
ADAM10 Site-Dependent Biology: Keeping Control of a Pervasive Protease



Abstract
:1. Introduction
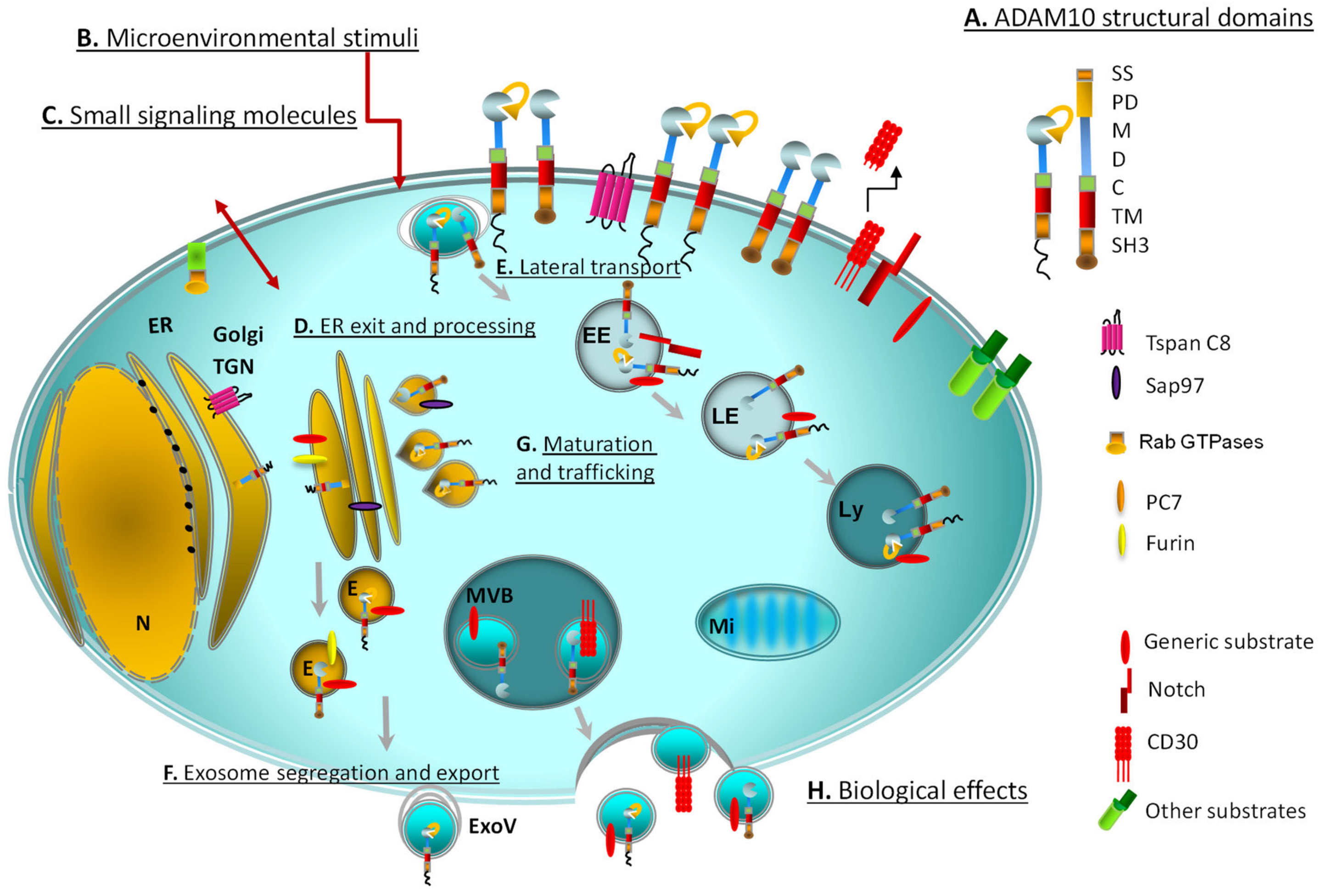
2. ADAM10 Processing, Maturation and Trafficking
2.1. Structural and Spatial Determinants of ADAM10 Function
2.2. Regulation of ADAM10 Trafficking and Export
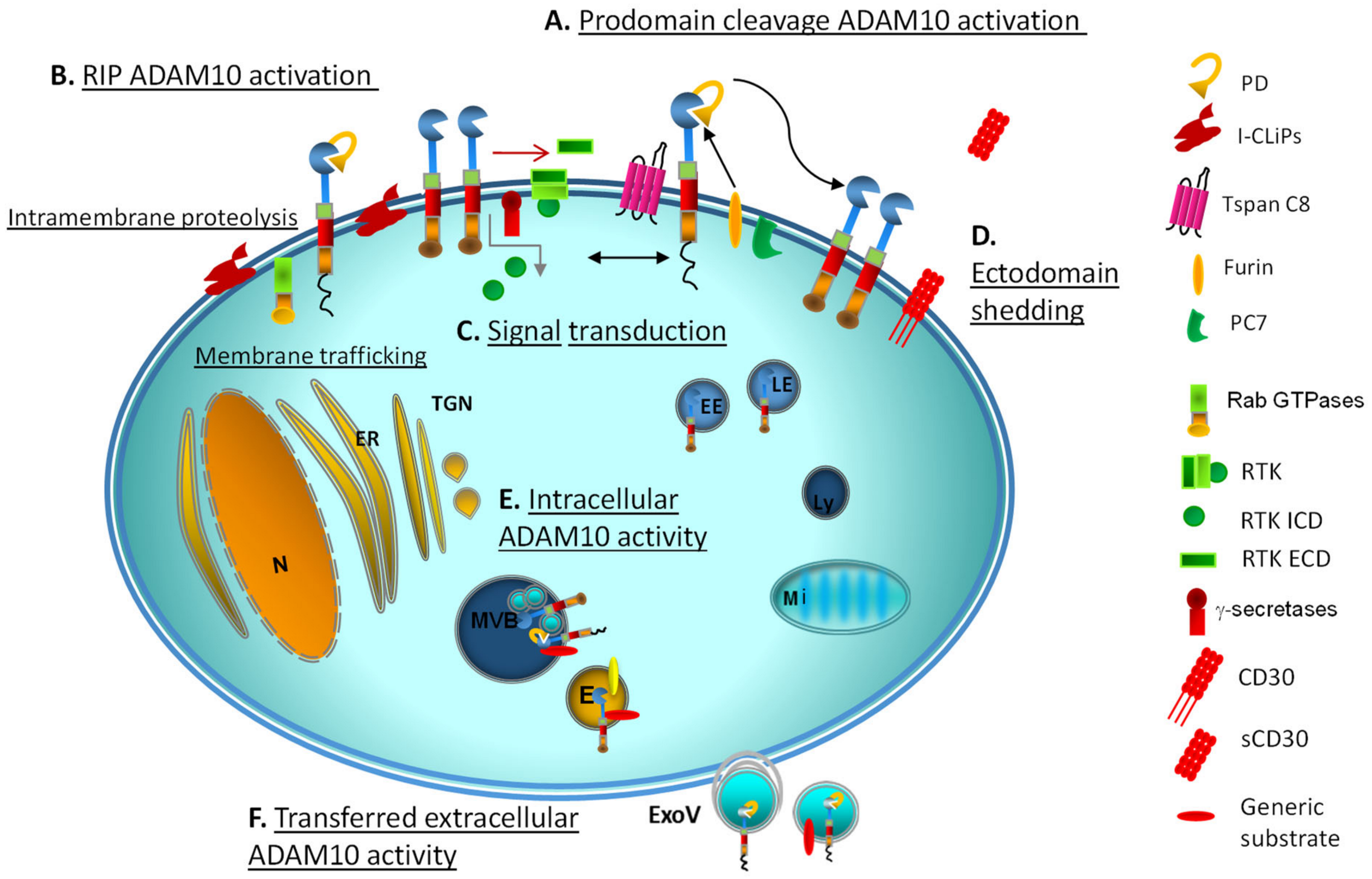
2.3. ADAM10 Activation and Proteolytic Ensemble
3. ADAM10 as a Bridge between Extracellular and Intracellular Environments
3.1. ADAM10 Activity and Intracellular Signalling
3.2. Small Signaling Molecules as Metabolic Indicators of ADAM Activity
4. ADAMs Substrates and Partners
4.1. Multimolecular Complexes and Subcellular Compartments
4.2. Early Clues and Recent Evidence of Active Intracellular ADAM10
5. ADAM10 Relocation and Related Function
5.1. Redistribution Induced by Regulatory Proteins
5.2. ADAM10 Activity in Extracellular Vesicles and in Unexpected Cell Sites
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Forneris, F.; Mattevi, A. Enzymes Without Borders: Mobilizing Substrates, Delivering Products. Science 2008, 321, 213–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freeman, M. Rhomboids, signalling and cell biology. Biochem. Soc. Trans. 2016, 44, 945–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimoda, M.; Khokha, R. Metalloproteinases in extracellular vesicles. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1989–2000. [Google Scholar] [CrossRef] [PubMed]
- Lambrecht, B.N.; Vanderkerken, M.; Hammad, H. The emerging role of ADAM metalloproteinases in immunity. Nat. Rev. Immunol. 2018, 18, 745–758. [Google Scholar] [CrossRef]
