1. Introduction
Metastasis is a process by which cancer cells leave their primary site and spread to other tissues and organs to form new tumors [
1]. Ninety percent of deaths from solid tumors are due to metastasis. Therefore, understanding and targeting this process is critical. Key features during epithelial cancer cell metastasis are detachment, adaptation and prevention of detachment-induced cell death (anoikis) in the absence of extracellular matrix (ECM), and subsequent reattachment [
1]. This is a highly complex process regulated by numerous genes including adhesion molecules, receptor tyrosine kinases, and pathways such as the mechanistic target of rapamycin (mTOR) and RhoA signalling [
1].
Animal models have been essential in order to understand the mechanisms of metastasis. However, these models have a variety of limitations including a long latency for the development of metastasis, very high costs and ethical implications. This often makes their use impractical, particularly for preliminary screens of novel candidate genes or treatment compounds.
Several advanced in vitro cancer metastasis models have also been established in recent years [
2]. Typically they involve specific culture conditions, including serum-free conditions and 3D matrix systems that allow for the culture of tumor spheres [
3,
4] and organoids [
5]. Some studies have used these approaches to specifically compare detached cells to that of the standard adherent cell population. For example, glioblastoma cells have the capacity to form neurospheres, and these show distinctive features from adherent counterparts [
6]. Notably, in a recent study of ovarian cancer dissemination, a multi-omic approach was used to identify and screen genes associated with anoikis resistance by comparing adherent cells and cells forced to survive in suspension in polyHEMA-treated culture dishes [
7].
Some studies have also investigated the naturally occurring floating cell population observed under standard culture conditions. Major changes in gene and protein expression patterns are likely to occur in adherent cells that detach and float in suspension as single cells or small clusters. Indeed, several studies indicate that cells grown as spheres in suspension have certain unique properties such as resistance to anoikis-mediated cell death, changes in the cell cycle, regulation of surface and adhesion molecules, cancer stem cell characteristics and the process of epithelial to mesenchymal transition [
8]. For example, stimulation of the UP-LN1 carcinoma cell line with IFN-y promoted the appearance of cancer stem cells (CSCs) specifically in the floating cell population but not the adherent population, concomitant with induction of CXCR4 and enhanced migratory and invasive potential [
9]. When culturing the gastric cell line SGC-7901 in serum-free medium, a subpopulation of cells formed spheres in suspension that were more tumorigenic and showed upregulation of several genes related to stemness [
10]. Increased CSC marker expression was also observed in the spheroid cell population compared to adherent cells derived from ovarian serous carcinomas [
11]. Primary gallbladder carcinomas grown under serum-free conditions also produce floating spheres, with increased expression of the CSC marker CD133(+), higher tumorigenic potential, and increased resistance to chemotherapeutic reagents [
12]. Indeed, there are many links between stem cell properties and resistance to chemotherapy and radiotherapy. In one study, endometrial cancer cells made resistant to cisplatin became smaller and grew in a floating state, concomitant with increased expression of several drug-resistance genes. Curiously, although these cells were also more resistant to mitomycin and adriamycin, they were more sensitive to etoposide and 5-fluorouracil [
13]. It is unclear why nearly all small cell lung cancer (SCLC) cell lines grow in suspension [
7]. One SCLC cell line, NCI-H446, has been shown to grow with coexisting floating and adherent subpopulations, where the floating cells express much higher levels of neuron-specific enolase as well as the neuronal cell adhesion molecule NCAM [
14]. The SCLC cell lines NCI-H69 and NCI-N592 also grow in suspension, but surprisingly the adriamycin-resistant subclones develop an adherent phenotype [
15]. Using the non-small cell lung cancer (NSCLC) lines, which unlike SCLC typically grow as adherent cells, Gomez-Casal et al. [
16] showed that A549 and H460 cells that survived ionizing radiation started to grow in spheres and expressed CSC markers, concomitant with increased levels of epithelial–mesenchymal transition (EMT) markers. In stark contrast, however, EMT was associated with the adherent subpopulation of the SCLC cell lines [
17]. Other changes in gene expression during the transformation from adherent to a floating phenotype have been observed in sublines established from the rat hepatoma cell line AH7974, in which floating cells decreased the expression of extracellular matrix proteins, cell surface integrins and heparan sulphate proteoglycans [
18]. Thus, the interrelationship between adherent and suspension cells implicate key cancer features related to metastasis, linking EMT, CSCs and therapeutic resistance. Dissecting this complexity may reveal additional pathways and targets to prevent metastasis.
Few genes have been identified to date that drive the process of adherence towards suspension. One study showed that the free-floating organoids from the breast cancer cell line PMC42 became adherent upon addition of epidermal growth factor (EGF), concomitant with increased expression of the cell-adhesive proteins laminin and fibronectin [
19]. Identifying genes differentially expressed or driving this process may also reveal surprising new therapeutic targets. In this respect, Jensen et al. [
20] showed that floating thyroid cancer cells were more resistant to serum deprivation. The tumor spheres displayed constitutively activated AKT and had increased levels of the gap junction protein connexin 43. Silencing connexin 43 expression, or inhibiting gap junctions chemically, resulted in a loss of pAKT and induction of apoptosis in the spheres [
20]. The induction of anoikis (matrix deprivation-induced apoptosis) is one putative therapeutic target for treating metastatic tumors [
21].
