
BiFC Method Based on Intraorganellar Protein Crowding Detects Oleate-Dependent Peroxisomal Targeting of Pichia pastoris Malate Dehydrogenase


Abstract
:1. Introduction
2. Results
2.1. NADH-Shuttling Proteins in P. pastoris
2.2. Localization of PpMdhA-GFP, PpMdhB-GFP, and PpGpdA-GFP in Different Carbon Sources
2.3. An Assay to Improve Detection of Peroxisomally-Localized Proteins
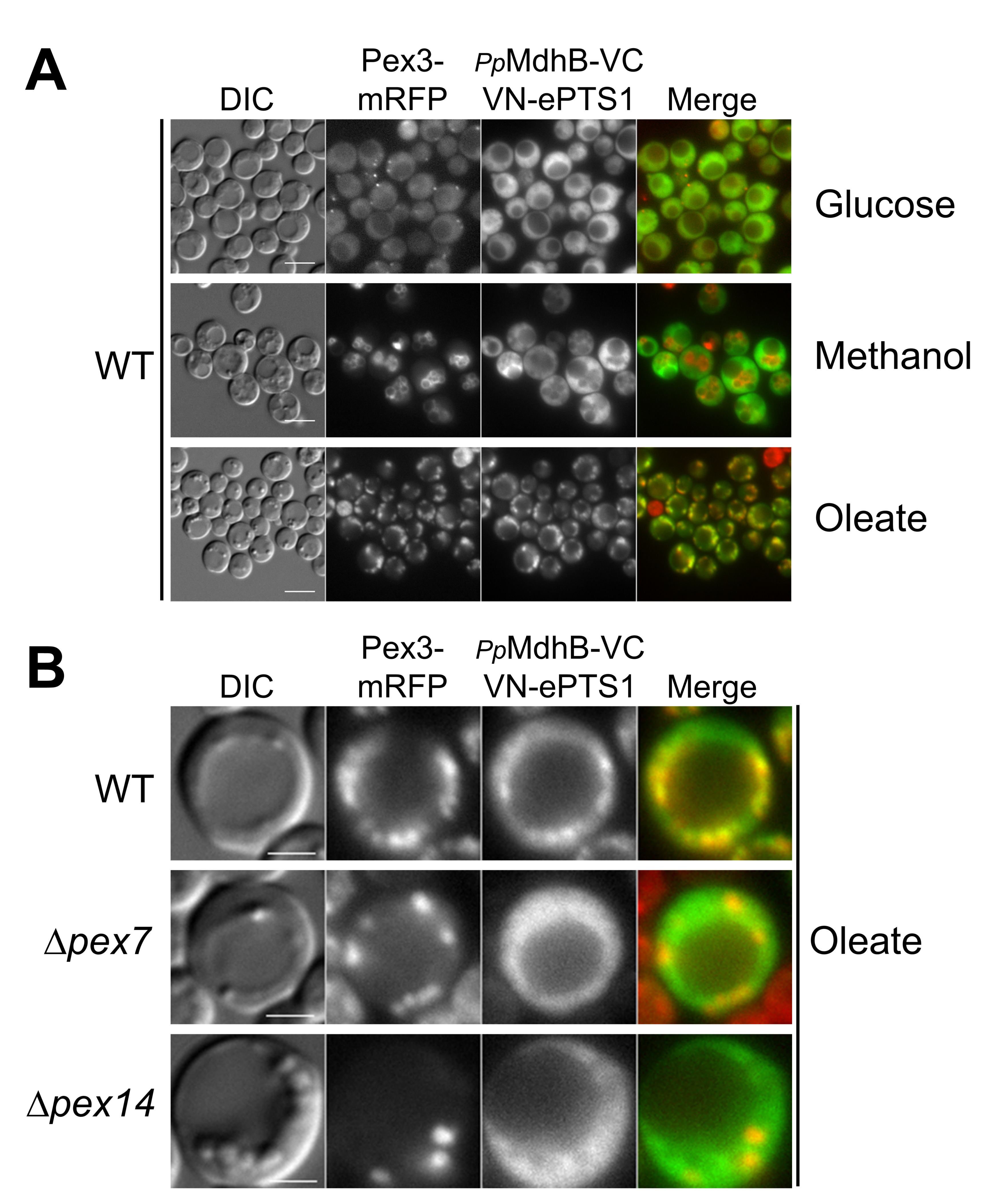
2.4. Testing the Localization of the NADH-Shuttling Proteins Using the BiFC Assay
2.5. VC Fused to the N-Terminus of PpMdhB Is Not Imported to Peroxisomes
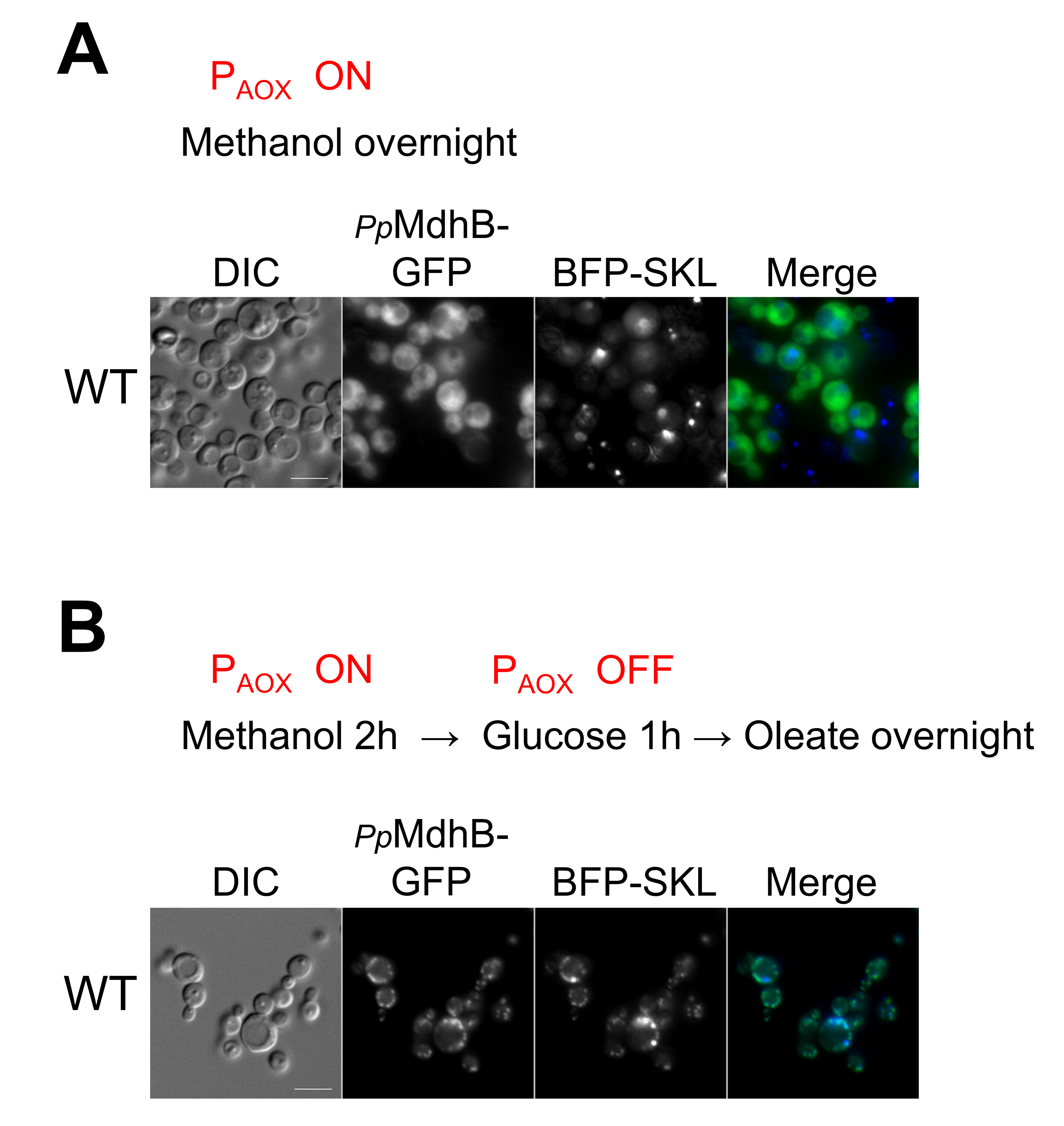
2.6. Independent Confirmation of the Peroxisomal Localization of PpMdhB-GFP in Oleate
3. Discussion
3.1. Physiological Roles of NADH-Shuttling Proteins in P. pastoris
3.2. Targeting of PpMdhB to Peroxisomes
4. Materials and Methods
4.1. Yeast Strains and Media
4.2. Plasmid Constructions
4.3. Yeast Strain Constructions
4.4. Fluorescence Microscopy
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| EYFP | enhanced yellow fluorescent protein |
| BiFC | Bimolecular Fluorescence Complementation |
| Mdh | malate dehydrogenase |
| Gpd | glycerol-3-phosphate dehydrogenase |
| PTS | peroxisomal targeting signal |
| VN | Venus N-terminal 1–158 amino acids |
| VC | Venus C-terminal 159–239 amino acids |
References
- Hu, C.-D.; Chinenov, Y.; Kerppola, T.K. Visualization of interactions among bZIP and Rel family proteins in living cells using bimolecular fluorescence complementation. Mol. Cell 2002, 9, 789–798. [Google Scholar] [CrossRef]
- Nagai, T.; Ibata, K.; Park, E.S.; Kubota, M.; Mikoshiba, K.; Miyawaki, A. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat. Biotechnol. 2002, 20, 87–90. [Google Scholar] [CrossRef]
- Goodman, R.P.; Calvo, S.E.; Mootha, V.K. Spatiotemporal compartmentalization of hepatic NADH and NADPH metabolism. J. Biol. Chem. 2018, 293, 7508–7516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakker, B.M.; Overkamp, K.M.; van Maris, A.J.; Kotter, P.; Luttik, M.A.; van Dijken, J.P.; Pronk, J.T. Stoichiometry and compartmentation of NADH metabolism in Saccharomyces cerevisiae. FEMS Microbiol. Rev. 2001, 25, 15–37. [Google Scholar] [CrossRef]
