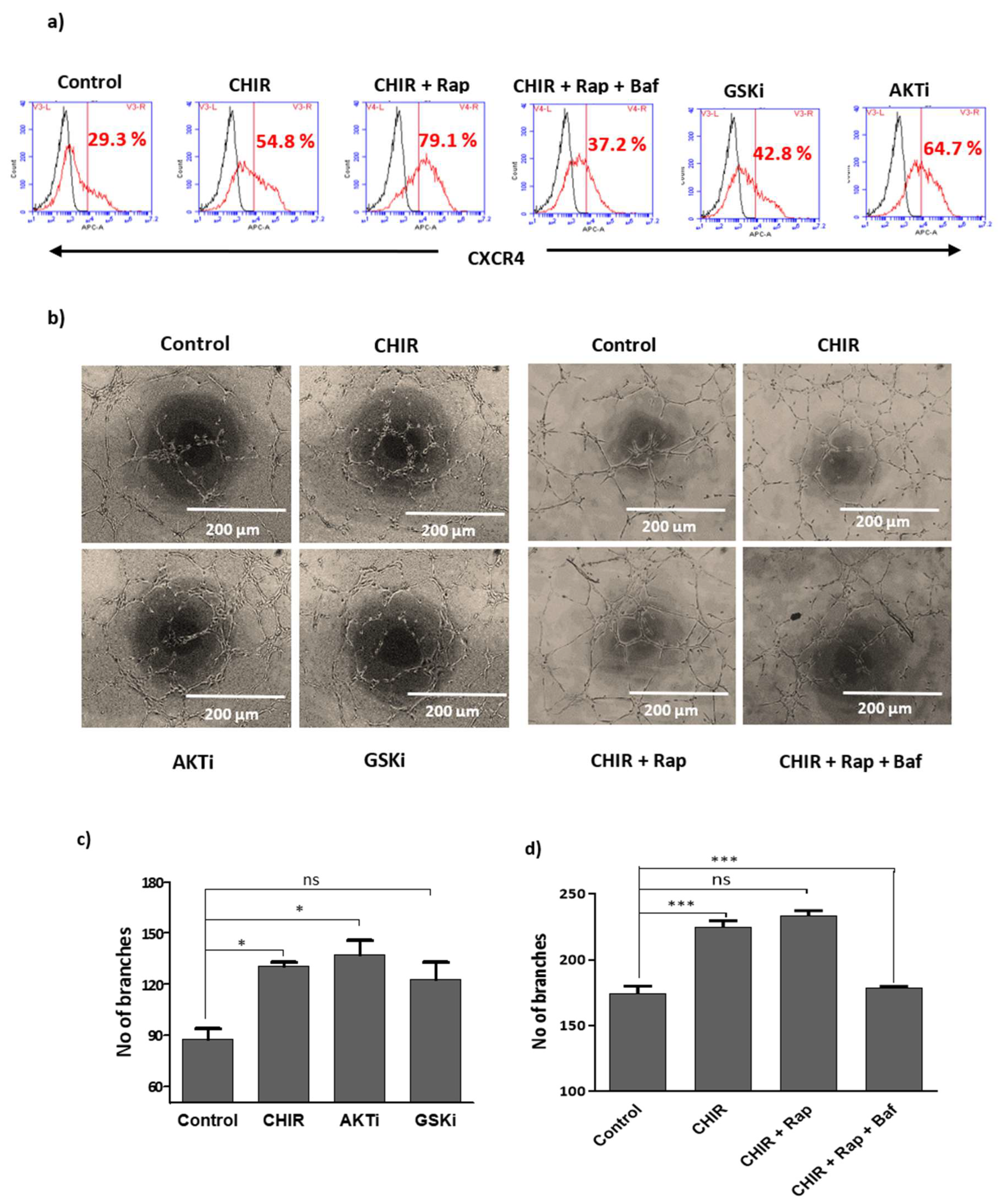
2.6. GSK-3β Inhibition by CHIR99021 Deregulates mTOR Signaling by Rheb-Dependent and Beta-Catenin-Independent signaling
3. Discussion
4. Materials and Methods
4.1. Isolation of Late EPCs and Cell Culture
4.2. Flow Cytometer Analysis
4.3. Cell Counting Kit-8 (CCK-8) Assay
4.4. BrdU Incorporation Assay
4.5. Transwell Migration Assay
4.6. Tube Formation Assay
4.7. Immunoblots
4.8. Immunofluorescence
4.9. Transfections with EGFP-LC3 Plasmid
4.10. Senescence Associated β-Galactosidase Assay
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Urbich, C.; Dimmeler, S. Endothelial progenitor cells: Characterization and role in vascular biology. Circ. Res. 2004, 95, 343–353. [Google Scholar] [CrossRef]
- Dai, X.; Yan, X.; Zeng, J.; Chen, J.; Wang, Y.; Chen, J.; Li, Y.; Barati, M.T.; Wintergerst, K.A.; Pan, K.; et al. Elevating CXCR7 Improves Angiogenic Function of EPCs via Akt/GSK-3β/Fyn-Mediated Nrf2 Activation in Diabetic Limb Ischemia. Circ. Res. 2017, 120, e7–e23. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.Y.; Su, C.; Cao, Z.; Xu, S.Y.; Xia, W.H.; Xie, W.L.; Chen, L.; Yu, B.B.; Zhang, B.; Wang, Y.; et al. CXCR7 upregulation is required for early endothelial progenitor cell-mediated endothelial repair in patients with hypertension. Hypertension 2014, 63, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q. The impact of progenitor cells in atherosclerosis. Nat. Clin. Pract. Cardiovasc. Med. 2006, 3, 94–101. [Google Scholar] [CrossRef]
- Ii, M.; Takenaka, H.; Asai, J.; Ibusuki, K.; Mizukami, Y.; Maruyama, K.; Yoon, Y.S.; Wecker, A.; Luedemann, C.; Eaton, E.; et al. Endothelial progenitor thrombospondin-1 mediates diabetes-induced delay in reendothelialization following arterial injury. Circ. Res. 2006, 98, 697–704. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Liu, L.; Liu, H.; Wu, K.; Liu, Y.; Bai, L.; Wang, Q.; Qi, B.; Qi, B.; Zhang, L. Reduced NRF2 expression suppresses endothelial progenitor cell function and induces senescence during aging. Aging 2019, 11, 7021–7035. [Google Scholar] [CrossRef]
- Fang, X.; Yu, S.X.; Lu, Y.; Bast, R.C., Jr.; Woodgett, J.R.; Mills, G.B. Phosphorylation and inactivation of glycogen synthase kinase 3 by protein kinase A. Proc. Natl. Acad. Sci. USA 2000, 97, 11960–11965. [Google Scholar] [CrossRef]
- Cross, D.A.; Alessi, D.R.; Cohen, P.; Andjelkovich, M.; Hemmings, B.A. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 1995, 378, 785–789. [Google Scholar] [CrossRef]
- Nusse, R.; Clevers, H. Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef]