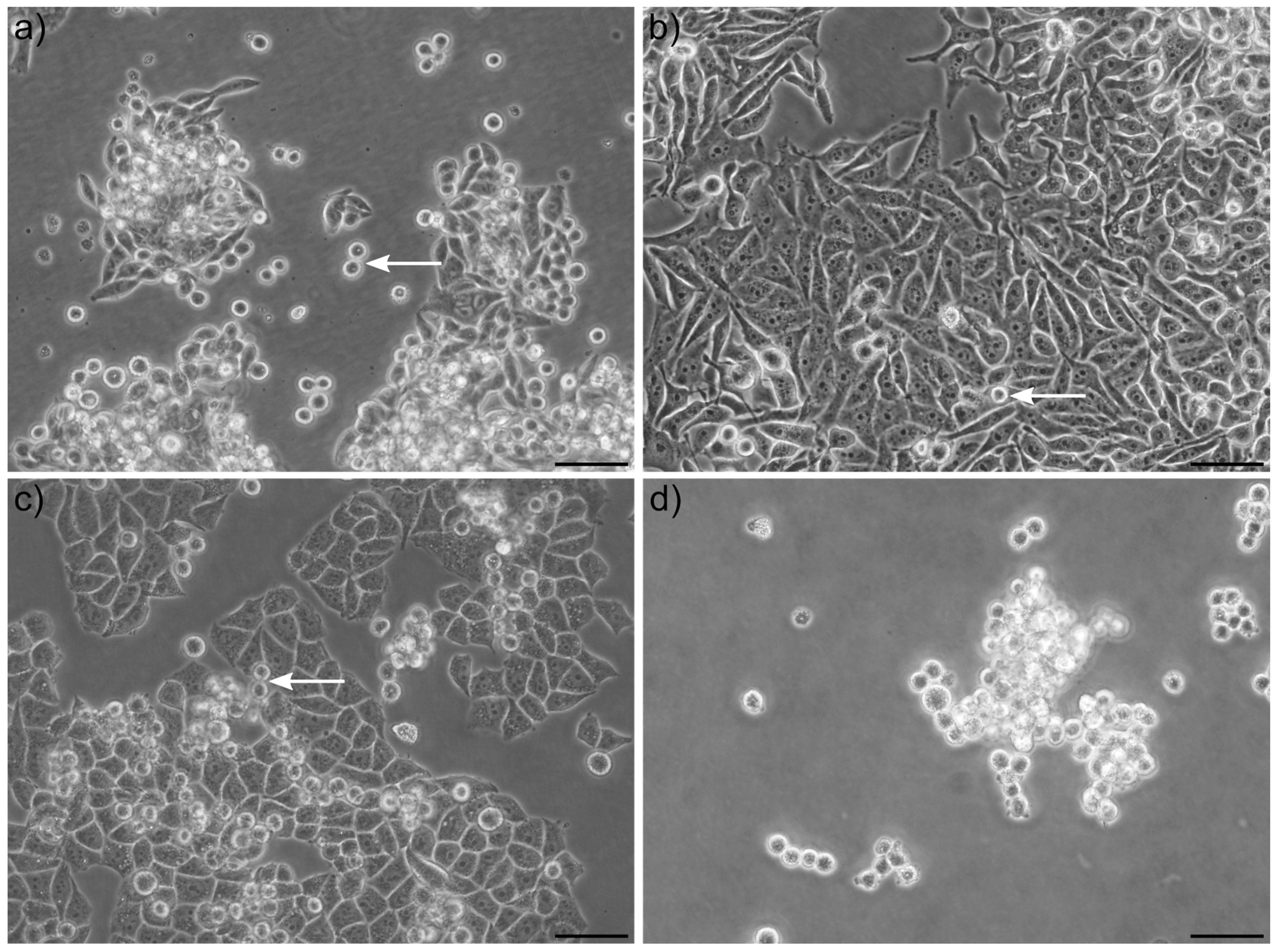
When culturing cancer cell lines on adherent tissue culture plates, the population of cells seen floating in suspension is by many researchers—especially when occurring as single cells and not clusters—considered as dead (or dying) cells and debris. Here, we show that in many cases, the majority (up to 99%) of these cells are alive, can proliferate and rapidly settle down again as adherent cells when transferred to a new tissue culture plate. Although most studies have focused on tumor spheres, we have observed that single floating cells, coexisting in typically adherent cell lines, also have unique properties. Our in vitro model of cells co-existing as adherent and spontaneously detached floating cells in many ways mimic, in a very simplistic way, some characteristics of the process of metastasis from the primary tumor to a secondary organ, including detachment, survival in suspension and reattachment. We postulated that naturally co-occurring populations of free-floating and adherent cells can be used as simplistic models to screen for genes, drugs and pathways that regulate cancer metastasis. To our knowledge, large-scale genome-wide differential transcriptomic analysis comparing adherent and floating subpopulations of the same cell line co-cultured under identical conditions has not been reported. To get a better understanding of the underlying genes and pathways associated with the transition from an adherent to a floating cell phenotype, we performed RNA sequencing in three different cancer cell lines isolated from solid epithelial tumors: HeLa (cervical carcinoma cell line), SW620 (metastatic colon carcinoma cell line isolated from a lymph node) and C33a (cervical carcinoma cell line with the potential to spontaneously metastasize). As proof of principle, we identified a group of genes and signaling pathways, which we verified to be implicated in the process of either detachment, anoikis or reattachment. We suggest this model allows for a cost-effective screening method for the identification of novel anti-metastatic compounds that either inhibit cell detachment or prevent survival in suspension.
3. Discussion
Metastasis is the major killer in patients with cancer. There is a lack of simple models of malignancy and metastasis that facilitate fast, affordable and scalable in vitro gene and compound analysis. Detachment, survival in suspension, and reattachment in vitro are properties that are, in a simplistic view, mimicking the process of metastasis from primary tumors to a secondary site. To our knowledge we have for the first time performed RNA sequencing in order to compare the gene-expression profile of adherent versus floating cells that co-exist in the same tissue culture dish. We analyzed these sequencing data in order to select some genes and pathways for further validation including by Western blot, knockdown, drug modulation and by using modified culture conditions. The analysis and proof-of-concept experiments have solidified our view that this simple model is useful for cancer research to identify and screen genes, drugs and pathways that regulate either cell detachment, survival in suspension, or reattachment, which are key events in cancer metastasis with potential for anti-metastatic drug target development.
