
Effective Drug Concentration and Selectivity Depends on Fraction of Primitive Cells

 , , , ,
, , , ,
Abstract
:
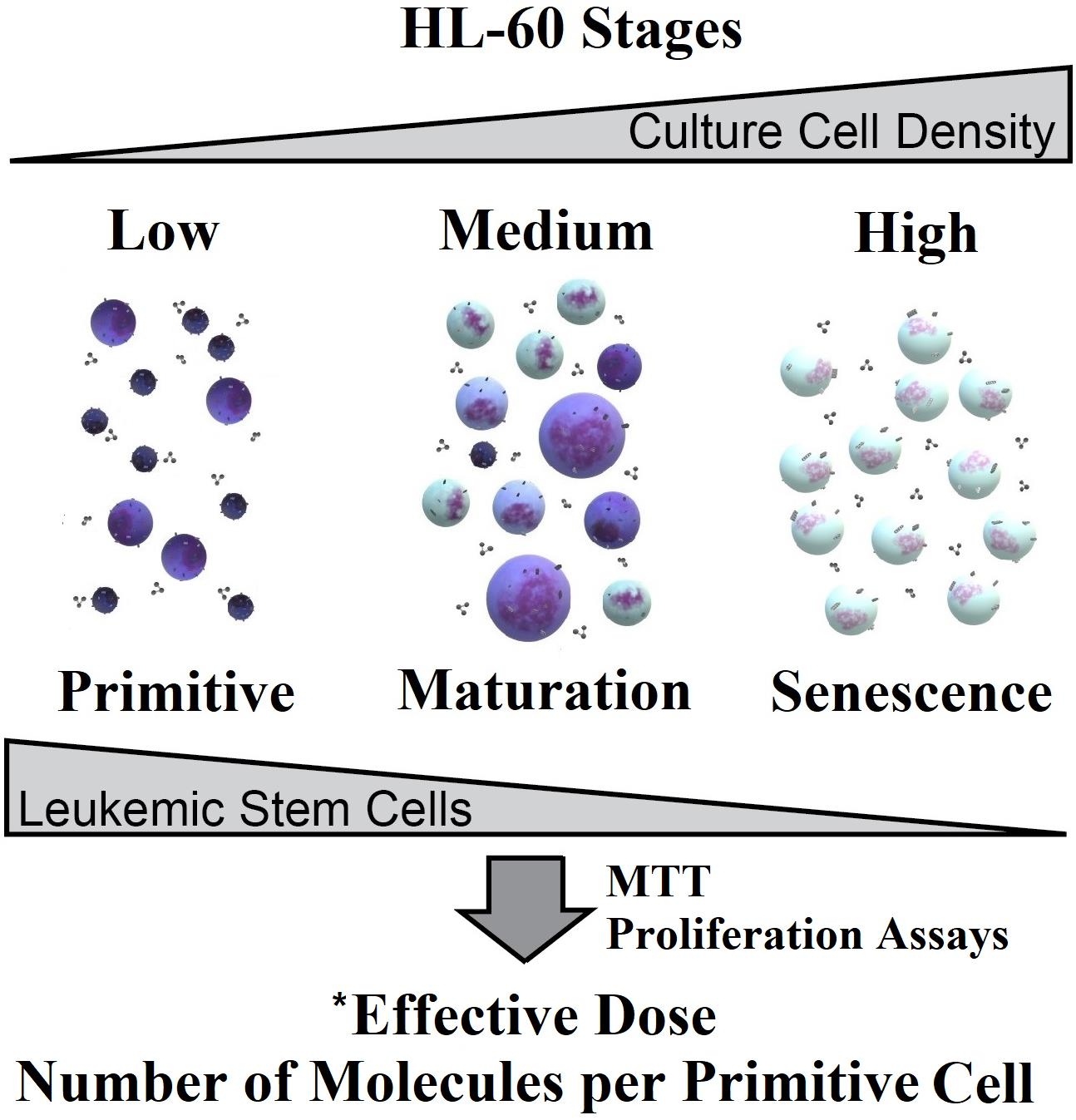
1. Introduction
Highlights
2. Results and Discussion
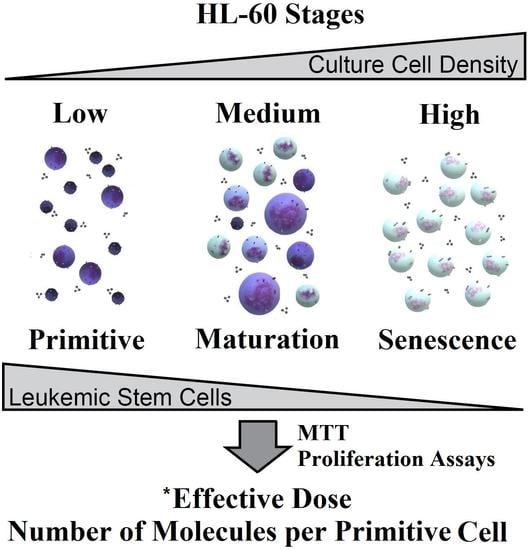
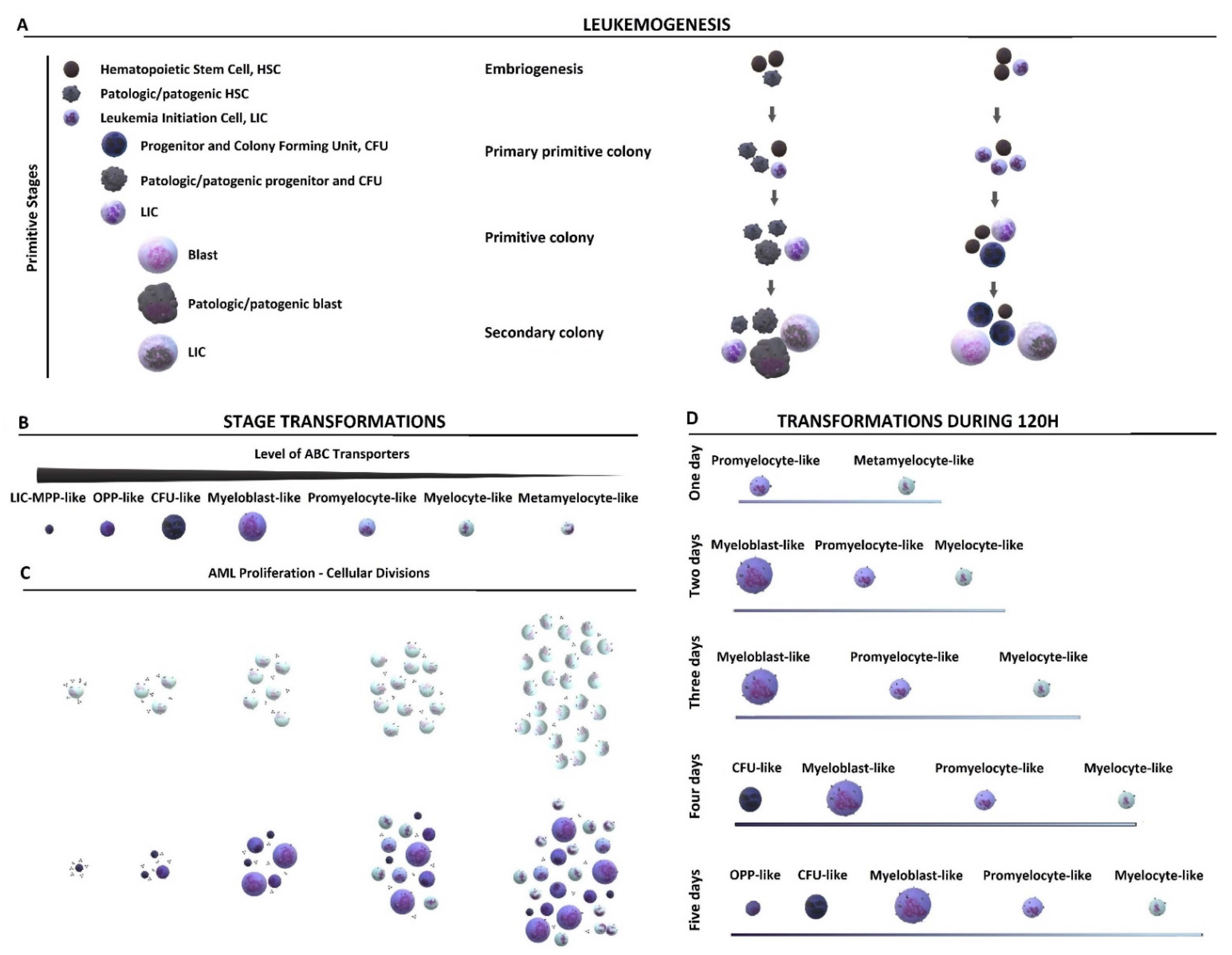
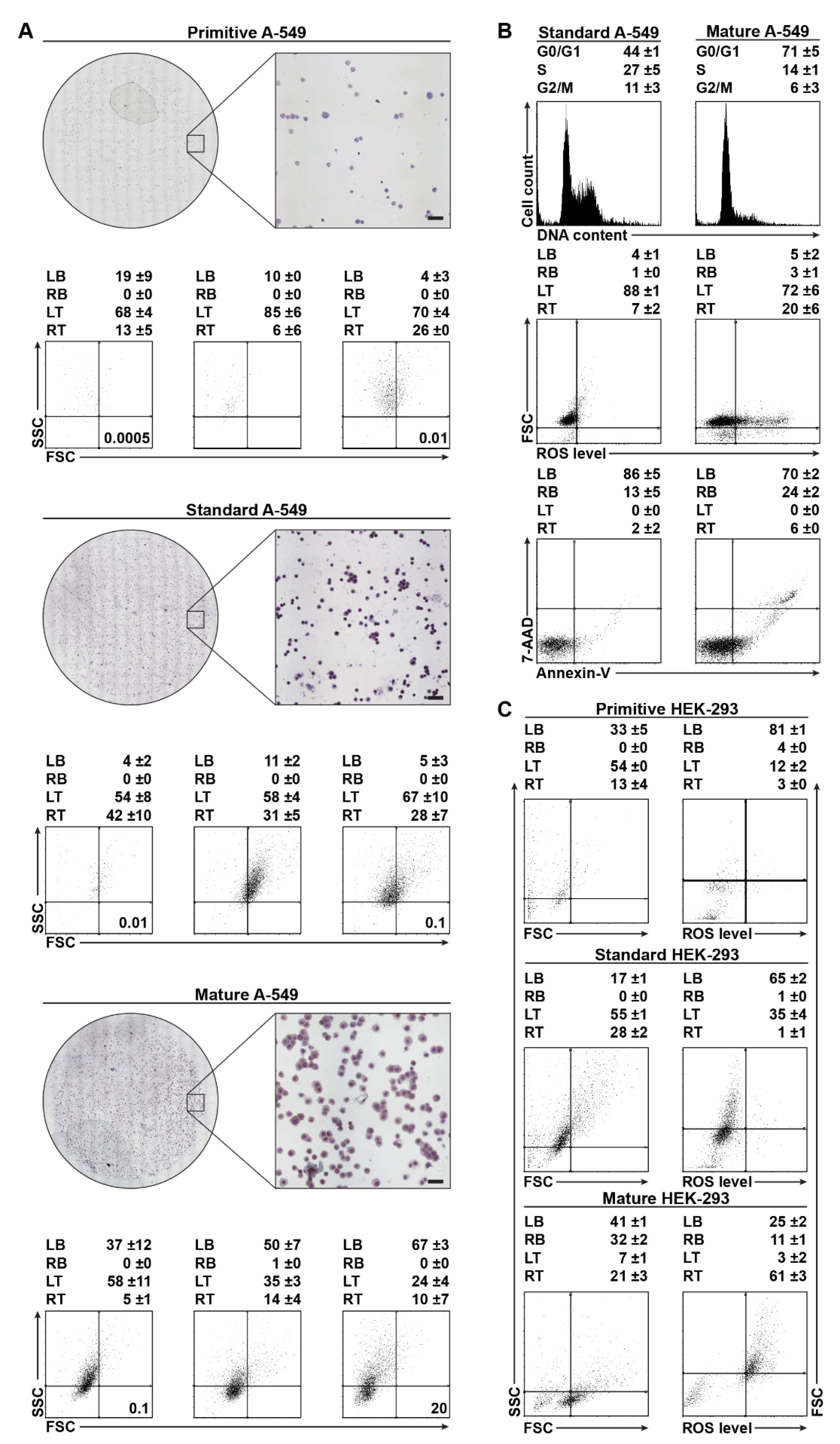
2.1. Cytological Culture Stages Profiles
2.1.1. HL-60
2.1.2. A-549 Cell-Density Cytological Stage Profile
2.1.3. HEK-293 Morphology and ROS
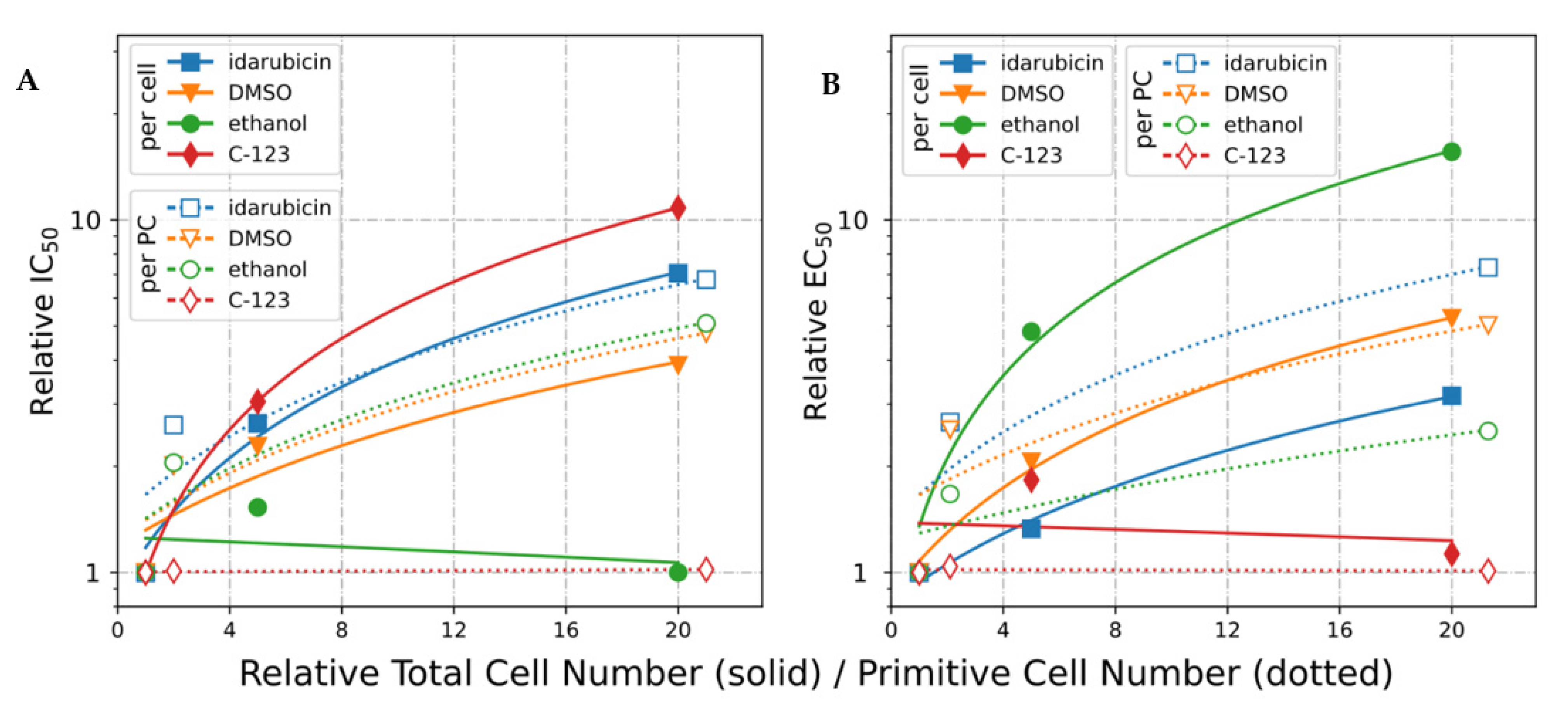
2.2. Effective Concentration as Molecules per (Primitive) Cell
2.2.1. Mechanism of Action of tested Drugs
2.2.2. Total and Primitive Cell Dependence of IC50 in HL-60 Cells
2.2.3. IC50 Normalized to Cell Number (EC50)
2.2.4. IC50 Normalized to Primitive Cell Number (*EC50)
2.2.5. IC50 and EC50 Calculated Using A-549 Cells
2.3. Selectivity Index Quantification
2.3.1. HL-60
2.3.2. A-549
2.3.3. Selectivity Index Summary
3. Materials and Methods
3.1. Software, Equipment, and Statistical Analysis
3.2. Drugs
3.3. Cell Cultures
3.4. Transmitted Light Microscopy
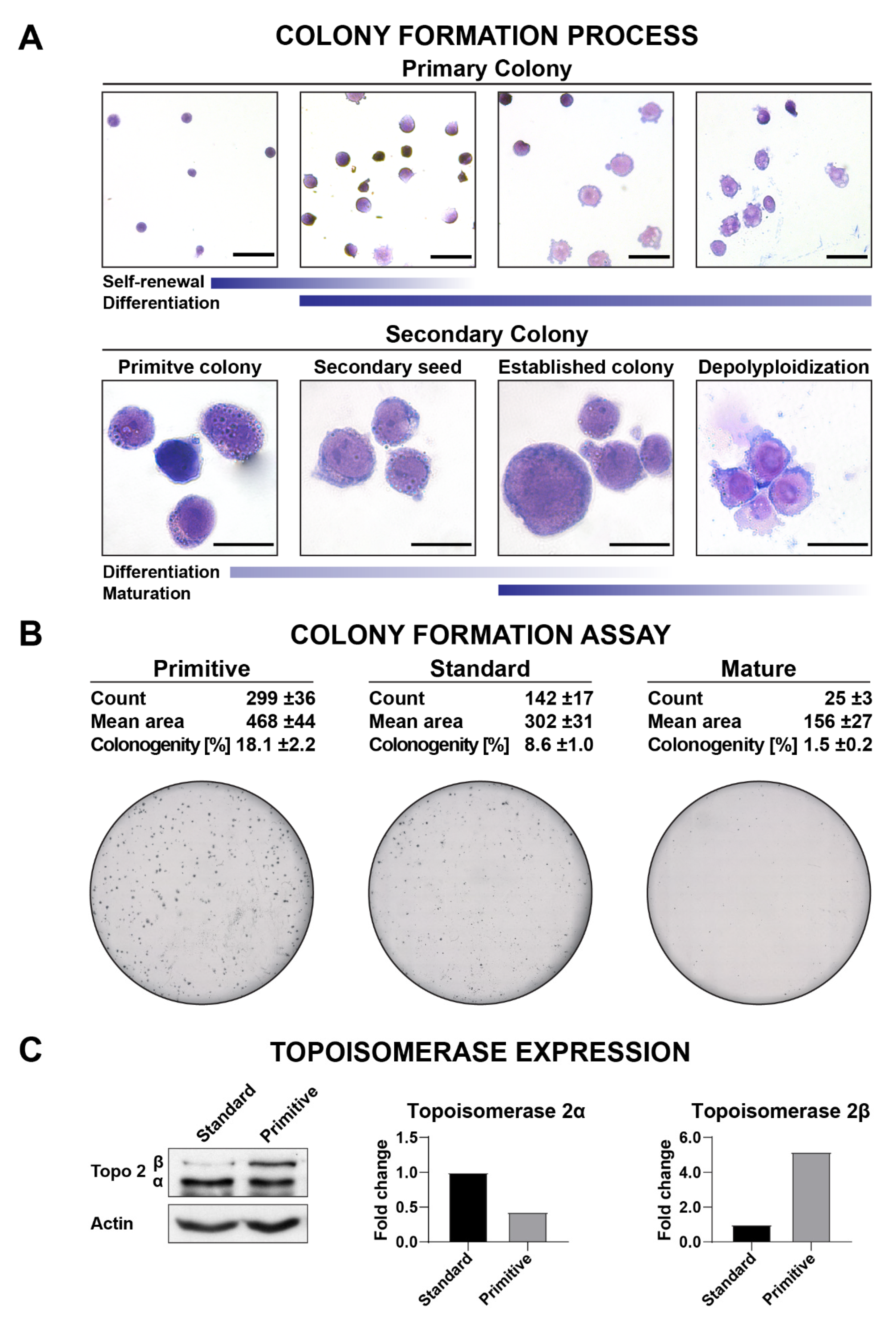
3.5. Clonogenicity
3.6. Drug Sensitivity Assay
3.7. Western Blotting
3.8. Topoisomerase Inhibition Assays
3.9. Proteasome Inhibition
3.10. Molecular Computational Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ABC | ATP-Binding Cassette |
| AML | Acute Myeloid Leukemia |
| A-549 | lung adenocarcinoma cell line |
| CFU-like | Colony Forming Unit-like |
| DMSO | dimethyl sulfoxide |
| EC | Effective Concentration |
| FSC | laser light forward scattering |
| HSC | Hematopoietic Stem Cell |
| HEK-293 | human embryonic kidney cells |
| HL-60 | human myeloid leukemia cells |
| IC | Inhibitory Concentration |
| LB | left bottom |
| LIC | Leukemia Initiation Cell |
| LT | left top |
| LSC HSC-like | Leukemic Stem Cell HSC-like |
| MDR | Multi-Drug Resistance |
| MGG | May-Grunwald-Giemsa |
| OPP-like | Oligopotent Progenitor-like |
| PC | primitive cell |
| RB | right bottom |
| ROS | Reactive Oxygen Spices |
| RT | right top |
| SI | Selectivity Index |
| SSC | laser light side scattering |
References
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef]
- Gal, H.; Amariglio, N.; Trakhtenbrot, L.; Jacob-Hirsh, J.; Margalit, O.; Avigdor, A.; Nagler, A.; Tavor, S.; Ein-Dor, L.; Lapidot, T.; et al. Gene expression profiles of AML derived stem cells; similarity to hematopoietic stem cells. Leukemia 2006, 20, 2147–2154. [Google Scholar] [CrossRef]
- Walter, M.J.; Shen, D.; Ding, L.; Shao, J.; Koboldt, D.C.; Chen, K.; Larson, D.E.; McLellan, M.D.; Dooling, D.; Abbott, R.; et al. Clonal architecture of secondary acute myeloid leukemia. N. Engl. J. Med. 2012, 366, 1090–1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Jan, M.; Snyder, T.M.; Corces-Zimmerman, M.R.; Vyas, P.; Weissman, I.L.; Quake, S.R.; Majeti, R. Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Sci. Transl. Med. 2012, 4, 149ra118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shlush, L.I.; Zandi, S.; Mitchell, A.; Chen, W.C.; Brandwein, J.M.; Gupta, V.; Kennedy, J.A.; Schimmer, A.D.; Schuh, A.C.; Yee, K.W.; et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature 2014, 506, 328–333. [Google Scholar] [CrossRef]
- Goardon, N.; Marchi, E.; Atzberger, A.; Quek, L.; Schuh, A.; Soneji, S.; Woll, P.; Mead, A.; Alford, K.A.; Rout, R.; et al. Coexistence of LMPP-like and GMP-like leukemia stem cells in acute myeloid leukemia. Cancer Cell 2011, 19, 138–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majeti, R.; Weissman, I.L. Human acute myelogenous leukemia stem cells revisited: There’s more than meets the eye. Cancer Cell 2011, 19, 9–10. [Google Scholar] [CrossRef] [Green Version]
