
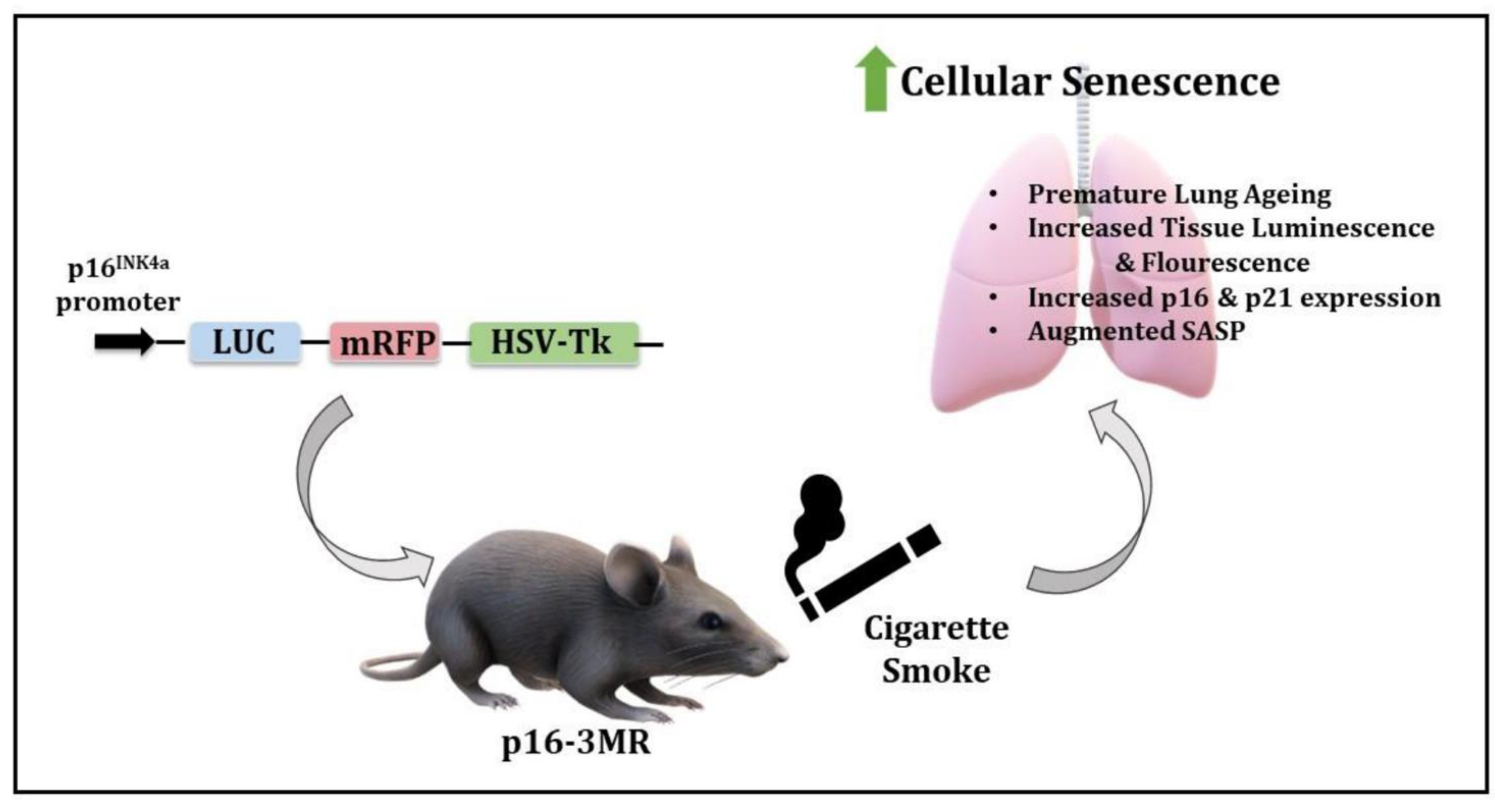
p16-3MR: A Novel Model to Study Cellular Senescence in Cigarette Smoke-Induced Lung Injuries



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
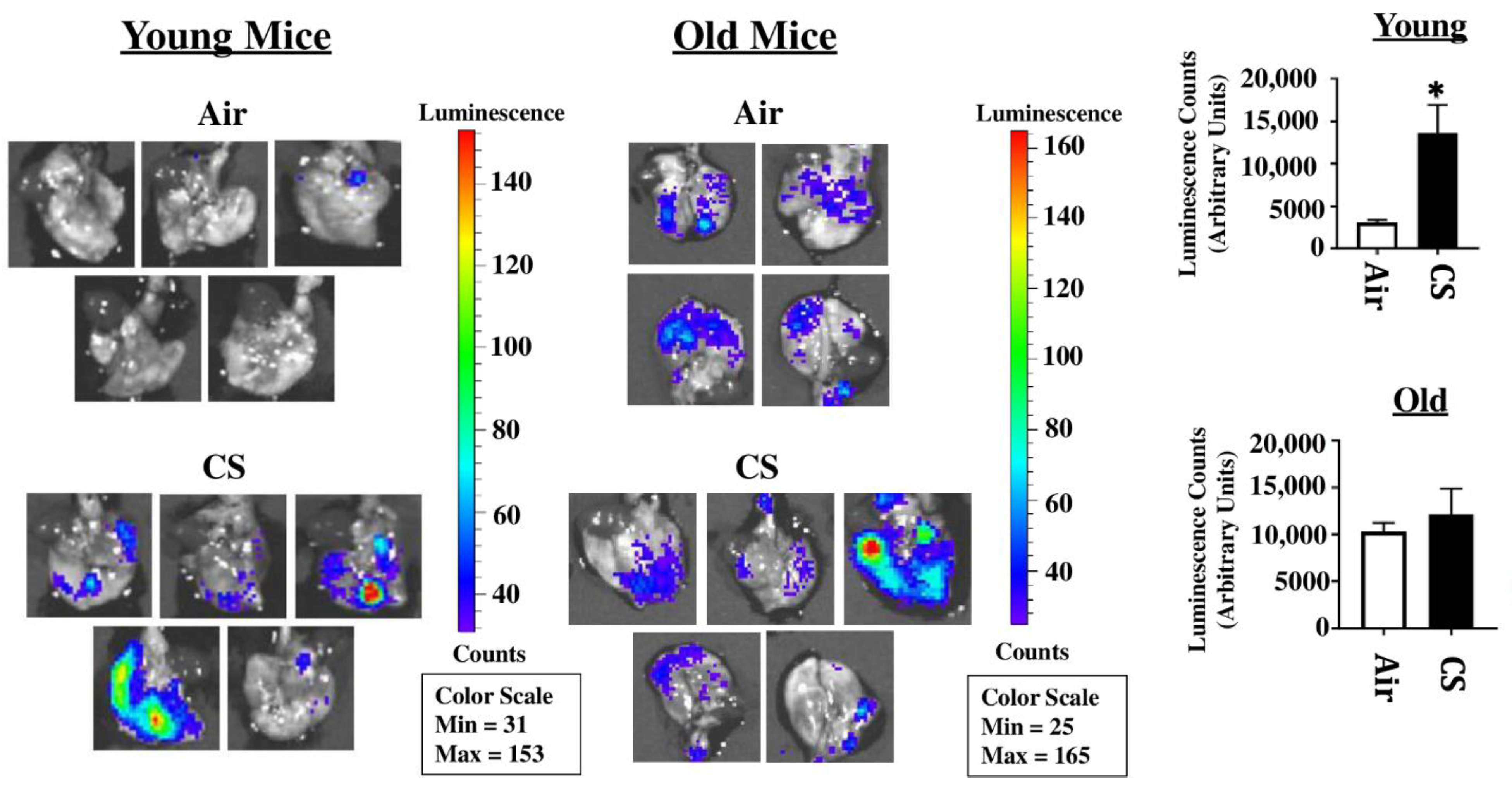
2.1. Sub-Chronic CS Exposure Augments Luminescence Indicative of p16 Expression in the Lungs of p16-3MR Reporter Mice
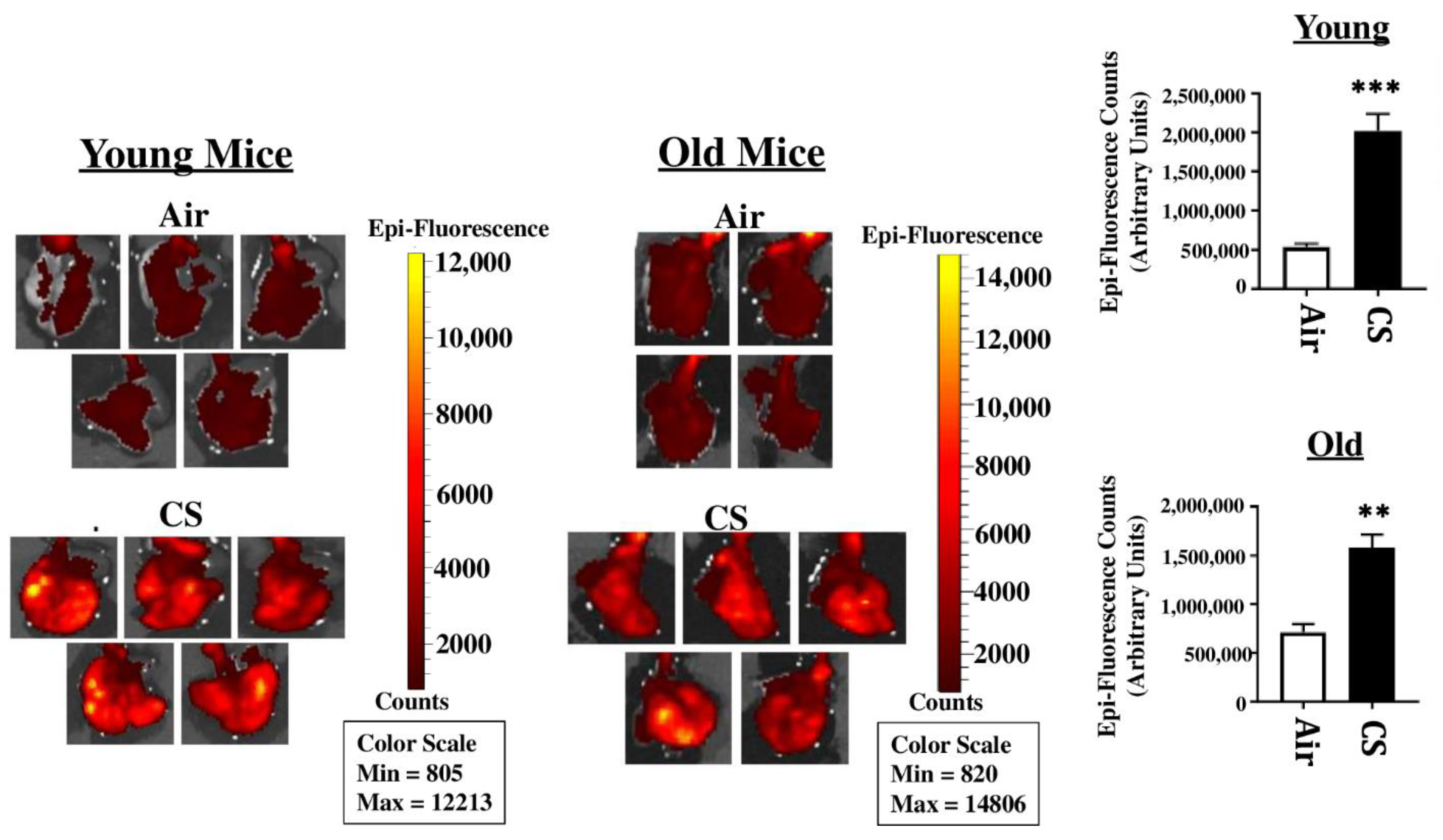
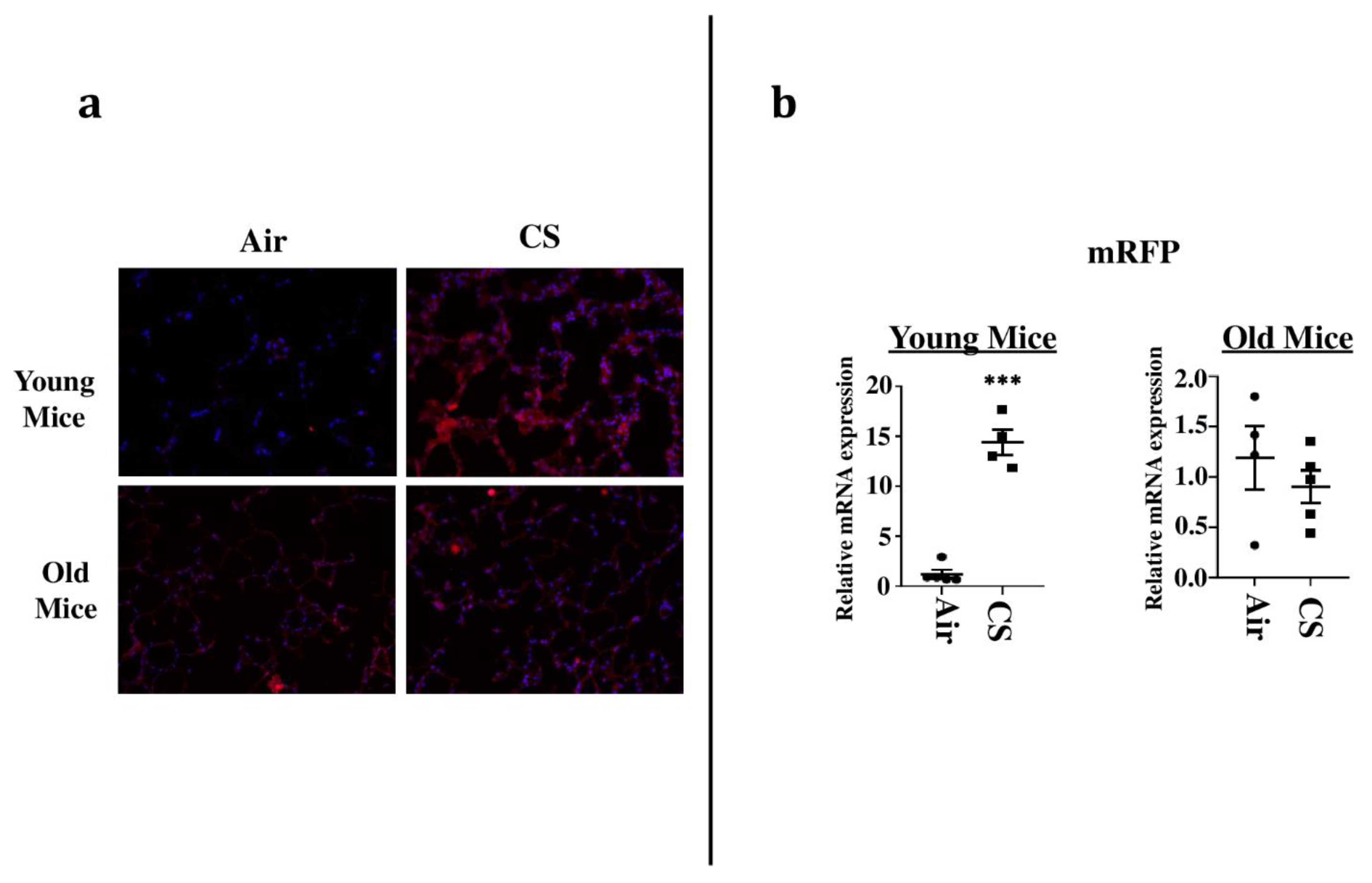
2.2. Sub-Chronic Exposure to CS Results in a Significant Increase in the mRFP Expression in the Lungs of Young p16-3MR Mice
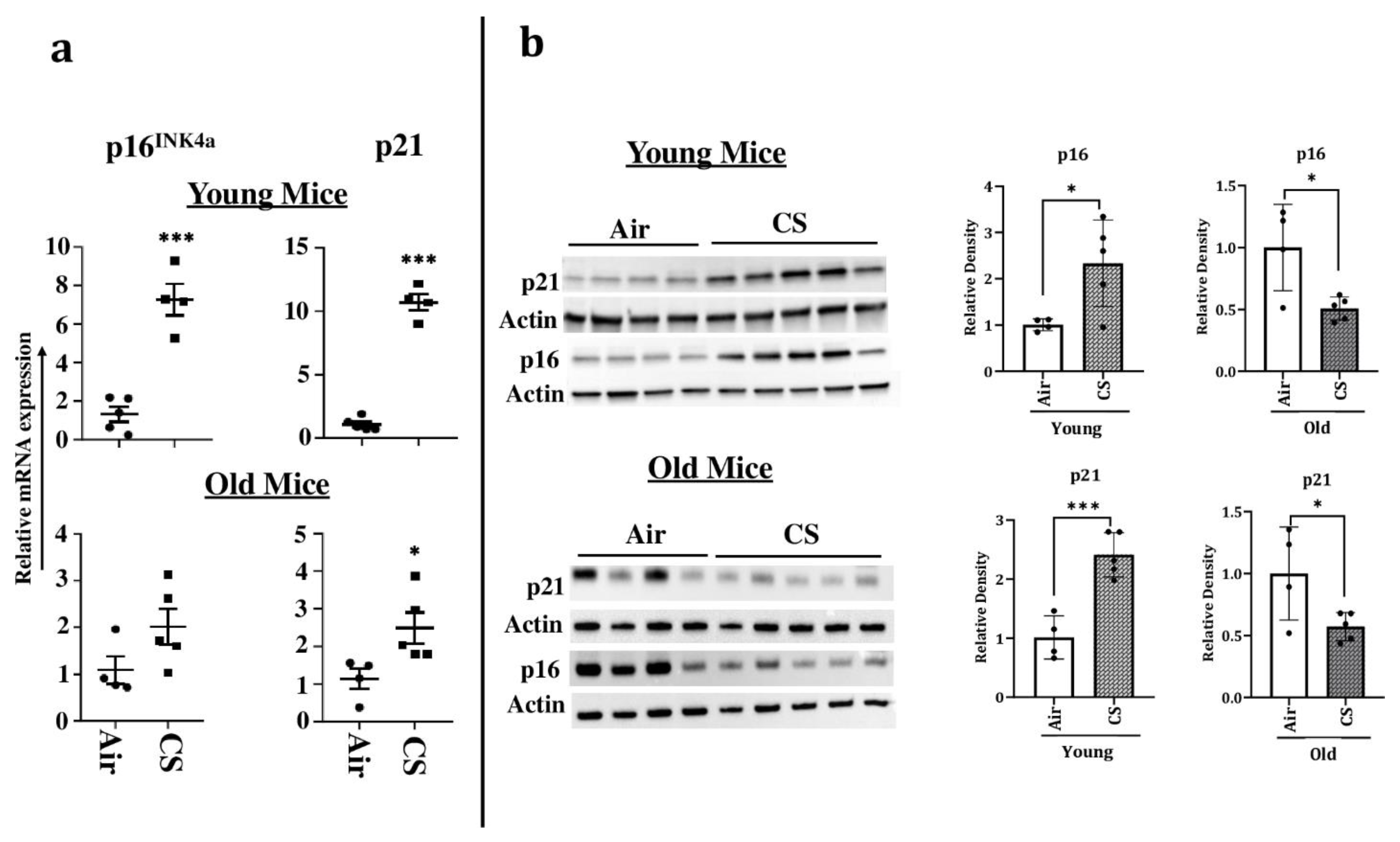
2.3. Increased Expression of Cellular-Senescence Markers (p16 and p21) Following Sub-Chronic CS Exposure in Young p16-3MR Mice
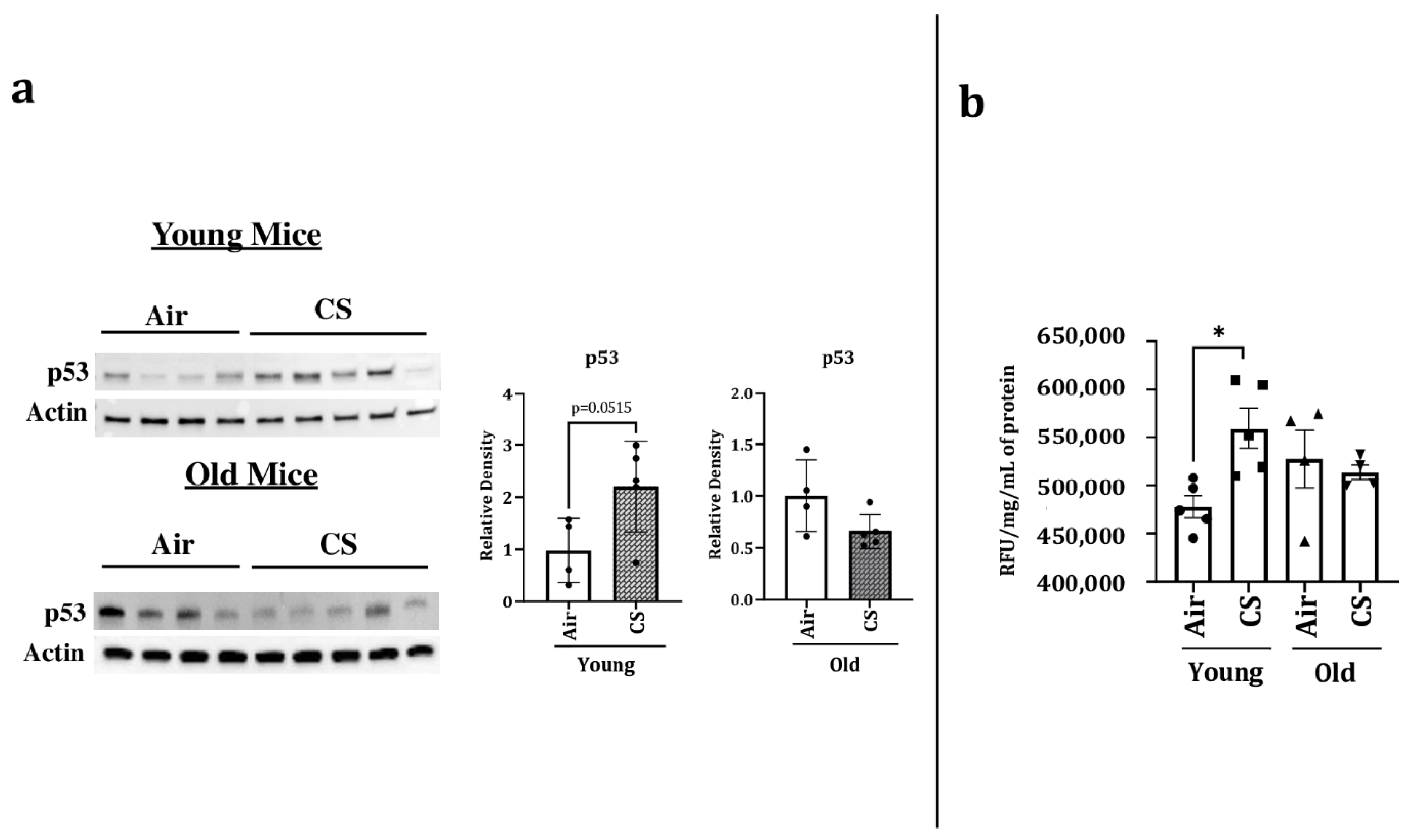
2.4. Sub-Chronic CS Exposure Contributes towards the Augmentation of p53 Protein Expression and SA-β-gal Activity in the Lungs of p16-3MR Mice
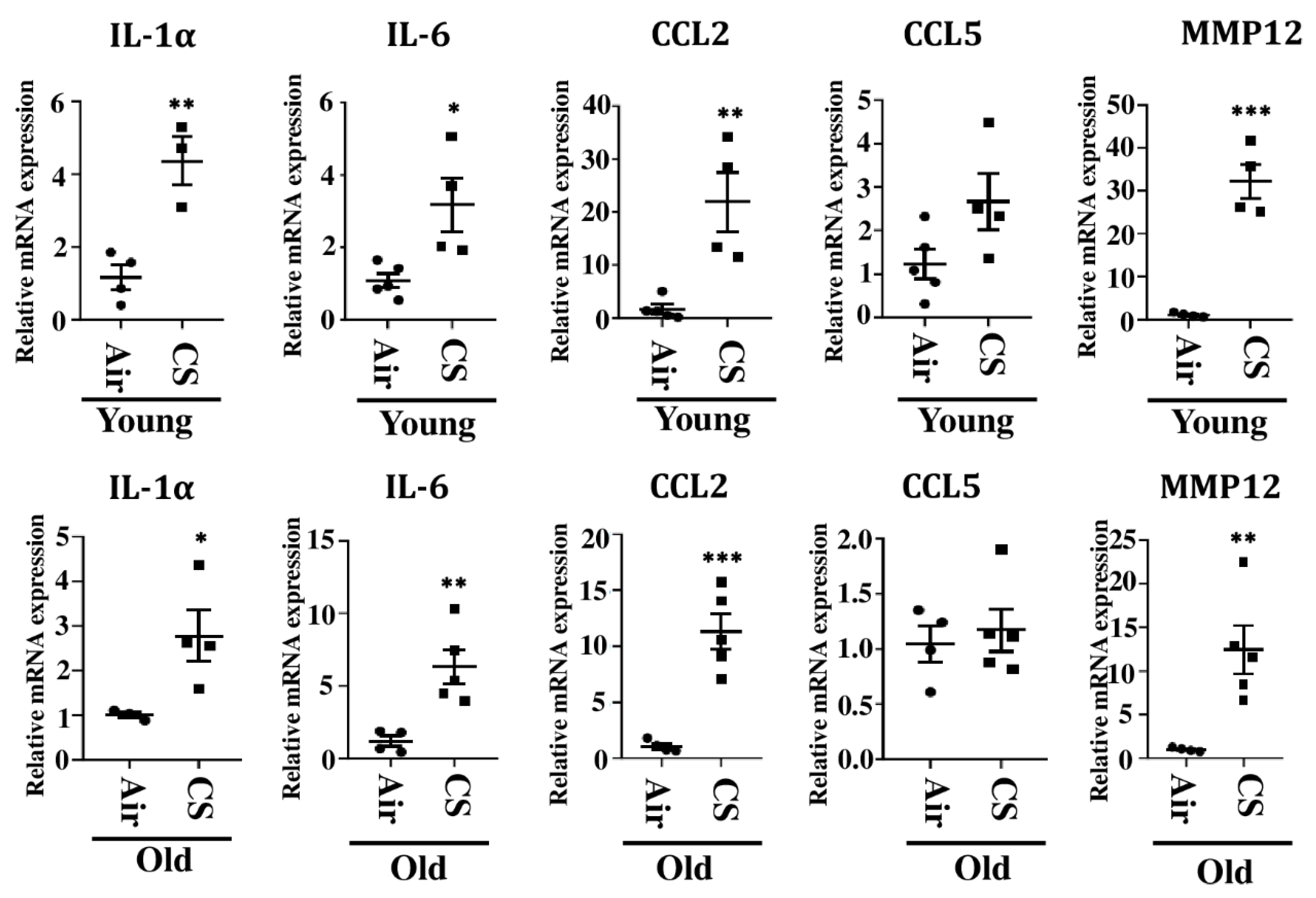
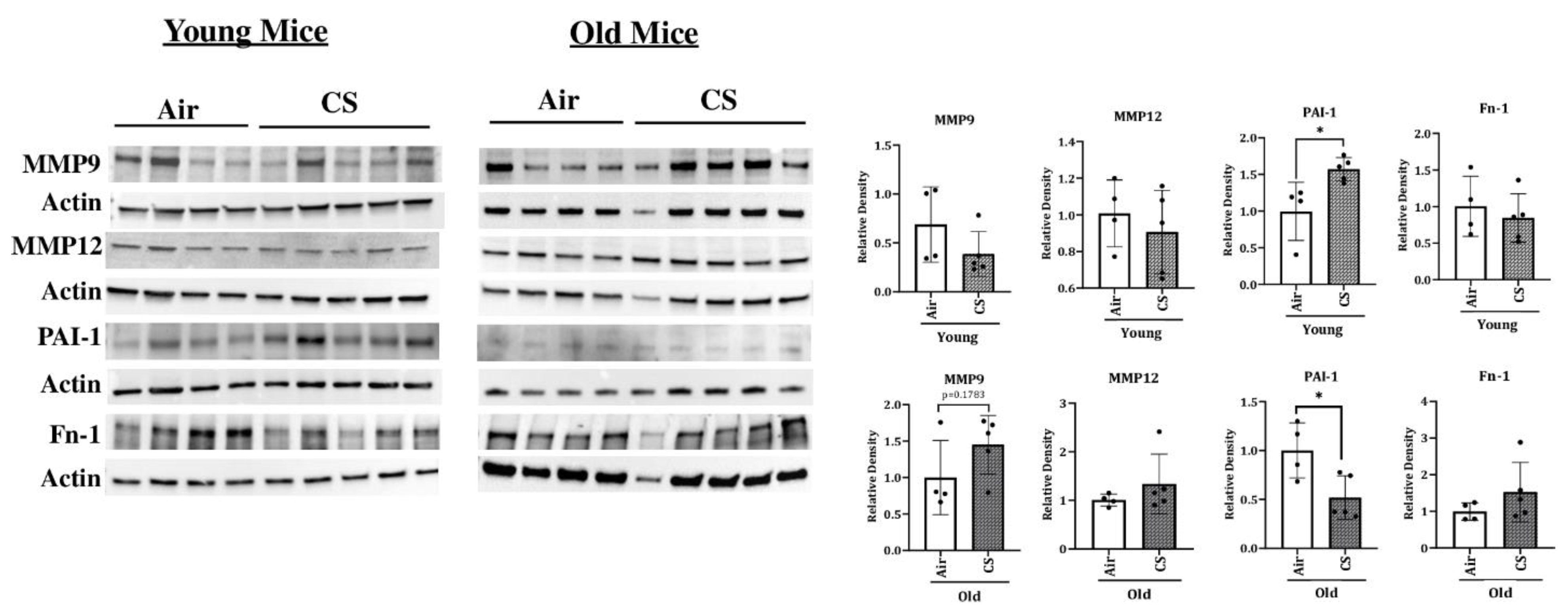
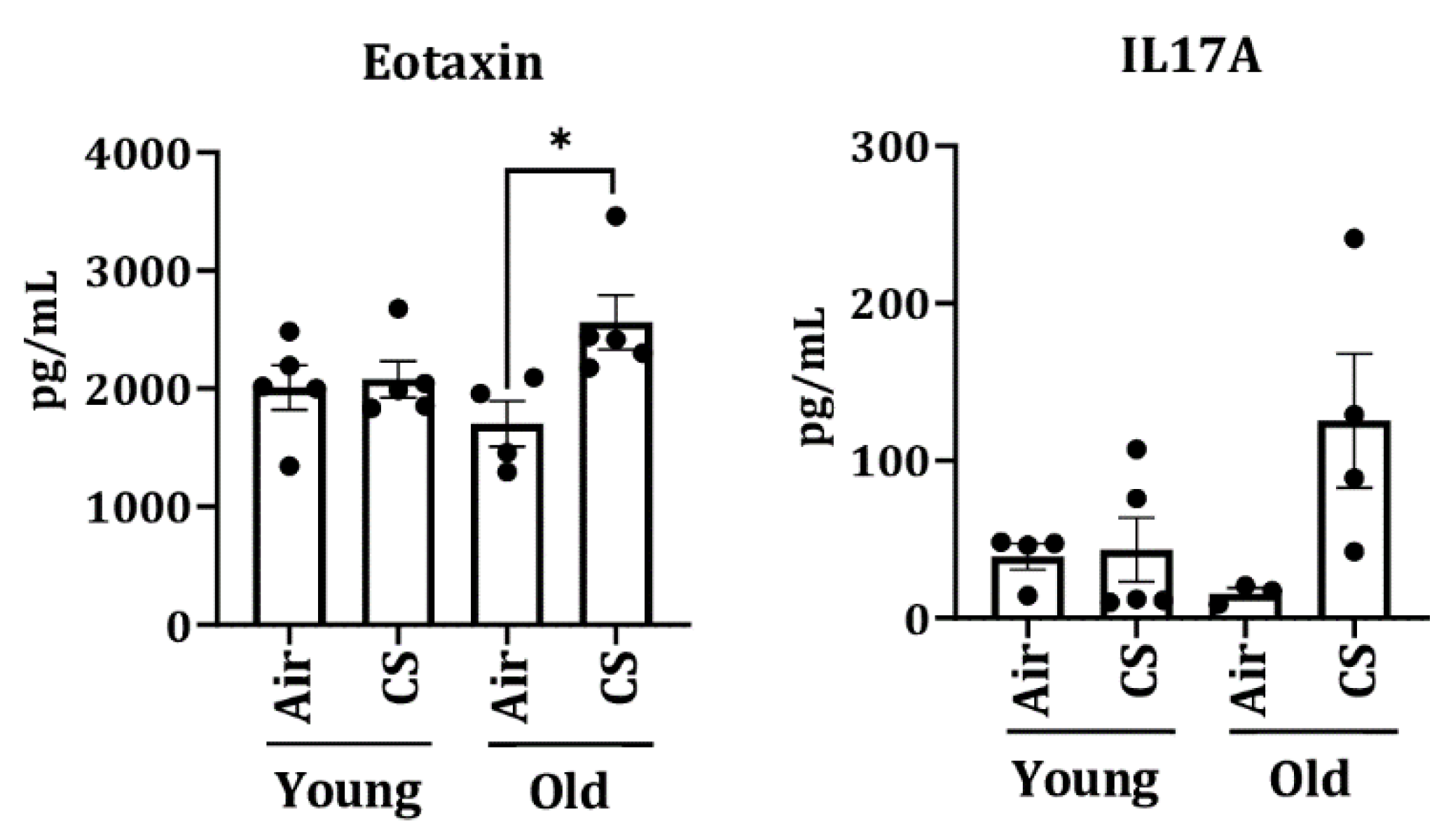
2.5. Alterations in the mRNA and Protein Expression of SASP-Associated Genes in CS-Exposed p16-3MR Mice
3. Discussion
4. Materials and Methods
4.1. Ethics Statement and Scientific Rigor/Reproducibility
4.2. p16-3MR Mouse Model
4.3. Sub-Chronic CS Exposure
4.4. Tissue Luminescence and Fluorescence Using IVIS Imaging
4.5. Fluorescence Microscopy in Lung Tissue Section
4.6. mRNA Expression Analyses Using qPCR
4.7. Immunoblot Analysis