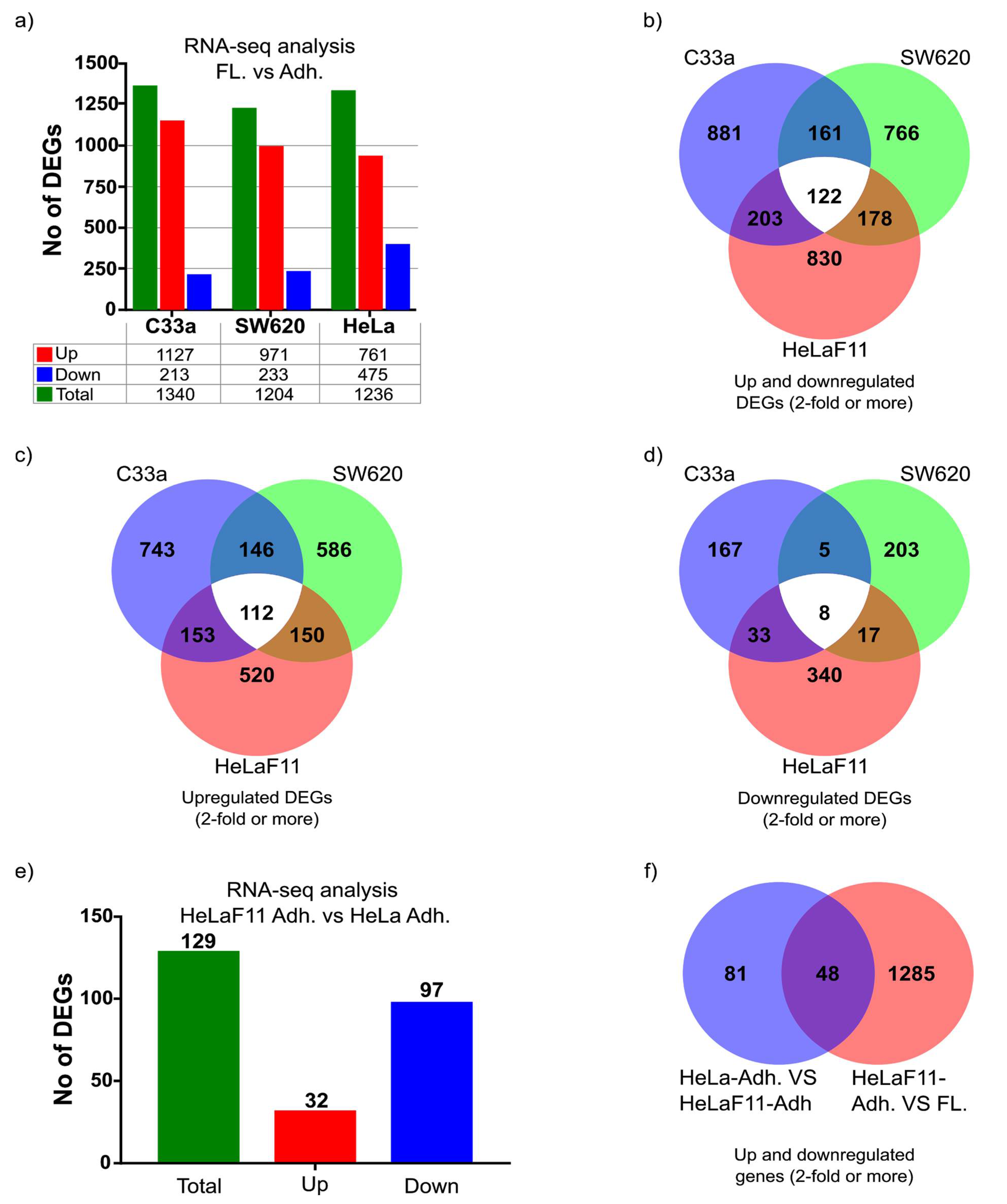
Our analysis suggests there are significant differences between cell lines in their tendency to release floating cells. Moreover, a significant amount of C33a and SW620 floating cells appears to be present despite ample space (e.g., 50%) for cells to settle down, which suggests there is an interplay between adherent and floating cells that can regulate this balance. Further analysis of DEGs from our study, particularly those of secreted factors, may provide clues towards these regulatory mechanisms.
In general, we observed a higher presence of floating cells in aggressive tumor cells (and less or none in primary cells and non-malignant cell lines). C33a has been shown to metastasize [
34] and SW620 is a well-known metastatic cell line isolated from the lymph node. Moreover, we have observed that non-malignant primary fibroblasts and immortalized HaCaT keratinocytes release very few cells with the capacity to settle down and grow again, whereas a more malignant cell line such as the well-known breast cancer cell line MDA-MB-231 seems to behave more as our SW620 and C33a cell models. On the other hand, malignant MCF-7 and BT-20 breast cancer cells seem much less likely to release cells that can settle down and form new colonies. We have also observed that floating cells can form more clonogenic colonies than the equivalent amount of adherent cells, one of many features that should be explored in the future to clarify if there is a direct correlation between the ratio of adherent and floating cells to malignancy. Comparing the gene-expression profile and malignant potential of HeLa cells and HeLaF11, cells may provide interesting clues towards this unexplored hypothesis. Such comparison can also provide useful insight into which genes regulate cell detachment. As an example, from the RNA-seq analysis, we observed significantly reduced gene expression of laminin subunit beta 3 (HeLaF11 compared to HeLa cells), a well-known regulator of cell adhesion (
Supplementary Table S6).