- te Boekhorst, P.A.; de Leeuw, K.; Schoester, M.; Wittebol, S.; Nooter, K.; Hagemeijer, A.; Löwenberg, B.; Sonneveld, P. Predominance of functional multidrug resistance (MDR-1) phenotype in CD34+ acute myeloid leukemia cells. Blood 1993, 82, 3157–3162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kornblau, S.M.; Womble, M.; Qiu, Y.H.; Jackson, C.E.; Chen, W.; Konopleva, M.; Estey, E.H.; Andreeff, M. Simultaneous activation of multiple signal transduction pathways confers poor prognosis in acute myelogenous leukemia. Blood 2006, 108, 2358–2365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Øyan, A.; Bø, T.; Jonassen, I.; Gjertsen, B.; Bruserud, Ø.; Kalland, K.-H. cDNA microarray analysis of non-selected cases of acute myeloid leukemia demonstrates distinct clustering independent of cytogenetic aberrations and consistent with morphological signs of differentiation. Int. J. Oncol. 2006, 28, 1065–1080. [Google Scholar] [CrossRef] [PubMed]
- Misaghian, N.; Ligresti, G.; Steelman, L.S.; Bertrand, F.E.; Bäsecke, J.; Libra, M.; Nicoletti, F.; Stivala, F.; Milella, M.; Tafuri, A.; et al. Targeting the leukemic stem cell: The Holy Grail of leukemia therapy. Leukemia 2009, 23, 25–42. [Google Scholar] [CrossRef] [Green Version]
- Stiehl, T.; Baran, N.; Ho, A.D.; Marciniak-Czochra, A. Cell division patterns in acute myeloid leukemia stem-like cells determine clinical course: A model to predict patient survival. Cancer Res. 2015, 75, 940–949. [Google Scholar] [CrossRef] [Green Version]
- Tang, L.; Bergevoet, S.M.; Gilissen, C.; de Witte, T.; Jansen, J.H.; van der Reijden, B.A.; Raymakers, R.A.P. Hematopoietic stem cells exhibit a specific ABC transporter gene expression profile clearly distinct from other stem cells. BMC Pharmacol. 2010, 10, 12. [Google Scholar] [CrossRef] [Green Version]
- De Jonge-Peeters, S.D.P.W.M.; Kuipers, F.; de Vries, E.G.E.; Vellenga, E. ABC transporter expression in hematopoietic stem cells and the role in AML drug resistance. Crit. Rev. Oncol. Hematol. 2007, 62, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Bunting, K.D. ABC transporters as phenotypic markers and functional regulators of stem cells. Stem Cells 2002, 20, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Borowski, E.; Bontemps-Gracz, M.M.; Piwkowska, A. Strategies for overcoming ABC-transporters-mediated multidrug resistance (MDR) of tumor cells. Acta Biochim. Pol. 2005, 52, 609–627. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, K.; Yamada, K.; Egashira, M.; Yazaki, Y.; Hirai, H.; Kikuchi, A.; Oshimi, K. Temporal and spatial distribution of DNA topoisomerase II alters during proliferation, differentiation, and apoptosis in HL-60 cells. Blood 1998, 91, 1407–1417. [Google Scholar] [CrossRef] [Green Version]
- Aoyama, M.; Grabowski, D.R.; Isaacs, R.J.; Krivacic, K.A.; Rybicki, L.A.; Bukowski, R.M.; Ganapathi, M.K.; Hickson, L.D.; Ganapathi, R. Altered expression and activity of topoisomerases during all-trans retinoic acid-induced differentiation of HL-60 cells. Blood 1998, 92, 2863–2870. [Google Scholar] [CrossRef] [PubMed]
- Michelson, A.P.; McDonough, S.; Willman, C.L.; Koegle, E.R.; Godwin, J.E.; Petersdorf, S.H.; List, A.F.; Othus, M.; Appelbaum, F.R.; Radich, J.P.; et al. Association of immunophenotype with expression of topoisomerase II α and β in adult acute myeloid leukemia. Sci. Rep. 2020, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Harker, W.G.; Slade, D.L.; Parr, R.L.; Drake, F.H. Mitoxantrone resistance in HL-60 leukemia cells: Reduced nuclear topoisomerase II catalytic activity and drug-induced DNA cleavage in association with reduced expression of the topoisomerase II β isoform. Biochemistry 1991, 30, 9953–9961. [Google Scholar] [CrossRef]