4.8. Measurement of SA-β-gal Activity
4.9. Assessment of Pro-Inflammatory Mediators Using Luminex
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tuder, R.M.; Kern, J.A.; Miller, Y.E. Senescence in chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc. 2012, 9, 62–63. [Google Scholar] [CrossRef] [PubMed]
- Karrasch, S.; Holz, O.; Jörres, R.A. Aging and induced senescence as factors in the pathogenesis of lung emphysema. Respir. Med. 2008, 102, 1215–1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundar, I.K.; Rashid, K.; Gerloff, J.; Li, D.; Rahman, I. Genetic Ablation of p16INK4a Does Not Protect against Cellular Senescence in Mouse Models of Chronic Obstructive Pulmonary Disease/Emphysema. Am. J. Respir. Cell Mol. Biol. 2018, 59, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Sandford, A.; Man, P.; Sin, D.D. Is the aging process accelerated in chronic obstructive pulmonary disease? Curr. Opin. Pulm. Med. 2011, 17, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Onizawa, S.; Nagai, A.; Aoshiba, K. Epithelial cell senescence impairs repair process and exacerbates inflammation after airway injury. Respir. Res. 2011, 12, 78. [Google Scholar] [CrossRef] [Green Version]
- Tsuji, T.; Aoshiba, K.; Nagai, A. Alveolar cell senescence in patients with pulmonary emphysema. Am. J. Respir. Crit. Care Med. 2006, 174, 886–893. [Google Scholar] [CrossRef]