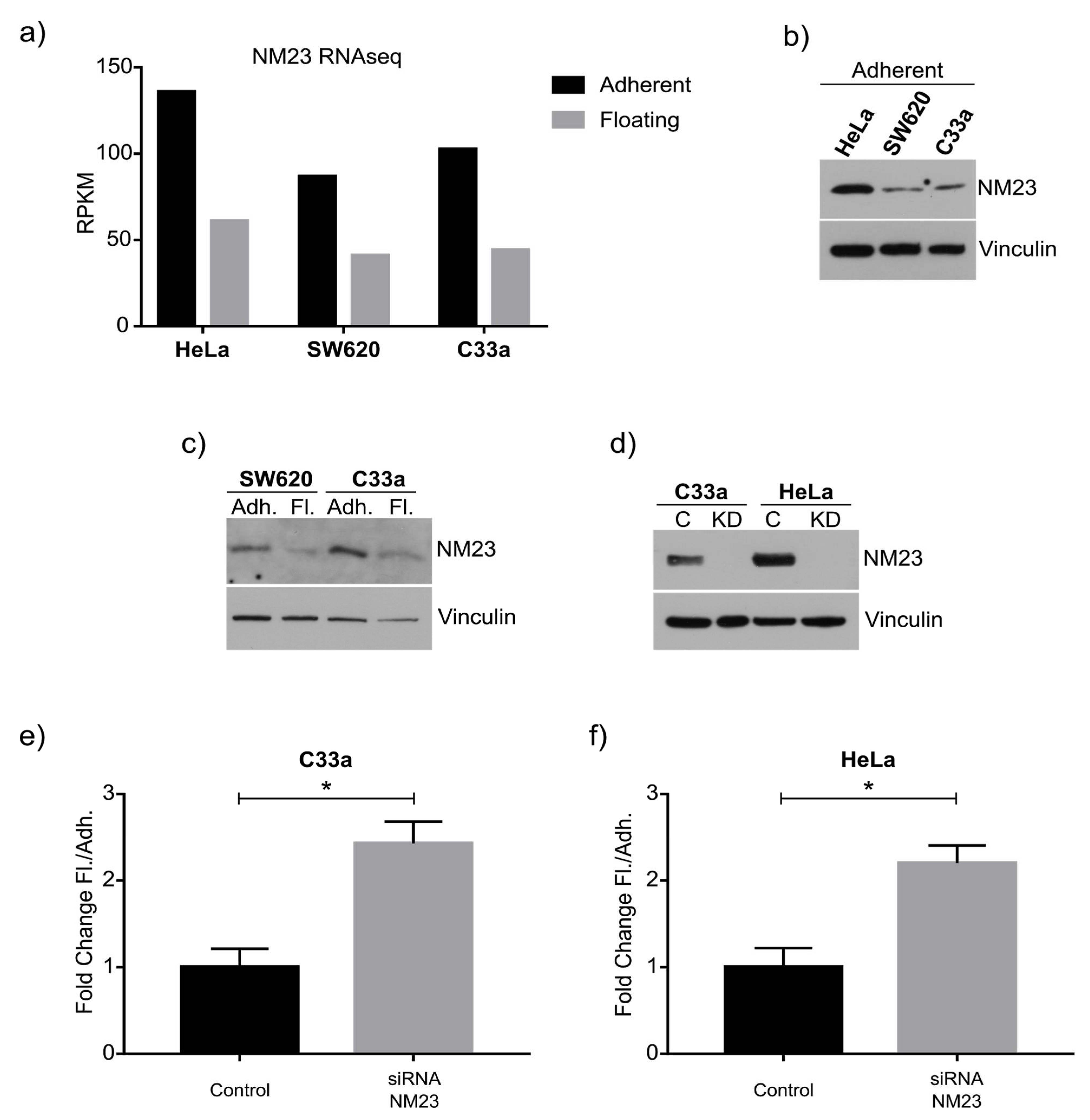
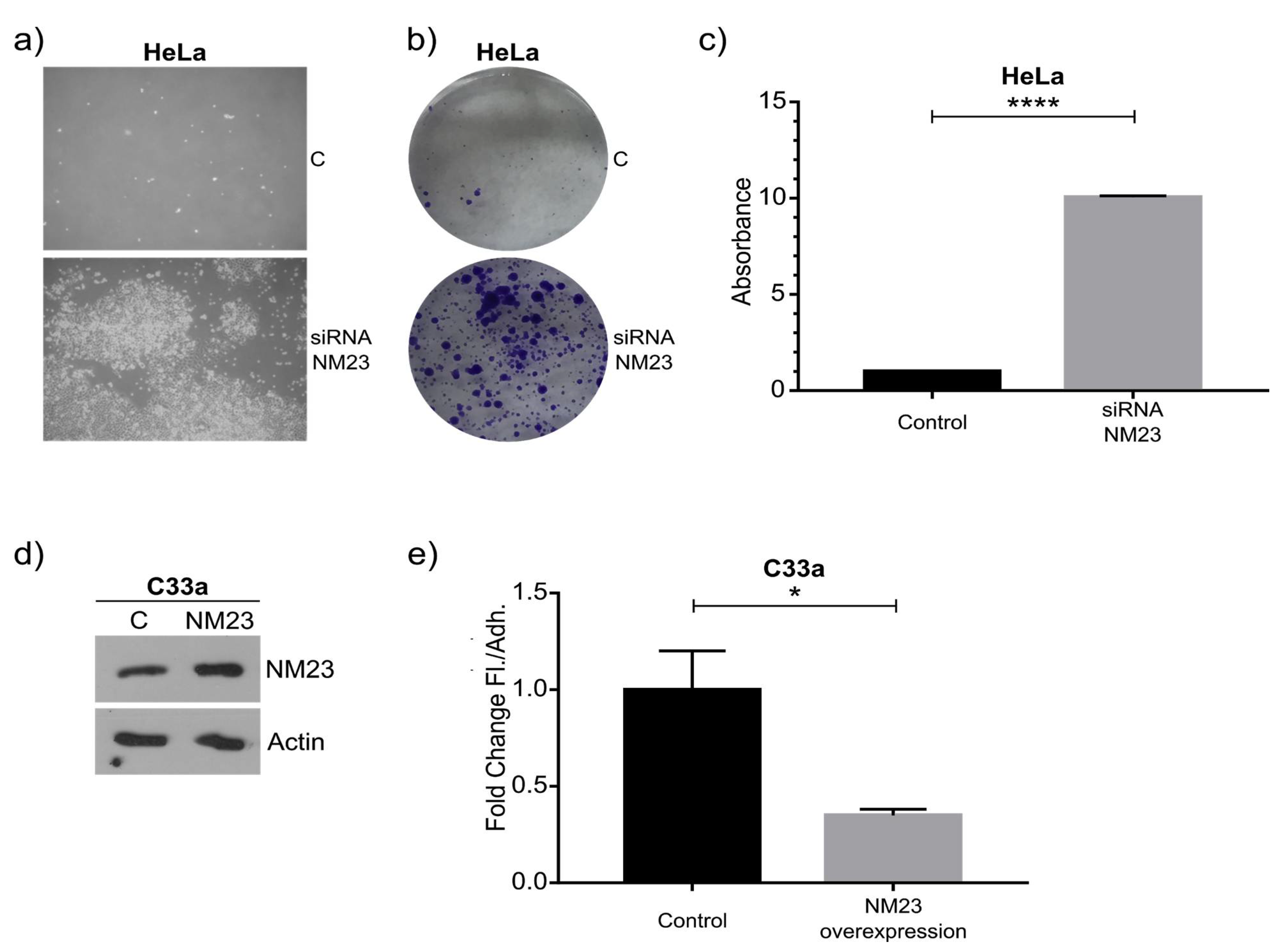
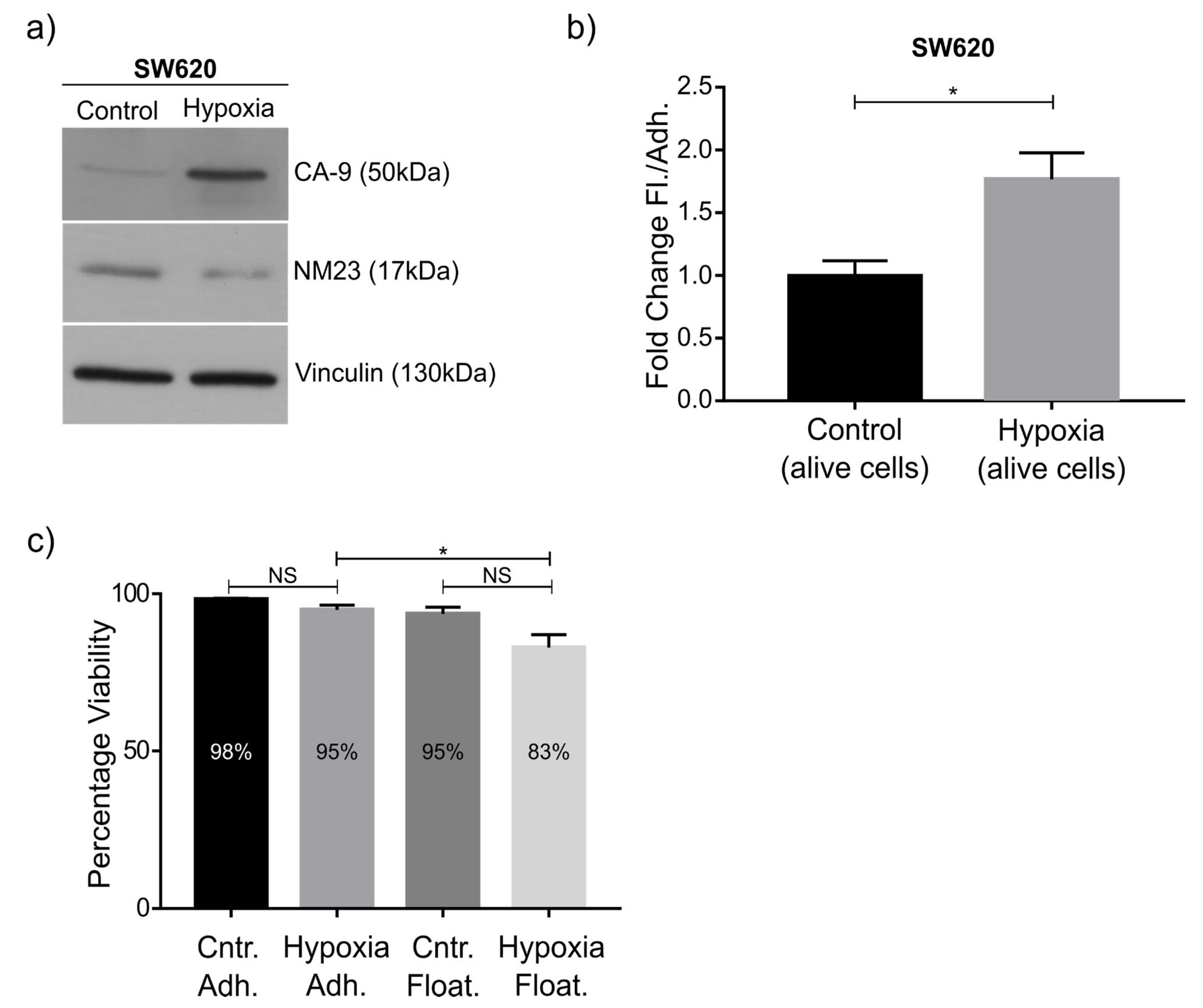
As a proof of principle of our model, we focused on NM23-H1, as this anti-metastatic gene has been robustly linked to metastasis, and was downregulated in the floating cell population of all three cell lines. Western blot confirmed the RNAseq data and knockdown and overexpression confirmed its role in regulating the ratio between adherent and floating cells. The mechanism behind this remains to be studied in detail, although previous studies have shown NM23-H1 to regulate adherens junctions [
25] as well as cell–substrate adhesion [
26]. Consistent with previous reports [
29], NM23-H1 was also downregulated upon hypoxia, a condition that induced the ratio of floating cells. In the future it would be of interest to see whether the amount of extracellular NM23-H1 also regulates the ratio between floating and adherent cells, seeing that most floating cells seem to rapidly adhere upon transfer to a new tissue culture plate. In addition to NM23-H1, other gene candidates are being investigated. For example, in relation to EMT, we observe a significant downregulation of E-cadherin protein expression in floating SW620 and HeLaF11 cells, whereas C33a cells are E-cadherin negative. There is upregulation of several proteins associated with metastasis, including VEGF. Notably, a significant upregulation of MMP14 (Matrix Metallopeptidase 14) is seen in the floating population of SW620 and C33a cells. In that respect, MMP14 has been shown to decrease cell adhesion and provide anoikis resistance in SW620 cells [
35].