- Sanz, M.A.; Montesinos, P.; Kim, H.T.; Ruiz-Argüelles, G.J.; Undurraga, M.S.; Uriarte, M.R.; Martínez, L.; Jacomo, R.H.; Gutiérrez-Aguirre, H.; Melo, R.A.M.; et al. All-trans retinoic acid with daunorubicin or idarubicin for risk-adapted treatment of acute promyelocytic leukaemia: A matched-pair analysis of the PETHEMA LPA-2005 and IC-APL studies. Ann. Hematol. 2015, 94, 1347–1356. [Google Scholar] [CrossRef] [PubMed]
- McNamara, S.; Wang, H.; Hanna, N.; Miller, W.H. Topoisomerase IIβ negatively modulates retinoic acid receptor α function: A novel mechanism of retinoic acid resistance. Mol. Cell. Biol. 2008, 28, 2066–2077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichikawa, Y.; Pluznik, D.H.; Sachs, L. Feedback inhibition of the development of macrophage and granulocyte colonies. I. Inhibition by macrophage. Proc. Natl. Acad. Sci. USA 1967, 58, 1480–1486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Fujii, H.; Kim, H.J.; Hermans, K.; Usenko, T.; Xie, S.; Luo, Z.J.; Ma, J.; Celso, C.L.; Dick, J.E.; et al. Enhanced human hematopoietic stem and progenitor cell engraftment by blocking donor T cell–mediated TNF signaling. Sci. Transl. Med. 2017, 9, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, D.B.; Frismantas, V.; Milton, Y.; David, R.; Pop-Damkov, P.; Ferguson, D.; MacDonald, A.; Vargel Bölükbaşı, Ö.; Joyce, C.E.; Moreira Teixeira, L.S.; et al. On-chip recapitulation of clinical bone marrow toxicities and patient-specific pathophysiology. Nat. Biomed. Eng. 2020, 4, 394–406. [Google Scholar] [CrossRef] [PubMed]
- Berry, J.D.; Godara, P.; Liovic, P.; Haylock, D.N. Predictions for optimal mitigation of paracrine inhibitory signalling in haemopoietic stem cell cultures. Stem Cell Res. Ther. 2015, 6, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Mahadik, B.; Hannon, B.; Harley, B.A.C. A computational model of feedback-mediated hematopoietic stem cell differentiation in vitro. PLoS ONE 2019, 14, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, A.C.; Ishida, R.; Nakauchi, H.; Yamazaki, S. Long-term ex vivo expansion of mouse hematopoietic stem cells. Nat. Protoc. 2020, 15, 628–648. [Google Scholar] [CrossRef]
- Csaszar, E.; Kirouac, D.C.; Yu, M.; Wang, W.; Qiao, W.; Cooke, M.P.; Boitano, A.E.; Ito, C.; Zandstra, P.W. Rapid expansion of human hematopoietic stem cells by automated control of inhibitory feedback signaling. Cell Stem Cell 2012, 10, 218–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Jones, B.; Zhang, Y.; Vinardell, T.; Pei, M. Low-density expansion protects human synovium-derived stem cells from replicative senescence: A preliminary study. Drug Deliv. Transl. Res. 2012, 2, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Lica, J.J.; Grabe, G.J.; Heldt, M.; Misiak, M.; Bloch, P.; Serocki, M.; Switalska, M.; Wietrzyk, J.; Baginski, M.; Hellmann, A.; et al. Cell density-dependent cytological stage profile and its application for a screen of cytostatic agents active toward leukemic stem cells. Stem Cells Dev. 2018, 27, 488–513. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.; Pusztai, L.; Swanton, C. Cancer heterogeneity: Implications for targeted therapeutics. Br. J. Cancer 2013, 108, 479–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barile, F.A.; Dierickx, P.J.; Kristen, U. In vitro cytotoxicity testing for prediction of acute human toxicity. Cell Biol. Toxicol. 1994, 10, 155–162. [Google Scholar] [CrossRef]
- Doskey, C.M.; Van ’T Erve, T.J.; Wagner, B.A.; Buettner, G.R. Moles of a substance per cell is a highly informative dosing metric in cell culture. PLoS ONE 2015, 10, 1–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gülden, M.; Mörchel, S.; Seibert, H. Factors influencing nominal effective concentrations of chemical compounds in vitro: Cell concentration. Toxicol. Vitr. 2001, 15, 233–243. [Google Scholar] [CrossRef]