- Nyunoya, T.; Monick, M.M.; Klingelhutz, A.; Yarovinsky, T.O.; Cagley, J.R.; Hunninghake, G.W. Cigarette smoke induces cellular senescence. Am. J. Respir. Cell Mol. Biol. 2006, 35, 681–688. [Google Scholar] [CrossRef] [Green Version]
- Amsellem, V.; Gary-Bobo, G.; Marcos, E.; Maitre, B.; Chaar, V.; Validire, P.; Stern, J.-B.; Noureddine, H.; Sapin, E.; Rideau, D.; et al. Telomere dysfunction causes sustained inflammation in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2011, 184, 1358–1366. [Google Scholar] [CrossRef]
- Campisi, J.; Di Fagagna, F.D. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef]
- Wallis, R.; Mizen, H.; Bishop, C.L. The bright and dark side of extracellular vesicles in the senescence-associated secretory phenotype. Mech. Ageing Dev. 2020, 189, 111263. [Google Scholar] [CrossRef]
- Franceschi, C.; Campisi, J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2014, 69 (Suppl. 1), S4–S9. [Google Scholar] [CrossRef]
- Liu, R.-M.; Liu, G. Cell senescence and fibrotic lung diseases. Exp. Gerontol. 2020, 132, 110836. [Google Scholar] [CrossRef]
- Van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Childs, B.G.; Gluscevic, M.; Baker, D.J.; Laberge, R.-M.; Marquess, D.; Dananberg, J.; Van Deursen, J.M. Senescent cells: An emerging target for diseases of ageing. Nat. Rev. Drug Discov. 2017, 16, 718–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patil, P.; Dong, Q.; Wang, D.; Chang, J.; Wiley, C.; DeMaria, M.; Lee, J.; Kang, J.; Niedernhofer, L.J.; Robbins, P.D.; et al. Systemic clearance of p16(INK4a)-positive senescent cells mitigates age-associated intervertebral disc degeneration. Aging Cell 2019, 18, e12927. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Chang, J.; Iyer, S.; Han, L.; Campisi, J.; Manolagas, S.C.; Zhou, D.; Almeida, M. Elimination of senescent osteoclast progenitors has no effect on the age-associated loss of bone mass in mice. Aging Cell 2019, 18, e12923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rocha, L.R.; Huu, V.A.N.; La Torre, C.P.; Xu, Q.; Jabari, M.; Krawczyk, M.; Weinreb, R.N.; Skowronska-Krawczyk, D. Early removal of senescent cells protects retinal ganglion cells loss in experimental ocular hypertension. Aging Cell 2019, 19, e13089. [Google Scholar] [CrossRef] [Green Version]
- DeMaria, M.; Ohtani, N.; Youssef, S.A.; Rodier, F.; Toussaint, W.; Mitchell, J.R.; Laberge, R.-M.; Vijg, J.; Van Steeg, H.; Dollé, M.E.; et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev. Cell 2014, 31, 722–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.-Y.; Souroullas, G.P.; Diekman, B.O.; Krishnamurthy, J.; Hall, B.M.; Sorrentino, J.A.; Parker, J.S.; Sessions, G.A.; Gudkov, A.V.; Sharpless, N.E. Cells exhibiting strong p16(INK4a) promoter activation in vivo display features of senescence. Proc. Natl. Acad. Sci. USA 2019, 116, 2603–2611. [Google Scholar] [CrossRef] [Green Version]
- Rashid, K.; Sundar, I.K.; Gerloff, J.; Li, D.; Rahman, I. Lung cellular senescence is independent of aging in a mouse model of COPD/emphysema. Sci. Rep. 2018, 8, 9023. [Google Scholar] [CrossRef] [Green Version]