- Barcelos, L.S.; Duplaa, C.; Kränkel, N.; Graiani, G.; Invernici, G.; Katare, R.; Siragusa, M.; Meloni, M.; Campesi, I.; Monica, M.; et al. Human CD133+ progenitor cells promote the healing of diabetic ischemic ulcers by paracrine stimulation of angiogenesis and activation of Wnt signaling. Circ. Res. 2009, 104, 1095–1102. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Li, L.; Chen, J.X. Overexpression of angiopoietin-1 increases CD133+/c-kit+ cells and reduces myocardial apoptosis in db/db mouse infarcted hearts. PLoS ONE 2012, 7, e35905. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Ouyang, H.; Zhu, T.; Lindvall, C.; Wang, Y.; Zhang, X.; Yang, Q.; Bennett, C.; Harada, Y.; Stankunas, K.; et al. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell 2006, 126, 955–968. [Google Scholar] [CrossRef] [PubMed]
- Menon, S.; Dibble, C.C.; Talbott, G.; Hoxhaj, G.; Valvezan, A.J.; Takahashi, H.; Cantley, L.C.; Manning, B.D. Spatial control of the TSC complex integrates insulin and nutrient regulation of mTORC1 at the lysosome. Cell 2014, 156, 771–785. [Google Scholar] [CrossRef]
- Pal, R.; Xiong, Y.; Sardiello, M. Abnormal glycogen storage in tuberous sclerosis complex caused by impairment of mTORC1-dependent and -independent signaling pathways. Proc. Natl. Acad. Sci. USA 2019, 116, 2977–2986. [Google Scholar] [CrossRef]
- Avrahami, L.; Paz, R.; Dominko, K.; Hecimovic, S.; Bucci, C.; Eldar-Finkelman, H. GSK-3-TSC axis governs lysosomal acidification through autophagy and endocytic pathways. Cell Signal. 2020, 71, 109597. [Google Scholar] [CrossRef] [PubMed]
- Acebron, S.P.; Karaulanov, E.; Berger, B.S.; Huang, Y.L.; Niehrs, C. Mitotic wnt signaling promotes protein stabilization and regulates cell size. Mol. Cell 2014, 54, 663–674. [Google Scholar] [CrossRef]
- Leeman, D.S.; Hebestreit, K.; Ruetz, T.; Webb, A.E.; McKay, A.; Pollina, E.A.; Dulken, B.W.; Zhao, X.; Yeo, R.W.; Ho, T.T.; et al. Lysosome activation clears aggregates and enhances quiescent neural stem cell activation during aging. Science 2018, 359, 1277–1283. [Google Scholar] [CrossRef]
- Zhou, J.; Freeman, T.A.; Ahmad, F.; Shang, X.; Mangano, E.; Gao, E.; Farber, J.; Wang, Y.; Ma, X.L.; Woodgett, J.; et al. GSK-3α is a central regulator of age-related pathologies in mice. J. Clin. Investig. 2013, 123, 1821–1832. [Google Scholar] [CrossRef]
- Zhai, P.; Sciarretta, S.; Galeotti, J.; Volpe, M.; Sadoshima, J. Differential roles of GSK-3β during myocardial ischemia and ischemia/reperfusion. Circ. Res. 2011, 109, 502–511. [Google Scholar] [CrossRef]
- Tai, H.; Wang, Z.; Gong, H.; Han, X.; Zhou, J.; Wang, X.; Wei, X.; Ding, Y.; Huang, N.; Qin, J.; et al. Autophagy impairment with lysosomal and mitochondrial dysfunction is an important characteristic of oxidative stress-induced senescence. Autophagy 2017, 13, 99–113. [Google Scholar] [CrossRef]
- Revuelta, M.; Matheu, A. Autophagy in stem cell aging. Aging Cell 2017, 16, 912–915. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Sharpless, N.E. Senescence in Health and Disease. Cell 2017, 169, 1000–1011. [Google Scholar] [CrossRef]