- Quinn, J.J.; Jones, M.G.; Okimoto, R.A.; Nanjo, S.; Chan, M.M.; Yosef, N.; Bivona, T.G.; Weissman, J.S. Single-cell lineages reveal the rates, routes, and drivers of metastasis in cancer xenografts. Science 2021, 371, eabc1944. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, M.; Kenmotsu, H.; Koh, Y.; Yoshino, T.; Yoshikawa, T.; Naito, T.; Takahashi, T.; Murakami, H.; Nakamura, Y.; Tsuya, A.; et al. Size-based isolation of circulating tumor cells in lung cancer patients using a microcavity array system. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saglietti, C.; Volante, M.; La Rosa, S.; Letovanec, I.; Pusztaszeri, M.; Gatti, G.; Bongiovanni, M. Cytology of primary salivary gland-type tumors of the lower respiratory tract: Report of 15 cases and review of the literature. Front. Med. 2017, 4. [Google Scholar] [CrossRef] [Green Version]
- Hofman, V.; Bonnetaud, C.; Ilie, M.I.; Vielh, P.; Vignaud, J.M.; Fléjou, J.F.; Lantuejoul, S.; Piaton, E.; Mourad, N.; Butori, C.; et al. Preoperative circulating tumor cell detection using the isolation by size of epithelial tumor cell method for patients with lung cancer is a new prognostic biomarker. Clin. Cancer Res. 2011, 17, 827–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhar, M.; Pao, E.; Renier, C.; Go, D.E.; Che, J.; Montoya, R.; Conrad, R.; Matsumoto, M.; Heirich, K.; Triboulet, M.; et al. Label-free enumeration, collection and downstream cytological and cytogenetic analysis of circulating tumor cells. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Jaccard, N.; Griffin, L.D.; Keser, A.; Macown, R.J.; Super, A.; Veraitch, F.S.; Szita, N. Automated method for the rapid and precise estimation of adherent cell culture characteristics from phase contrast microscopy images. Biotechnol. Bioeng. 2014, 111, 504–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, T.; Chandimali, N.; Huynh, D.L.; Zhang, J.J.; Kim, N.; Bak, Y.; Yoon, D.Y.; Yu, D.Y.; Lee, J.C.; Gera, M.; et al. BRM270 inhibits cancer stem cell maintenance via microRNA regulation in chemoresistant A-549 lung adenocarcinoma cells article. Cell Death Dis. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Udoh, K.; Parte, S.; Carter, K.; Mack, A.; Kakar, S.S. Targeting of lung cancer stem cell self-renewal pathway by a small molecule verrucarin J. Stem Cell Rev. Rep. 2019, 15, 601–611. [Google Scholar] [CrossRef]
- Halim, N.H.A.; Zakaria, N.; Satar, N.A.; Yahaya, B.H. Isolation and characterization of cancer stem cells of the non-small-cell lung cancer (A-549) cell line. Methods Mol. Biol. 2016, 1516, 371–388. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jiang, M.; Du, C.; Yu, Y.; Liu, Y.; Li, M.; Luo, F. Utilization of lung cancer cell lines for the study of lung cancer stem cells. Oncol. Lett. 2018, 15, 6791–6798. [Google Scholar] [CrossRef]
- Sabisz, M.; Skladanowski, A. Cancer stem cells and escape from drug-induced premature senescence in human lung tumor cells: Implications for drug resistance and in vitro drug screening models. Cell Cycle 2009, 8, 3208–3217. [Google Scholar] [CrossRef] [Green Version]
- Gu, H.; Wu, X.-Y.; Fan, R.-T.; Wang, X.; Guo, Y.-Z.; Wang, R. Side population cells from long-term passage non-small cell lung cancer cells display loss of cancer stem cell-like properties and chemoradioresistance. Oncol. Lett. 2016, 12, 2886–2893. [Google Scholar] [CrossRef]