- Paez-Ribes, M.; González-Gualda, E.; Doherty, G.J.; Muñoz-Espín, D. Targeting senescent cells in translational medicine. EMBO Mol. Med. 2019, 11, e10234. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Barnes, P.J. COPD as a disease of accelerated lung aging. Chest 2009, 135, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Selman, M.; López-Otín, C.; Pardo, A. Age-driven developmental drift in the pathogenesis of idiopathic pulmonary fibrosis. Eur. Respir. J. 2016, 48, 538–552. [Google Scholar] [CrossRef] [PubMed]
- Nash, S.H.; Liao, L.M.; Harris, T.B.; Freedman, N.D. Cigarette Smoking and Mortality in Adults Aged 70 Years and Older: Results From the NIH-AARP Cohort. Am. J. Prev. Med. 2017, 52, 276–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schafer, M.J.; White, T.A.; Iijima, K.; Haak, A.J.; Ligresti, G.; Atkinson, E.J.; Oberg, A.L.; Birch, J.; Salmonowicz, H.; Zhu, Y.; et al. Cellular senescence mediates fibrotic pulmonary disease. Nat. Commun. 2017, 8, 14532. [Google Scholar] [CrossRef]
- Wu, J.; Dong, F.; Wang, R.-A.; Wang, J.; Zhao, J.; Yang, M.; Gong, W.; Cui, R.; Dong, L. Central role of cellular senescence in TSLP-induced airway remodeling in asthma. PLoS ONE 2013, 8, e77795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandsma, C.-A.; De Vries, M.; Costa, R.; Woldhuis, R.R.; Königshoff, M.; Timens, W. Lung ageing and COPD: Is there a role for ageing in abnormal tissue repair? Eur. Respir. Rev. 2017, 26, 170073. [Google Scholar] [CrossRef] [Green Version]