- Al-Saryi, N.A.; Al-Hejjaj, M.Y.; Van Roermund, C.W.T.; Hulmes, G.E.; Ekal, L.; Payton, C.; Wanders, R.J.A.; Hettema, E.H. Two NAD-linked redox shuttles maintain the peroxisomal redox balance in Saccharomyces cerevisiae. Sci. Rep. 2017, 7, 11868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neuberger, G.; Maurer-Stroh, S.; Eisenhaber, B.; Hartig, A.; Eisenhaber, F. Prediction of peroxisomal targeting signal 1 containing proteins from amino acid sequence. J. Mol. Biol. 2003, 328, 581–592. [Google Scholar] [CrossRef]
- Petriv, O.I.; Tang, L.; Titorenko, V.I.; Rachubinski, R.A. A new definition for the consensus sequence of the peroxisome targeting signal type 2. J. Mol. Biol. 2004, 341, 119–134. [Google Scholar] [CrossRef]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Claros, M.G.; Vincens, P. Computational method to predict mitochondrially imported proteins and their targeting sequences. J. Biol. Inorg. Chem. 1996, 241, 779–786. [Google Scholar] [CrossRef]
- Shyu, Y.J.; Suarez, C.D.; Hu, C.D. Visualization of ternary complexes in living cells by using a BiFC-based FRET assay. Nat. Protoc. 2008, 3, 1693–1702. [Google Scholar] [CrossRef]
- DeLoache, W.C.; Russ, Z.N.; Dueber, J.E. Towards repurposing the yeast peroxisome for compartmentalizing heterologous metabolic pathways. Nat. Commun. 2016, 7, 11152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elgersma, Y.; Elgersma-Hooisma, M.; Wenzel, T.; McCaffery, J.M.; Farquhar, M.G.; Subramani, S. A mobile PTS2 receptor for peroxisomal protein import in Pichia pastoris. J. Cell Biol. 1998, 140, 807–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nazarko, T.Y.; Farré, J.-C.; Subramani, S. Peroxisome size provides insights into the function of autophagy-related proteins. Mol. Biol. Cell 2009, 20, 3828–3839. [Google Scholar] [CrossRef] [Green Version]
- Al Saryi, N.A.; Hutchinson, J.D.; Al-Hejjaj, M.Y.; Sedelnikova, S.; Baker, P.; Hettema, E.H. Pnc1 piggy-back import into peroxisomes relies on Gpd1 homodimerisation. Sci. Rep. 2017, 7, 42579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofhuis, J.; Schueren, F.; Nötzel, C.; Lingner, T.; Gärtner, J.; Jahn, O.; Thoms, S. The functional readthrough extension