- Tirino, V.; Camerlingo, R.; Bifulco, K.; Irollo, E.; Montella, R.; Paino, F.; Sessa, G.; Carriero, M.V.; Normanno, N.; Rocco, G.; et al. TGF-β1 exposure induces epithelial to mesenchymal transition both in CSCs and non-CSCs of the A-549 cell line, leading to an increase of migration ability in the CD133+ A-549 cell fraction. Cell Death Dis. 2013, 4, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Reka, A.K.; Kuick, R.; Kurapati, H.; Standiford, T.J.; Omenn, G.S.; Keshamouni, V.G. Identifying inhibitors of epithelial-mesenchymal transition by connectivity map-based systems approach. J. Thorac. Oncol. 2011, 6, 1784–1792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yıldırım, M.; Atmaca, Z.E.; Kayalar, O.; Oztay, F. Prevention the TGF-β induced epithelial-mesenchymal transition with atorvastatin in human A-549 cell line. Eur. Respir. Soc. 2017, 50, PA1024. [Google Scholar]
- Gong, L.; Liu, G.; Zhu, H.; Li, C.; Li, P.; Liu, C.; Tang, H.; Wu, K.; Wu, J.; Liu, D.; et al. IL-32 induces epithelial-mesenchymal transition by triggering endoplasmic reticulum stress in A-549 cells. BMC Pulm. Med. 2020, 20, 1–9. [Google Scholar] [CrossRef]
- Zhang, Z.; Liang, Z.; Li, H.; Li, C.; Yang, Z.; Li, Y.; She, D.; Cao, L.; Wang, W.; Liu, C.; et al. Perfluorocarbon reduces cell damage from Blast injury by inhibiting signal paths of NF-κB, MAPK and Bcl-2/Bax signaling pathway in A-549 cells. PLoS ONE 2017, 12, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Cooper, J.R.; Abdullatif, M.B.; Burnett, E.C.; Kempsell, K.E.; Conforti, F.; Tolley, H.; Collins, J.E.; Davies, D.E. Long term culture of the A-549 cancer cell line promotes multilamellar body formation and differentiation towards an alveolar type II Pneumocyte phenotype. PLoS ONE 2016, 11, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, R.; Li, G.; Zhang, Q.; Tang, Q.; Huang, J.; Hu, C.; Liu, Y.; Wang, Q.; Liu, W.; Gao, N.; et al. Hirsutine induces mPTP-dependent apoptosis through ROCK1/PTEN/PI3K/GSK3β pathway in human lung cancer cells article. Cell Death Dis. 2018, 9. [Google Scholar] [CrossRef]
- Park, C.W.; Bak, Y.; Kim, M.J.; Srinivasrao, G.; Hwang, J.; Sung, N.K.; Kim, B.Y.; Yu, J.H.; Hong, J.T.; Yoon, D.Y. The novel small molecule STK899704 promotes senescence of the human A-549 NSCLC cells by inducing DNA damage responses and cell cycle arrest. Front. Pharmacol. 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nova, Z.; Skovierova, H.; Strnadel, J.; Halasova, E.; Calkovska, A. Short-term versus long-term culture of A-549 cells for evaluating the effects of lipopolysaccharide on oxidative stress, surfactant proteins and cathelicidin LL-37. Int. J. Mol. Sci. 2020, 21, 1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.C.; Boone, M.; Meuris, L.; Lemmens, I.; Van Roy, N.; Soete, A.; Reumers, J.; Moisse, M.; Plaisance, S.; Drmanac, R.; et al. Genome dynamics of the human embryonic kidney 293 lineage in response to cell biology manipulations. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [Green Version]
- Malm, M.; Saghaleyni, R.; Lundqvist, M.; Giudici, M.; Chotteau, V.; Field, R.; Varley, P.G.; Hatton, D.; Grassi, L.; Svensson, T.; et al. Evolution from adherent to suspension: Systems biology of HEK293 cell line development. Sci. Rep. 2020, 10, 1–15. [Google Scholar] [CrossRef]
- Misiak, M.; Heldt, M.; Szeligowska, M.; Mazzini, S.; Scaglioni, L.; Grabe, G.J.; Serocki, M.; Lica, J.; Switalska, M.; Wietrzyk, J.; et al. Molecular basis for the DNA damage induction and anticancer activity of asymmetrically substituted anthrapyridazone PDZ-7. Oncotarget 2017, 8, 105137–105154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacayo, N.J.; Duran, G.E.; Sikic, B.I. Modulation of resistance to idarubicin by the cyclosporin PSC 833 (valspodar) in multidrug-resistant cells. J. Exp. Ther. Oncol. 2003, 3, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Smeets, M.E.; Raymakers, R.A.; Vierwinden, G.; Pennings, A.H.; Boezeman, J.; Minderman, H.; de Witte, T.M. Idarubicin DNA intercalation is reduced by MRP1 and not Pgp. Leukemia 1999, 13, 1390–1398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Roozendaal, K.E.P.; Darling, D.; Farzaneh, F. DMSO and retinoic acid induce HL-60 differentiation by different but converging pathways. Exp. Cell Res. 1990, 190, 137–140. [Google Scholar] [CrossRef]