- Durham, A.; Adcock, I. The relationship between COPD and lung cancer. Lung Cancer 2015, 90, 121–127. [Google Scholar] [CrossRef] [Green Version]
- Sorrentino, J.A.; Krishnamurthy, J.; Tilley, S.; Alb, J.G.; Burd, C.E.; Sharpless, N.E. p16INK4a reporter mice reveal age-promoting effects of environmental toxicants. J. Clin. Investig. 2014, 124, 169–173. [Google Scholar] [CrossRef]
- Burd, C.E.; Sorrentino, J.A.; Clark, K.S.; Darr, D.B.; Krishnamurthy, J.; Deal, A.M.; Bardeesy, N.; Castrillon, D.H.; Beach, D.H.; Sharpless, N.E. Monitoring Tumorigenesis and Senescence In Vivo with a p16INK4a-Luciferase Model. Cell 2013, 152, 340–351. [Google Scholar] [CrossRef] [Green Version]
- Omori, S.; Wang, T.-W.; Johmura, Y.; Kanai, T.; Nakano, Y.; Kido, T.; Susaki, E.A.; Nakajima, T.; Shichino, S.; Ueha, S.; et al. Generation of a p16 Reporter Mouse and Its Use to Characterize and Target p16high Cells In Vivo. Cell Metab. 2020, 32, 814–828.e6. [Google Scholar] [CrossRef]
- Folgueras, A.R.; Freitas-Rodríguez, S.; Velasco, G.; López-Otín, C. Mouse Models to Disentangle the Hallmarks of Human Aging. Circ. Res. 2018, 123, 905–924. [Google Scholar] [CrossRef] [PubMed]
- Oh, C.K.; Ariue, B.; Alban, R.F.; Shaw, B.; Cho, S.H. PAI-1 promotes extracellular matrix deposition in the airways of a murine asthma model. Biochem. Biophys. Res. Commun. 2002, 294, 1155–1160. [Google Scholar] [CrossRef]