- Caescu, C.I.; Jeschke, G.R.; Turk, B.E. Active-site determinants of substrate recognition by the metalloproteinases TACE and ADAM10. Biochem. J. 2009, 424, 79–88. [Google Scholar] [CrossRef] [Green Version]
- Lichtenthaler, S.F.; Lemberg, M.K.; Fluhrer, R. Proteolytic ectodomain shedding of membrane proteins in mammals—hardware, concepts, and recent developments. EMBO J. 2018, 37, e99456. [Google Scholar] [CrossRef]
- Duffy, M.J.; McKiernan, E.; O’Donovan, N.; McGowan, P.M. Role of ADAMs in Cancer Formation and Progression. Clin. Cancer Res. 2009, 15, 1140–1144. [Google Scholar] [CrossRef] [Green Version]
- Murphy, G. The ADAMs: Signalling scissors in the tumour microenvironment. Nat. Rev. Cancer 2008, 8, 932–941. [Google Scholar] [CrossRef]
- Escalona, R.M.; Chan, E.; Kannourakis, G.; Findlay, J.K.; Ahmed, N. The Many Facets of Metzincins and Their Endogenous Inhibitors: Perspectives on Ovarian Cancer Progression. Int. J. Mol. Sci. 2018, 19, 450. [Google Scholar] [CrossRef] [Green Version]
- Matthews, A.L.; Szyroka, J.; Collier, R.; Noy, P.J.; Tomlinson, M.G. Scissor sisters: Regulation of ADAM10 by the TspanC8 tetraspanins. Biochem. Soc. Trans. 2017, 45, 719–730. [Google Scholar] [CrossRef] [Green Version]
- Saha, N.; Robev, D.; Himanen, J.P.; Nikolov, D.B. ADAM proteases: Emerging role and targeting of the non-catalytic domains. Cancer Lett. 2019, 467, 50–57. [Google Scholar] [CrossRef]
- Edwards, D.R.; Handsley, M.M.; Pennington, C.J. The ADAM metalloproteinases. Mol. Asp. Med. 2008, 29, 258–289. [Google Scholar] [CrossRef]
- Postina, R. Activation of α-secretase cleavage. J. Neurochem. 2011, 120, 46–54. [Google Scholar] [CrossRef]