- Cook, R.T.; Keiner, J.A.; Yen, A. Ethanol causes accelerated G1 arrest in differentiating HL-60 cells. Alcohol. Clin. Exp. Res. 1990, 14, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Aroor, A.R.; Baker, R.C. Ethanol-induced apoptosis in human HL-60 cells. Life Sci. 1997, 61, 2345–2350. [Google Scholar] [CrossRef]
- Wesierska-Gadek, J.; Schloffer, D.; Gueorguieva, M.; Uhl, M.; Skladanowski, A. Increased susceptibility of poly(ADP-ribose) polymerase-1 knockout cells to antitumor triazoloacridone C-1305 is associated with permanent G2 cell cycle arrest. Cancer Res. 2004, 64, 4487–4497. [Google Scholar] [CrossRef] [Green Version]
- Lemke, K.; Poindessous, V.; Skladanowski, A.; Larsen, A.K. The antitumor triazoloacridone C-1305 is a topoisomerase II poison with unusual properties. Mol. Pharmacol. 2004, 66, 1035–1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemke, K.; Wojciechowski, M.; Laine, W.; Bailly, C.; Colson, P.; Baginski, M.; Larsen, A.K.; Skladanowski, A. Induction of unique structural changes in guanine-rich DNA regions by the triazoloacridone C-1305, a topoisomerase II inhibitor with antitumor activities. Nucleic Acids Res. 2005, 33, 6034–6047. [Google Scholar] [CrossRef] [Green Version]
- Olewniak, J.; Lemke, K.; Wojciechowski, M.; Składanowski, A. Interaction of antitumor triazoloacridone C-1305 and its analogs with telomeric DNA. Acta Biochim. Pol. 2006, 53, 157–158. [Google Scholar]
- Bram, E.E.; Ifergan, I.; Grimberg, M.; Lemke, K.; Skladanowski, A.; Assaraf, Y.G. C421 allele-specific ABCG2 gene amplification confers resistance to the antitumor triazoloacridone C-1305 in human lung cancer cells. Biochem. Pharmacol. 2007, 74, 41–53. [Google Scholar] [CrossRef]
- Kaminsky, R.; Schmid, C.; Brun, R. An “in vitro selectivity index” for evaluation of cytotoxicity of antitrypanosomal compounds. In Vitro Toxicol. 1996, 9, 315–324. [Google Scholar]
- Badisa, R.B.; Darling-Reed, S.F.; Joseph, P.; Cooperwood, J.S.; Latinwo, L.M.; Goodman, C.B. Selective cytotoxic activities of two novel synthetic drugs on human breast carcinoma MCF-7 cells. Anticancer Res. 2009, 29, 2993–2996. [Google Scholar] [PubMed]
- De Oliveira, P.F.; Alves, J.M.; Damasceno, J.L.; Oliveira, R.A.; Dias, H., Jr.; Crotti, A.E.; Tavares, D.C. Cytotoxicity screening of essential oils in cancer cell lines. Rev. Bras. Farmacogn. 2015, 25, 183–188. [Google Scholar] [CrossRef] [Green Version]
- Peña-Morán, O.A.; Villarreal, M.L.; Álvarez-Berber, L.; Meneses-Acosta, A.; Rodríguez-López, V. Cytotoxicity, Post-Treatment Recovery, and Selectivity Analysis of Naturally Occurring Podophyllotoxins from Bursera fagaroides var. fagaroides on Breast Cancer Cell Lines. Molecules 2016, 21, 1013. [Google Scholar] [CrossRef]
- Yoo, Y.D.; Lee, D.; Cha-Molstad, H.; Kim, H.; Mun, S.R.; Ji, C.; Park, S.H.; Sung, K.S.; Choi, S.A.; Hwang, J.; et al. Glioma-derived cancer stem cells are hypersensitive to proteasomal inhibition. EMBO Rep. 2017, 18, 150–168. [Google Scholar] [CrossRef] [Green Version]
- Mortenson, M.M.; Schlieman, M.G.; Virudachalam, S.; Bold, R.J. Effects of the proteasome inhibitor bortezomib alone and in combination with chemotherapy in the A-549 non-small-cell lung cancer cell line. Cancer Chemother. Pharmacol. 2004, 54, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general Amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Allen, A.E.A.; Payne, M.C.; Cole, D.J. Harmonic force constants for molecular mechanics force fields via hessian matrix projection. J. Chem. Theory Comput. 2018, 14, 274–281. [Google Scholar] [CrossRef]