- Freitas-Rodríguez, S.; Folgueras, A.R.; López-Otín, C. The role of matrix metalloproteinases in aging: Tissue remodeling and beyond. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2017, 1864, 2015–2025. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, J.J.; Lutey, B.A.; Suzuki, Y.; Toennies, H.M.; Kelley, D.G.; Kobayashi, D.K.; Ijem, W.G.; Deslee, G.; Moore, C.H.; Jacobs, M.E.; et al. The role of matrix metalloproteinase-9 in cigarette smoke-induced emphysema. Am. J. Respir. Crit. Care Med. 2011, 183, 876–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirkland, J.L.; Tchkonia, T. Senolytic drugs: From discovery to translation. J. Intern. Med. 2020, 288, 518–536. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Edirisinghe, I.; Rajendrasozhan, S.; Yang, S.-R.; Caito, S.; Adenuga, D.; Rahman, I. Cigarette smoke-mediated inflammatory and oxidative responses are strain-dependent in mice. Am. J. Physiol. Cell. Mol. Physiol. 2008, 294, L1174–L1186. [Google Scholar] [CrossRef] [PubMed]
- Rajendrasozhan, S.; Chung, S.; Sundar, I.K.; Yao, H.; Rahman, I. Targeted disruption of NF-{kappa}B1 (p50) augments cigarette smoke-induced lung inflammation and emphysema in mice: A critical role of p50 in chromatin remodeling. Am. J. Physiol. Cell. Mol. Physiol. 2010, 298, L197–L209. [Google Scholar] [CrossRef] [Green Version]