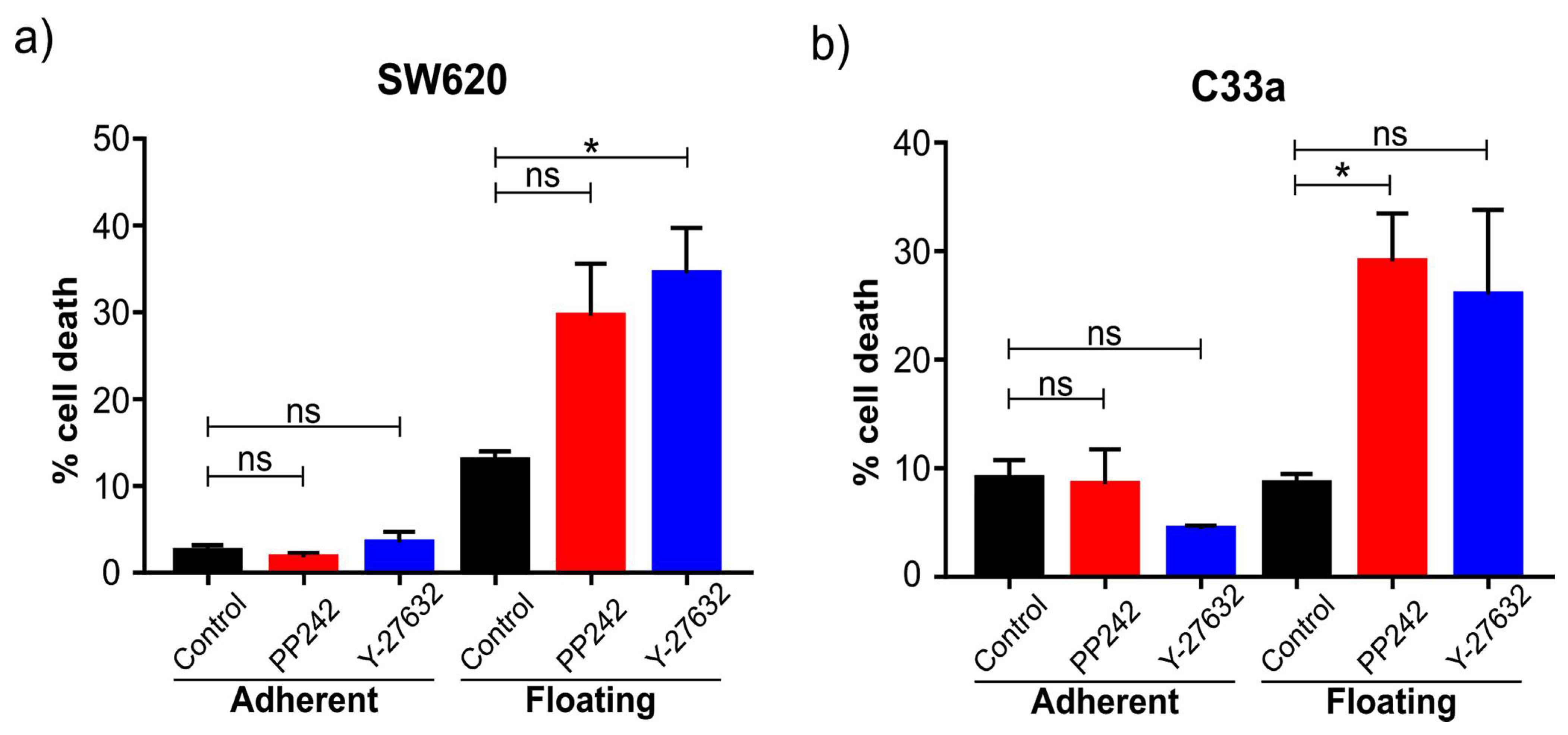
Several different signaling pathways were shown to be significantly different between floating and adherent cells. Our trials of inhibiting mTOR and ROCK signaling showed the floating cells were more sensitive to these drugs. Clearly, additional work is needed to understand the underlying molecular mechanism behind this. However, it is encouraging and suggests this model can be used as a very fast preliminary screening method to test how specific inhibitors affect the two cell populations differently, which may give insight into their putative promise in cancer and metastasis in particular. Inhibitors of mTOR, for example, are currently being evaluated in the clinic, and the observation that floating cells are particularly sensitive is of interest, as we suggest it can be a potent inhibitor of metastasis if administered during the time window of cell detachment in a tumor. Both in vivo and in vitro studies also implicate ROCK as a potential target for cancer treatment, especially to prevent metastasis. The fact that both pathways are significantly involved in metastasis and their chemical inhibition affects floating cells specifically suggest that the “floating cell model” studied here may have the potential for fast and cheaper preliminary screenings of anti-metastatic drugs. Our observation that mTOR and ROCK inhibitors are more effective in the floating subpopulation of cells is supported by the fact that cell detachment has been shown to prime SW620 cells to anoikis [
36]. However, additional work will determine the robustness of this model.
Changes in pathways related to apoptosis were also observed. We analyzed the percentage of cells undergoing Caspase-3/7 apoptosis and apoptosis with the externalization of phosphatidylserine. In both cases, the percentage was lower than 4% and with no differences between floating and adherent cells. These results suggest very low levels of apoptosis under normal cell culture conditions. In the case of the floating cells, these results suggest that they are resistant to anoikis because they do not present programmed cell death when they detach. Indeed, SW620 cells derived from a metastatic site are resistant to anoikis, whereas the SW420 cell line derived from the primary tumor was sensitive [
36]. However, ROCK and mTOR inhibition significantly induced both caspase-dependent and caspase-independent apoptosis specifically in the floating cell population. Further screening of drugs may identify links between cell detachment and sensitivity to certain drugs. Another observation was the enhanced clustering of floating cells culturing in “suspension culture dishes” that prevents adherence to the dish. Whether major gene-expression changes occur under these conditions, and whether this further affects drug sensitivity and anoikis, would be an interesting future question to address.