- Lian, X.; Zhang, J.; Azarin, S.M.; Zhu, K.; Hazeltine, L.B.; Bao, X.; Hsiao, C.; Kamp, T.J.; Palecek, S.P. Directed cardiomyocyte differentiation from human pluripotent stem cells by modulating Wnt/β-catenin signaling under fully defined conditions. Nat. Protoc. 2013, 8, 162–175. [Google Scholar] [CrossRef] [PubMed]
- Lian, X.; Bao, X.; Al-Ahmad, A.; Liu, J.; Wu, Y.; Dong, W.; Dunn, K.K.; Shusta, E.V.; Palecek, S.P. Efficient differentiation of human pluripotent stem cells to endothelial progenitors via small-molecule activation of WNT signaling. Stem Cell Rep. 2014, 3, 804–816. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Ai, Z.; Yao, K.; Cao, L.; Du, J.; Shi, X.; Guo, Z.; Zhang, Y. CHIR99021 promotes self-renewal of mouse embryonic stem cells by modulation of protein-encoding gene and long intergenic non-coding RNA expression. Exp. Cell Res. 2013, 319, 2684–2699. [Google Scholar] [CrossRef]
- Heusch, G. Myocardial Ischemia: Lack of Coronary Blood Flow or Myocardial Oxygen Supply/Demand Imbalance? Circ. Res. 2016, 119, 194–196. [Google Scholar] [CrossRef]
- Werner, N.; Kosiol, S.; Schiegl, T.; Ahlers, P.; Walenta, K.; Link, A.; Böhm, M.; Nickenig, G. Circulating endothelial progenitor cells and cardiovascular outcomes. N. Engl. J. Med. 2005, 353, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Rafii, S.; Lyden, D. Therapeutic stem and progenitor cell transplantation for organ vascularization and regeneration. Nat. Med. 2003, 9, 702–712. [Google Scholar] [CrossRef]
- Flügel, D.; Görlach, A.; Kietzmann, T. GSK-3β regulates cell growth, migration, and angiogenesis via Fbw7 and USP28-dependent degradation of HIF-1α. Blood 2012, 119, 1292–1301. [Google Scholar] [CrossRef]
- Zhai, P.; Sadoshima, J. Glycogen synthase kinase-3β controls autophagy during myocardial ischemia and reperfusion. Autophagy 2012, 8, 138–139. [Google Scholar] [CrossRef]
- Lal, H.; Ahmad, F.; Woodgett, J.; Force, T. The GSK-3 family as therapeutic target for myocardial diseases. Circ. Res. 2015, 116, 138–149. [Google Scholar] [CrossRef]
- Desbois-Mouthon, C.; Cadoret, A.; Blivet-Van Eggelpoël, M.J.; Bertrand, F.; Cherqui, G.; Perret, C.; Capeau, J. Insulin and IGF-1 stimulate the beta-catenin pathway through two signalling cascades involving GSK-3beta inhibition and Ras activation. Oncogene 2001, 20, 252–259. [Google Scholar] [CrossRef]
- Su, F.; Zhao, L.; Zhang, S.; Wang, J.; Chen, N.; Gong, Q.; Tang, J.; Wang, H.; Yao, J.; Wang, Q.; et al. Cardioprotection by PI3K-mediated signaling is required for anti-arrhythmia and myocardial repair in response to ischemic preconditioning in infarcted pig hearts. Lab. Investig. 2015, 95, 860–871. [Google Scholar] [CrossRef]
- Dibble, C.C.; Cantley, L.C. Regulation of mTORC1 by PI3K signaling. Trends Cell Biol. 2015, 25, 545–555. [Google Scholar] [CrossRef]