- Sundar, I.K.; Rahman, I. Gene expression profiling of epigenetic chromatin modification enzymes and histone marks by cigarette smoke: Implications for COPD and lung cancer. Am. J. Physiol. Cell. Mol. Physiol. 2016, 311, L1245–L1258. [Google Scholar] [CrossRef] [Green Version]
- Sundar, I.K.; Rashid, K.; Gerloff, J.; Rangel-Moreno, J.; Li, D.; Rahman, I. Genetic ablation of histone deacetylase 2 leads to lung cellular senescence and lymphoid follicle formation in COPD/emphysema. FASEB J. 2018, 32, 4955–4971. [Google Scholar] [CrossRef] [PubMed] [Green Version]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaur, G.; Sundar, I.K.; Rahman, I. p16-3MR: A Novel Model to Study Cellular Senescence in Cigarette Smoke-Induced Lung Injuries. Int. J. Mol. Sci. 2021, 22, 4834. https://doi.org/10.3390/ijms22094834
Kaur G, Sundar IK, Rahman I. p16-3MR: A Novel Model to Study Cellular Senescence in Cigarette Smoke-Induced Lung Injuries. International Journal of Molecular Sciences. 2021; 22(9):4834. https://doi.org/10.3390/ijms22094834
Chicago/Turabian StyleKaur, Gagandeep, Isaac K. Sundar, and Irfan Rahman. 2021. "p16-3MR: A Novel Model to Study Cellular Senescence in Cigarette Smoke-Induced Lung Injuries" International Journal of Molecular Sciences 22, no. 9: 4834. https://doi.org/10.3390/ijms22094834
APA StyleKaur, G., Sundar, I. K., & Rahman, I. (2021). p16-3MR: A Novel Model to Study Cellular Senescence in Cigarette Smoke-Induced Lung Injuries. International Journal of Molecular Sciences, 22(9), 4834. https://doi.org/10.3390/ijms22094834