In the tumor microenvironment, alteration in the expression of many pro and antimetastatic genes takes place at the molecular level, and this is often due to cellular stress that further aggravates the metastatic cascade and advancement of the disease. Hypoxia is one of these stress factors that promotes metastatic progression [
27]. Again, this condition increased the ratio of floating cells, further consolidating the validity of this model. Mechanistically, hypoxia has been shown to influence the invasive and migratory behavior of cancer cells via EMT (trans-differentiation of cells in order to acquire plastic and mobile abilities), a process that alters their gene expression prior to migration. Indeed, we see downregulation of the EMT marker E-cadherin in floating cells, which may in part be due to downregulated NM23-H1 expression, a feature we also observed in hypoxia [
29,
30]. Using the RNAseq data, there is now an opportunity to screen some of the other hypoxia-associated genes that are differentially expressed in floating cells to assay their causative implication in the process of detachment.
This work has demonstrated that many of the characteristics of the floating cells resemble critical features of metastatic cells, such as detachment or anoikis resistance. It would also be of interest to compare the gene expression of these floating cells with that of typical circulating tumor cells, such as leukemia cells. However, obviously, a model of metastasis based on these cells has some limitations. On the one hand, it is a highly simplistic in vitro 2-D model that does not accurately resemble the particularities of a tumor microenvironment such as native morphology and physiological processes of tumor cells, features that more complex 3-D models are trying to address [
4]. While in vitro models offer clear advantages over in vivo models of metastasis, most notably cost, reproducibility and high throughput applications, they remain surrogate assays of metastatic function that merely provide a rapid, simplified system that can be manipulated to investigate factors regulating specific steps of the metastatic cascade or test the efficacy of inhibitors. Yet, these features are critical in the early steps of evaluating drugs and pathways. The sheer simplicity of this system allows any lab to perform similar experiments or validate any gene or pathway of interest such as those disclosed here from the RNAseq analysis. It would be necessary to validate the results in other models in vitro and in vivo that fulfil some of the characteristics that this model of floating cells lacks, such as cooperation with other cells and tissues, and subsequently also in clinical specimens. We believe however that this model, and the associated RNAseq data, in combination with other more complex models and systems, will be useful in the critical search for novel anti-metastatic targets.
4. Materials and Methods
4.1. Cell Culture
C33a (ATCC® HTB-31) cervical carcinoma cells, SW620 (ATCC® CCL-227) colorectal adenocarcinoma cells and HeLa (ATCC® CCL-2) cervical adenocarcinoma cells were obtained from ATCC (LGC Standards, Barcelona, Spain) and cultured as recommended using standard procedures. Briefly, cells were cultured in Dulbecco’s modified Eagle’s minimum essential medium (DMEM, Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS, Labclinics, Barcelona, Spain) and 1% penicillin/streptomycin (P/S, Thermo Fisher Scientific, Waltham, MA, USA). Cells were cultured in a 37 ºC humidified incubator in an atmosphere of 5% CO2. TrypLE Express (Thermo Fisher Scientific, Waltham, MA, USA) was added to detach cells from the plate.
4.2. RNA Isolation and Sequencing
The floating cell population and the adherents cell population from three 10 cm dishes were isolated and pooled for each cell line. RNA was isolated using an RNAeasy Plus Mini Kit (Qiagen, (Werfen, Barcelona, Spain) Cat No: 74134) that specifically removes any genomic DNA contamination using gEliminator Spin-columns. The quantity was analyzed by Nanodrop ND-1000 Analyzer and the quality was analyzed with a Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA, USA). Minimum 5 ug of RNA, with a minimum RIN (RNA integrity number) of 7, was shipped to Beijing Genomics Institute (BGI), Shenzhen, China, using RNA stable tubes (Biomatrica (San Diego, CA, USA) Cat. No 93220-001). BGI Beijing performed further internal quality control assays. Gene expression profiling by RNA sequencing was performed entirely by BGI as previously described [
37,
38]. Briefly: After extracting the total RNA from the samples, mRNA is enriched by using the oligo(dT) magnetic beads. Adding the fragmentation buffer, the mRNA is interrupted to short fragments (about 200 bp), then the first strand cDNA is synthesized by random hexamer-primer using the mRNA fragments as templates. Buffer, dNTPs, RNase H and DNA polymerase I are added to synthesize the second strand. The double-strand cDNA is purified with QiaQuick PCR extraction kit (Werfen, Barcelona, Spain) and washed with EB buffer for end repair and single nucleotide A (adenine) addition. Finally, sequencing adaptors are ligated to the fragments. The required fragments are purified by agarose gel electrophoresis and enriched by PCR amplification. The library products are ready for sequencing analysis via Illumina HiSeq™ 2000. Clean reads were mapped to reference sequences using SOAPaligner/soap2 [
39]. Mismatches no more than 2 bases were allowed in the alignment. The gene expression level is calculated by using the RPKM [
40] method (Reads Per kb per Million reads).
4.3. Timelapse Imaging
IncuCyte ZOOM Live-Cell Imaging system (Essen Bioscience, Hertfordshire, UK) was used to obtain images of adherent cells detaching from the plate and floating cells reattaching to the plate. Cells were seeded in a 24-well plate with increasing numbers of cells, from 10,000 per well to 50,000 per well. Then, 24 h later, floating cells were collected from the media and put in another well. The 24-well plate was put in the IncuCyte inside the tissue culture incubator and phase-contrast images were taken every 5 min for 48 h.
4.4. Drug Treatments
A total of 500,000 cells/well were seeded in 6-well plates 48 h before the treatment. Drug treatment was applied 6 h before the viability assay was performed. The mTOR pathway inhibitor PP242 (Tocris, Bristol, UK) was used at 100 nM (1:4000), and the ROCK inhibitor Y-27632 (Tocris, Bristol, UK) was used at 20 mM (1:2000). DMSO diluent was used as a control.