- Yu, H.; Littlewood, T.; Bennett, M. Akt isoforms in vascular disease. Vascul. Pharmacol. 2015, 71, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Shiojima, I.; Walsh, K. Regulation of cardiac growth and coronary angiogenesis by the Akt/PKB signaling pathway. Genes Dev. 2006, 20, 3347–3365. [Google Scholar] [CrossRef] [PubMed]
- Bravo-San Pedro, J.M.; Kroemer, G.; Galluzzi, L. Autophagy and Mitophagy in Cardiovascular Disease. Circ. Res. 2017, 120, 1812–1824. [Google Scholar] [CrossRef] [PubMed]
- Sciarretta, S.; Forte, M.; Frati, G.; Sadoshima, J. New Insights Into the Role of mTOR Signaling in the Cardiovascular System. Circ. Res. 2018, 122, 489–505. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Luo, L.; Chen, J. Roles of mTOR Signaling in Tissue Regeneration. Cells 2019, 8, 1075. [Google Scholar] [CrossRef]
- Sciarretta, S.; Volpe, M.; Sadoshima, J. Mammalian target of rapamycin signaling in cardiac physiology and disease. Circ. Res. 2014, 114, 549–564. [Google Scholar] [CrossRef]
- Yu, L.; McPhee, C.K.; Zheng, L.; Mardones, G.A.; Rong, Y.; Peng, J.; Mi, N.; Zhao, Y.; Liu, Z.; Wan, F.; et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 2010, 465, 942–946. [Google Scholar] [CrossRef] [PubMed]
- Munson, M.J.; Ganley, I.G. MTOR, PIK3C3, and autophagy: Signaling the beginning from the end. Autophagy 2015, 11, 2375–2376. [Google Scholar] [CrossRef] [PubMed]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR signaling in growth and metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.H.; Lipovsky, A.I.; Dibble, C.C.; Sahin, M.; Manning, B.D. S6K1 regulates GSK3 under conditions of mTOR-dependent feedback inhibition of Akt. Mol. Cell 2006, 24, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Li, Y.; Zhu, T.; Wu, J.; Guan, K.L. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 2002, 4, 648–657. [Google Scholar] [CrossRef]
- Carroll, B.; Maetzel, D.; Maddocks, O.D.; Otten, G.; Ratcliff, M.; Smith, G.R.; Dunlop, E.A.; Passos, J.F.; Davies, O.R.; Jaenisch, R.; et al. Control of TSC2-Rheb signaling axis by arginine regulates mTORC1 activity. eLife 2016, 5, e11058. [Google Scholar] [CrossRef] [PubMed]
- Niehrs, C. The complex world of WNT receptor signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 767–779. [Google Scholar] [CrossRef]
- Foulquier, S.; Daskalopoulos, E.P.; Lluri, G.; Hermans, K.C.M.; Deb, A.; Blankesteijn, W.M. WNT Signaling in Cardiac and Vascular Disease. Pharmacol. Rev. 2018, 70, 68–141. [Google Scholar] [CrossRef]
- Acebron, S.P.; Niehrs, C. β-Catenin-Independent Roles of Wnt/LRP6 Signaling. Trends Cell Biol. 2016, 26, 956–967. [Google Scholar] [CrossRef]
- Savini, M.; Wang, M.C. Does Autophagy Promote Longevity? It Depends. Cell 2019, 177, 221–222. [Google Scholar] [CrossRef]