- Colasanti, A.V.; Lu, X.J.; Olson, W.K. Analyzing and building nucleic acid structures with 3DNA. J. Vis. Exp. 2013. [Google Scholar] [CrossRef] [PubMed]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; Van Der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Ivani, I.; Dans, P.D.; Noy, A.; Pérez, A.; Faustino, I.; Hospital, A.; Walther, J.; Andrio, P.; Goñi, R.; Balaceanu, A.; et al. Parmbsc1: A refined force field for DNA simulations. Nat. Methods 2015, 13, 55–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eslami, H.; Mojahedi, F.; Moghadasi, J. Molecular dynamics simulation with weak coupling to heat and material baths. J. Chem. Phys. 2010, 133. [Google Scholar] [CrossRef] [PubMed]
- Tribello, G.A.; Bonomi, M.; Branduardi, D.; Camilloni, C.; Bussi, G. PLUMED 2: New feathers for an old bird. Comput. Phys. Commun. 2014, 185, 604–613. [Google Scholar] [CrossRef] [Green Version]
- Wieczór, M.; Hospital, A.; Bayarri, G.; Czub, J.; Orozco, M. Molywood: Streamlining the design and rendering of molecular movies. Bioinformatics 2020, 36, 4660–4661. [Google Scholar] [CrossRef] [PubMed]
- Hokland, P.; Woll, P.S.; Hansen, M.C.; Bill, M. The concept of leukaemic stem cells in acute myeloid leukaemia 25 years on: Hitting a moving target. Br. J. Haematol. 2019, 187, 144–156. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cellular Stage | HL-60 Percent of Cell Stage | ||
|---|---|---|---|
| Primitive | Standard | Mature | |
| * LSC-like, * Progenitor-like and * CFU-like | 20–25 | 5–10 | 1–1.5 |
| * ˚ Blast-like | 45–60 | 25–30 | 2–2.5 |
| ˚ Promyelocyte-like | 5–30 | 10–15 | 5–7 |
| ˚ Myelocyte-like | 2.5–5 | 40–50 | 10–20 |
| Senescent | 2.5–5 | 5–10 | 72–80 |
| Cellular Stage | A-549 Percent of Cell Stage | ||
|---|---|---|---|
| Primitive | Standard | Mature | |
| * CSC-like and * Progenitor-like | 70–90 | 30–70 | 20–35 |
| *˚ Blast-like | 10–25 | 30–60 | 10–15 |
| Senescent ROS negative | 0.5–5 | 5–2 | 25–35 |
| Senescent ROS positive | 0–0.5 | 5–8 | 25–35 |
| Compound | Main Targets | ||
|---|---|---|---|
| Strength of Interaction | Mechanism of Action | PC Activity | |
| C-123 | Strong | DNA Breaks DNA Intercalation Proteasome Inhibition | High |
| Bortezomib | Strong | Proteasome Inhibition | High |
| Idarubicin | Strong | Reactive Oxygen Species Topoisomerase IIα Inhibition DNA Breaks DNA Intercalation | Medium |
| Doxorubicin | Strong | Topoisomerase IIα Inhibition DNA Breaks DNA Intercalation | Medium |
| C-1305 | Strong | Topoisomerase IIα Inhibition DNA Intercalation | Medium |
| DMSO | Medium | Broad, Not fully determined | Medium |
| Ethanol | Weak | Broad, Not fully determined | Medium |
| A | Inhibitory Concentration IC50 | |||||||||||
| Culture Density [103 Cells per mL] | C-123 [nM] | Idarubicin [nM] | DMSO [Percent] | Ethanol [Percent] | ||||||||
| Primitive | Standard | Mature | Primitive | Standard | Mature | Primitive | Standard | Mature | Primitive | Standard | Mature | |
| 5 | 19.0 ±0.4 | 18.2 ±0.3 | 18.2 ±0.2 | 6.8 ±0.8 | 2.0 ±0.1 | 0.7 ±0.1 | 2.6 ±0.3 | 1.3 ±0.2 | 0.5 ±0.1 | 2.4 ±0.2 | 1.7 ±0.2 | 1.0 ±0.1 |
| 25 | 56.3 ±0.2 | 57.3 ±0.7 | 55.4 ±0.5 | 12.4 ±2.0 | 5.1 ±0.7 | 2.5 ±0.2 | 4.8 ±0.4 | 2.6 ±0.2 | 1.2 ±0.1 | 4.2 ±0.2 | 2.1 ±0.1 | 1.6 ±0.1 |
| 100 | 201.6 ±0.5 | 198.7 ±1.0 | 199.1 ±0.8 | 32.7 ±0.6 | 16.2 ±2.4 | 5.8 ±1.5 | 10.1 ±1.5 | 4.6 ±0.5 | 2.1 ±0.3 | 4.1 ±0.4 | 1.5 ±0.2 | 0.4 ±0.1 |
| B | IC50 average calculated for total cell number [103 cells per mL] | |||||||||||
| 5 | 25 | 100 | 5 | 25 | 100 | 5 | 25 | 100 | 5 | 25 | 100 | |
| 18.5 | 56.3 | 199.8 | 3.2 | 6.7 | 19.7 | 1.5 | 2.9 | 5.6 | 1.7 | 2.6 | 2.0 | |
| *IC50 average calculated for primitive cell number | ||||||||||||
| Primitive | Standard | Mature | Primitive | Standard | Mature | Primitive | Standard | Mature | Primitive | Standard | Mature | |