4.5. Viability Assays
Both cell viability and cell death were independently quantified after drug treatment by means of fluorescent staining, using the ReadyProbes Cell Viability Imaging Kit (Thermo Fisher Scientific, Waltham, MA, USA). After an overnight or 6 h treatment in 6-well plates, floating cells were collected and adherent cells were trypsinized and resuspended in PBS-BSA. One drop of propidium iodide stain (staining in red the nuclei of dead cells with compromised plasma membrane) was added to each 0.5 mL of cells with PBS-BSA. After 15 min incubation, samples were put in a 96-well (100 ul containing 50,000 cells per well) and observed using an Olympus FSX100 fluorescent microscope (Olympus Iberia, Barcelona, Spain) The percentage of dead cells was determined by counting the number of red cells in an area with 100 total cells (seen with phase contrast).
In addition to dead cells, apoptotic cells were determined. Caspase-3 is an early indicator of apoptosis, so CellEvent Caspase-3/7 Green Ready Probe (Thermo Fisher Scientific, Waltham, MA, USA) was used to detect apoptotic cells. CellEvent Caspase-3/7 is a 4-amino acid peptide (DEVD) conjugated with a stain that binds to DNA (this reagent is only fluorescent when bound to DNA). Two drops per mL of PBS-BSA were used and after 15 min of incubation at room temperature cells were viewed at the fluorescence microscope (FSX100 Olympus, (Olympus Iberia, Barcelona, Spain)). The percentage of dead cells due to Caspase-3/7 apoptosis was also determined by counting the number of green cells in an area with 100 total cells (seen with phase contrast). The Thermo Scientific Appliskan (Thermo Fisher Scientific, Waltham, MA, USA) was also used to determine the ratio between dead and total cells. To determine the number of dead cells, a first read was done to a 96-well plate with 100 μL of PBS-BSA with 50,000 per well with 10 ul of propidium iodide (1% diluted in PBS-BSA). Then, 10 ul of TritonX was added to each well, and a second read was done to determine the number of total cells.
4.6. Western Blotting and Antibodies
Cells were lysed in RIPA buffer (Santa Cruz Biotechnology, Heidelberg, Germany) and Western blotting was performed as previously described [
41]. Protein was quantified using the bicinchoninic acid (BCA) assay kit (Thermo Fisher Scientific, Waltham, MA, USA) and verified by Western blot using anti-actin and anti-vinculin antibodies. The following antibodies and dilutions were used: anti-NM23 (Abcam, Cambridge, UK, 1:700), anti-vinculin (Sigma-Prestige Antibodies, Merck Life Science, Madrid, Spain. 1:200), anti-β-actin (Calbiochem, (Merck Life Science, Madrid, Spain), 1:500), secondary goat anti-rabbit and goat anti-mouse HRP (Thermo Fisher Scientific, Waltham, MA, USA, 1:15,000). Quantification was performed using Image J.
4.7. NM23-H1 Knockdown and Overexpression
Cells in 6-well plates were transfected with 2 µg RNA using RNAiMax (Thermo Fisher Scientific, Waltham, MA, USA) following the manufacturer’s recommended standard procedure. The medium was changed 24 h after transfection and 72 h after transfection the amounts of floating and adherent cells were quantified by counting the cells in a Neubauer chamber and a Western blot was performed to check the efficiency of the knock-down. The floating cells obtained after transfection with siRNA control and siRNA NM23 were put in a new 6-well plate, and after 72 h the colony formation capacity was quantified using crystal violet. Then, 1 mL of crystal violet 5% (Sigma-Aldrich, Taufkirchen, Germany) was added to each well, and after 30 min, the cells were washed with PBS and then the crystal violet was quantified adding 100 μL of acetic acid. Then, 200 μL was loaded to a 96 well-plate and read at an absorbance of 590 nm using Epoch Biotek Spectrophotometer (Friedrichshall, Germany).
The retroviral plasmid for overexpression of NM23-H1 (pBABE-hygro NM23 H1 WT) was a gift from Ronald Kahn (Addgene plasmid # 11364; available on
http://n2t.net/addgene:11364 last accessed on 2 May 2021; RRID: Addgene_11364) and was generated as described [
42]. Retrovirus was produced and used to infect HeLa and C33a cells as previously described [
43].
4.8. Hypoxia Treatment
A total of 200,000 cells were seeded per well in a 6-well plate and after 48 h were put in an Invivo 200 hypoxia hood (Baker Ruskinn) with an oxygen concentration of 0.5% and a CO2 concentration of 5%. After 48 h protein extraction and cell counting was performed.
4.9. Flow Cytometry Analysis
106 C33a cells were seeded in 10 cm plates. After 4 days cells were treated with diluted DMSO (as a control), PP242 100 nM or Staurosporine 5 mM (as an apoptotic inducer) overnight. After this period, floating and adherent cells were collected, washed in cold PBS and treated with Annexin V-FITC Apoptosis Detection Kit (Thermo Fisher Scientific, Waltham, MA, USA). The cells were re-centrifuged and after discarding the supernatant, resuspended in 100 μL 1x Binding Buffer; 5 ul of Annexin V-FITC and 5 ul PI Staining Solution were added to each 100 ul of cell suspension. After a 15 min incubation at room temperature, 400 ul of 1x Binding Buffer was added. Finally, the stained cells were analyzed by BD LSRFortessa flow cytometry analyzer (Thermo Fisher Scientific, Waltham, MA, USA), measuring the fluorescence emission at 530 nm and >575 nm.
4.10. Statistical Analysis
Values are expressed as means. Statistical analyses and drawing of graphs were done using GraphPad Prism 6.0 (San Diego, CA, USA). Student’s t-test with Spearman correlation analysis or one way ANOVA with Tukey’s correction was performed as specified in the figure legends. A p-value < 0.05 was considered statistically significant and n.s. was reported for non-statistically significant results (p > 0.05). Unless specified, all the experiments were performed at least three independent times.


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