- Madeo, F.; Tavernarakis, N.; Kroemer, G. Can autophagy promote longevity? Nat. Cell Biol. 2010, 12, 842–846. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR signaling at a glance. J. Cell Sci. 2009, 122, 3589–3594. [Google Scholar] [CrossRef]
- Abu-Remaileh, M.; Wyant, G.A.; Kim, C.; Laqtom, N.N.; Abbasi, M.; Chan, S.H.; Freinkman, E.; Sabatini, D.M. Lysosomal metabolomics reveals V-ATPase- and mTOR-dependent regulation of amino acid efflux from lysosomes. Science 2017, 358, 807–813. [Google Scholar] [CrossRef] [PubMed]
- García-Prat, L.; Martínez-Vicente, M.; Perdiguero, E.; Ortet, L.; Rodríguez-Ubreva, J.; Rebollo, E.; Ruiz-Bonilla, V.; Gutarra, S.; Ballestar, E.; Serrano, A.L.; et al. Autophagy maintains stemness by preventing senescence. Nature 2016, 529, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.T.; Warr, M.R.; Adelman, E.R.; Lansinger, O.M.; Flach, J.; Verovskaya, E.V.; Figueroa, M.E.; Passegué, E. Autophagy maintains the metabolism and function of young and old stem cells. Nature 2017, 543, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Rethineswaran, V.K.; Kim, Y.J.; Jang, W.B.; Ji, S.T.; Kang, S.; Kim, D.Y.; Park, J.H.; Van, L.T.H.; Giang, L.T.T.; Ha, J.S.; et al. Enzyme-Aided Extraction of Fucoidan by AMG Augments the Functionality of EPCs through Regulation of the AKT/Rheb Signaling Pathway. Mar. Drugs 2019, 17, 392. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rethineswaran, V.K.; Kim, D.Y.; Kim, Y.-J.; Jang, W.; Ji, S.T.; Van, L.T.H.; Giang, L.T.T.; Ha, J.S.; Yun, J.; Jung, J.; et al. CHIR99021 Augmented the Function of Late Endothelial Progenitor Cells by Preventing Replicative Senescence. Int. J. Mol. Sci. 2021, 22, 4796. https://doi.org/10.3390/ijms22094796
Rethineswaran VK, Kim DY, Kim Y-J, Jang W, Ji ST, Van LTH, Giang LTT, Ha JS, Yun J, Jung J, et al. CHIR99021 Augmented the Function of Late Endothelial Progenitor Cells by Preventing Replicative Senescence. International Journal of Molecular Sciences. 2021; 22(9):4796. https://doi.org/10.3390/ijms22094796
Chicago/Turabian StyleRethineswaran, Vinoth Kumar, Da Yeon Kim, Yeon-Ju Kim, WoongBi Jang, Seung Taek Ji, Le Thi Hong Van, Ly Thanh Truong Giang, Jong Seong Ha, Jisoo Yun, Jinsup Jung, and et al. 2021. "CHIR99021 Augmented the Function of Late Endothelial Progenitor Cells by Preventing Replicative Senescence" International Journal of Molecular Sciences 22, no. 9: 4796. https://doi.org/10.3390/ijms22094796
APA StyleRethineswaran, V. K., Kim, D. Y., Kim, Y. -J., Jang, W., Ji, S. T., Van, L. T. H., Giang, L. T. T., Ha, J. S., Yun, J., Jung, J., & Kwon, S. -M. (2021). CHIR99021 Augmented the Function of Late Endothelial Progenitor Cells by Preventing Replicative Senescence. International Journal of Molecular Sciences, 22(9), 4796. https://doi.org/10.3390/ijms22094796