| 92.3 | 91.4 | 90.9 | 18.8 | 7.8 | 3.0 | 5.8 | 2.8 | 1.3 | 3.6 | 1.8 | 1.0 | |
| C | Effective Concentration EC50 | |||||||||||
| [fMper cell] | [fM per cell] | [µM per cell] | [µM per cell] | |||||||||
| 5 | 3.8 | 3.6 | 3.6 | 1.4 | 0.4 | 0.1 | 67.7 | 33.9 | 12.1 | 103.5 | 75.6 | 44.1 |
| 25 | 2.3 | 2.3 | 2.2 | 0.5 | 0.2 | 0.1 | 24.7 | 13.5 | 6.2 | 36.2 | 18.6 | 14.1 |
| 100 | 2.0 | 2.0 | 2.0 | 0.3 | 0.2 | 0.1 | 13.0 | 5.9 | 2.7 | 8.9 | 4.6 | 0.8 |
| D | EC50 average calculated for total cell number [103 cells per mL] | |||||||||||
| 5 | 25 | 100 | 5 | 25 | 100 | 5 | 25 | 100 | 5 | 25 | 100 | |
| 3.7 | 2.3 | 2.0 | 0.6 | 0.3 | 0.2 | 37.9 | 14.8 | 7.2 | 74.4 | 23.0 | 4.8 | |
| *EC50 average calculated for primitive cell number | ||||||||||||
| Primitive | Standard | Mature | Primitive | Standard | Mature | Primitive | Standard | Mature | Primitive | Standard | Mature | |
| 2.6 | 2.6 | 2.7 | 0.1 | 0.3 | 0.7 | 7.0 | 17.8 | 35.1 | 19.7 | 32.9 | 49.5 | |
| A | IC50 [nM] | ||||||
| Compound | A-549 | HEK-293 | Selectivity Index | ||||
| Primitive | Standard | Mature | Primitive | Primitive | Standard | Mature | |
| C-123 | 45.6 ± 1.8 | 39.8 ± 2.0 | 41.3 ± 1.3 | 27.6 ± 4.5 | 0.6 | 0.7 | 0.7 |
| Bortezomib | 13.5 ± 0.7 | 14.3 ± 0.9 | 15.3 ± 0.9 | 2.2 ± 0.1 | 0.2 | 0.1 | 0.1 |
| C-1305 | N.d. | 18.5 ± 1.4 | 33.3 ± 2.6 | 18.5 ± 2.8 | N.d. | 1 | 0.6 |
| Doxorubicin | N.d. | 39.7 ± 3.3 | 61.8 ± 3.8 | 51.7 ± 9.0 | N.d. | 1.3 | 0.8 |
| B | EC50 [fM per cell] | ||||||
| A-549 | HEK-293 | Selectivity Index | |||||
| Primitive | Standard | Mature | Primitive | Primitive | Standard | Mature | |
| C-123 | 36 ± 1 | 32 ± 2 | 33 ± 1 | 11 ± 2 | 0.3 | 0.3 | 0.3 |
| Bortezomib | 11 ± 1 | 11 ± 1 | 12 ± 1 | 1 ± 0 | 0.1 | 0.1 | 0.1 |
| C-1305 | N.d. | 15 ± 1 | 27 ± 2 | 7 ± 1 | N.d. | 0.5 | 0.3 |
| Doxorubicin | N.d. | 32 ± 3 | 49 ± 3 | 21 ± 4 | N.d. | 0.7 | 0.4 |
| A | Bortezomib Selectivity Index | |||
| Culture Density [103 Cells per mL] | Primitive HL-60 *IC50 | SIIC50 | Primitive HL-60 *EC50 | SIEC50 |
| 0.5 | 14.75 ± 1.02 | 0.15 | 2.95 ± 0.20 | 0.34 |
| B | C-123 Selectivity Index calculate per IC50 | |||
| Primitive HL-60 | Standard HL-60 | Mature HL-60 | ||
| 5 | 1.5 | 1.5 | 1.5 | |
| 25 | 0.5 | 0.5 | 0.5 | |
| 100 | 0.1 | 0.1 | 0.1 | |
| C | C-123 Selectivity Index calculate per EC50 | |||
| 5 | 14.6 | 15.2 | 15.2 | |
| 25 | 24.5 | 24.1 | 25.0 | |
| 100 | 27.4 | 27.8 | 27.8 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lica, J.J.; Wieczór, M.; Grabe, G.J.; Heldt, M.; Jancz, M.; Misiak, M.; Gucwa, K.; Brankiewicz, W.; Maciejewska, N.; Stupak, A.; et al. Effective Drug Concentration and Selectivity Depends on Fraction of Primitive Cells. Int. J. Mol. Sci. 2021, 22, 4931. https://doi.org/10.3390/ijms22094931
Lica JJ, Wieczór M, Grabe GJ, Heldt M, Jancz M, Misiak M, Gucwa K, Brankiewicz W, Maciejewska N, Stupak A, et al. Effective Drug Concentration and Selectivity Depends on Fraction of Primitive Cells. International Journal of Molecular Sciences. 2021; 22(9):4931. https://doi.org/10.3390/ijms22094931
Chicago/Turabian StyleLica, Jan Jakub, Miłosz Wieczór, Grzegorz Jan Grabe, Mateusz Heldt, Marta Jancz, Majus Misiak, Katarzyna Gucwa, Wioletta Brankiewicz, Natalia Maciejewska, Anna Stupak, and et al. 2021. "Effective Drug Concentration and Selectivity Depends on Fraction of Primitive Cells" International Journal of Molecular Sciences 22, no. 9: 4931. https://doi.org/10.3390/ijms22094931
APA StyleLica, J. J., Wieczór, M., Grabe, G. J., Heldt, M., Jancz, M., Misiak, M., Gucwa, K., Brankiewicz, W., Maciejewska, N., Stupak, A., Bagiński, M., Rolka, K., Hellmann, A., & Składanowski, A. (2021). Effective Drug Concentration and Selectivity Depends on Fraction of Primitive Cells. International Journal of Molecular Sciences, 22(9), 4931. https://doi.org/10.3390/ijms22094931