of malate dehydrogenase reveals a modification of the genetic code. Open Biol. 2016, 6, 160246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stiebler, A.C.; Freitag, J.; Schink, K.O.; Stehlik, T.; Tillmann, B.A.M.; Ast, J.; Bölker, M. Ribosomal readthrough at a short UGA stop codon context triggers dual localization of metabolic enzymes in fungi and animals. PLoS Genet. 2014, 10, e1004685. [Google Scholar] [CrossRef]
- Kabran, P.; Rossignol, T.; Gaillardin, C.; Nicaud, J.-M.; Neuvéglise, C. Alternative splicing regulates targeting of malate dehydrogenase in Yarrowia lipolytica. DNA Res. 2012, 19, 231–244. [Google Scholar] [CrossRef] [Green Version]
- Strijbis, K.; Burg, J.V.D.; Visser, W.F.; Berg, M.V.D.; Distel, B. Alternative splicing directs dual localization of Candida albicans 6-phosphogluconate dehydrogenase to cytosol and peroxisomes. FEMS Yeast Res. 2011, 12, 61–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freitag, J.; Ast, J.; Bölker, M. Cryptic peroxisomal targeting via alternative splicing and stop codon read-through in fungi. Nat. Cell Biol. 2012, 485, 522–525. [Google Scholar] [CrossRef] [PubMed]
- Szewczyk, E.; Andrianopoulos, A.; Davis, M.A.; Hynes, M.J. A single gene produces mitochondrial, cytoplasmic, and peroxisomal NADP-dependent isocitrate dehydrogenase in Aspergillus nidulans. J. Biol. Chem. 2001, 276, 37722–37729. [Google Scholar] [CrossRef] [Green Version]
- Wimmer, B.; Lottspeich, F.; Van Der Klei, I.; Veenhuis, M.; Gietl, C. The glyoxysomal and plastid molecular chaperones (70-kDa heat shock protein) of watermelon cotyledons are encoded by a single gene. Proc. Natl. Acad. Sci. USA 1997, 94, 13624–13629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marelli, M.; Smith, J.J.; Jung, S.; Yi, E.; Nesvizhskii, A.I.; Christmas, R.H.; Saleem, R.A.; Tam, Y.Y.C.; Fagarasanu, A.; Goodlett, D.R.; et al. Quantitative mass spectrometry reveals a role for the GTPase Rho1p in actin organization on the peroxisome membrane. J. Cell Biol. 2004, 167, 1099–1112. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.; Marelli, M.; Rachubinski, R.A.; Goodlett, D.R.; Aitchison, J.D. Dynamic changes in the subcellular distribution of Gpd1p in response to cell stress. J. Biol. Chem. 2010, 285, 6739–6749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Singh, R.; Williams, C.P.; van der Klei, I.J. Stress exposure results in increased peroxisomal levels of yeast Pnc1 and Gpd1, which are imported via a piggy-backing mechanism. Biochim. Biophys. Acta 2016, 1863, 148–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valadi, Å.; Granath, K.; Gustafsson, L.; Adler, L. Distinct intracellular localization of Gpd1p and Gpd2p, the two yeast isoforms of NAD+-dependent glycerol-3-phosphate dehydrogenase, explains their different contributions to redox-driven glycerol production. J. Biol. Chem. 2004, 279, 39677–39685. [Google Scholar] [CrossRef] [Green Version]
- Meyer, T.; Hölscher, C.; Schwöppe, C.; Von Schaewen, A. Alternative targeting of Arabidopsis plastidic glucose-6-phosphate dehydrogenase G6PD1 involves cysteine-dependent interaction with G6PD4 in the cytosol. Plant J. 2011, 66, 745–758. [Google Scholar] [CrossRef]
- Lin, S.-J.; Guarente, L. Nicotinamide adenine dinucleotide, a metabolic regulator of transcription, longevity and disease. Curr. Opin. Cell Biol. 2003, 15, 241–246. [Google Scholar] [CrossRef]
- Verleur, N.; Elgersma, Y.; Roermund, C.W.T.; Tabak, H.F.; Wanders, R.J.A. Cytosolic aspartate aminotransferase encoded by the AAT2 gene is targeted to the peroxisomes in oleate-grown Saccharomyces cerevisiae. JBIC J. Biol. Inorg. Chem. 1997, 247, 972–980. [Google Scholar] [CrossRef] [Green Version]
- Rosenthal, M.; Metzl-Raz, E.; Bürgi, J.; Yifrach, E.; Drwesh, L.; Fadel, A.; Peleg, Y.; Rapaport, D.; Wilmanns, M.; Barkai, N.; et al. Uncovering targeting priority to yeast peroxisomes using an in-cell competition assay. Proc. Natl. Acad. Sci. USA 2020, 117, 21432–21440. [Google Scholar] [CrossRef]
- Rymer, Ł.; Kempiński, B.; Chełstowska, A.; Skoneczny, M. The budding yeast Pex5p receptor directs Fox2p and Cta1p into peroxisomes via its N-terminal region near the FxxxW domain. J. Cell Sci. 2018, 131, jcs216986. [Google Scholar] [CrossRef] [Green Version]
- Kempinski, B.; Chelstowska, A.; Poznanski, J.; Krol, K.; Rymer, L.; Frydzinska, Z.; Girzalsky, W.; Skoneczna, A.; Erdmann, R.; Skoneczny, M. The peroxisomal targeting signal 3 (PTS3) of the budding yeast acyl-CoA oxidase is a signal patch. Front Cell Dev. Biol. 2020, 8, 198. [Google Scholar] [CrossRef] [PubMed]
- Riccio, V.; Demers, N.; Hua, R.; Vissa, M.; Cheng, D.T.; Strilchuk, A.W.; Wang, Y.; McQuibban, G.A.; Kim, P.K. Deubiquitinating enzyme USP30 maintains basal peroxisome abundance by regulating pexophagy. J. Cell Biol. 2019, 218, 798–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | C-Terminal | PTS1 Score | N-Terminal | MitoProt Score |
|---|---|---|---|---|
| ScMdh1 | NIEKGVNFVASK | −48.5 | MLSRVAKRAFSSTVANP | 0.9910 |
| ScMdh2 | GLEFVASRSASS | −38.2 | Not predictable | 0.0376 |
| ScMdh3 | KGKSFILDSSKL | 4.9 | Not predictable | 0.1859 |
| PpMdhA | IAKGQEFVKQNP | −37.7 | MLSTIAKRQFSSSASTA | 0.9549 |
| PpMdhB | NIAKGTAFIAGN | −52.6 | Not predictable | 0.1410 |
| ScGpd1 | PDMIEELDLHED | −101.6 | MSAAADRLNLTSGHLNAGRKRS | 0.3132 |
| ScGpd2 | PEMIEELDIDDE | −86.8 | MLAVRRLTRYTFLKRTH | 0.9932 |
| PpGpdA | FNKTEDVKHWED | −58.1 | MYLTSTVRALPVHFFRSRHCIRT | 0.6471 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Farré, J.-C.; Li, P.; Subramani, S. BiFC Method Based on Intraorganellar Protein Crowding Detects Oleate-Dependent Peroxisomal Targeting of Pichia pastoris Malate Dehydrogenase. Int. J. Mol. Sci. 2021, 22, 4890. https://doi.org/10.3390/ijms22094890
Farré J-C, Li P, Subramani S. BiFC Method Based on Intraorganellar Protein Crowding Detects Oleate-Dependent Peroxisomal Targeting of Pichia pastoris Malate Dehydrogenase. International Journal of Molecular Sciences. 2021; 22(9):4890. https://doi.org/10.3390/ijms22094890
Chicago/Turabian StyleFarré, Jean-Claude, Paul Li, and Suresh Subramani. 2021. "BiFC Method Based on Intraorganellar Protein Crowding Detects Oleate-Dependent Peroxisomal Targeting of Pichia pastoris Malate Dehydrogenase" International Journal of Molecular Sciences 22, no. 9: 4890. https://doi.org/10.3390/ijms22094890
APA StyleFarré, J. -C., Li, P., & Subramani, S. (2021). BiFC Method Based on Intraorganellar Protein Crowding Detects Oleate-Dependent Peroxisomal Targeting of Pichia pastoris Malate Dehydrogenase. International Journal of Molecular Sciences, 22(9), 4890. https://doi.org/10.3390/ijms22094890