- Saint-Pol, J.; Eschenbrenner, E.; Dornier, E.; Boucheix, C.; Charrin, S.; Rubinstein, E. Regulation of the trafficking and the function of the metalloprotease ADAM10 by tetraspanins. Biochem. Soc. Trans. 2017, 45, 937–944. [Google Scholar] [CrossRef]
- Seegar, T.C.; Blacklow, S.C. Domain integration of ADAM family proteins: Emerging themes from structural studies. Exp. Biol. Med. 2019, 244, 1510–1519. [Google Scholar] [CrossRef]
- Aljohmani, A.; Yildiz, D. A Disintegrin and Metalloproteinase—Control Elements in Infectious Diseases. Front. Cardiovasc. Med. 2020, 7, 8281. [Google Scholar] [CrossRef]
- Seegar, T.C.; Killingsworth, L.B.; Saha, N.; Meyer, P.A.; Patra, D.; Zimmerman, B.; Janes, P.W.; Rubinstein, E.; Nikolov, D.B.; Skiniotis, G.; et al. Structural Basis for Regulated Proteolysis by the α-Secretase ADAM10. Cell 2017, 171, 1638–1648.e7. [Google Scholar] [CrossRef] [Green Version]
- Xu, P.; Liu, J.; Sakaki-Yumoto, M.; Derynck, R. TACE Activation by MAPK-Mediated Regulation of Cell Surface Dimerization and TIMP3 Association. Sci. Signal. 2012, 5, ra34. [Google Scholar] [CrossRef] [Green Version]
- Scilabra, S.D.; Pigoni, M.; Pravatá, V.; Schätzl, T.; Müller, S.A.; Troeberg, L.; Lichtenthaler, S.F. Increased TIMP-3 expression alters the cellular secretome through dual inhibition of the metalloprotease ADAM10 and ligand-binding of the LRP-1 receptor. Sci. Rep. 2018, 8, 14697. [Google Scholar] [CrossRef]
- Deng, W.; Cho, S.; Su, P.-C.; Berger, B.W.; Li, R. Membrane-enabled dimerization of the intrinsically disordered cytoplasmic domain of ADAM10. Proc. Natl. Acad. Sci. USA 2014, 111, 15987–15992. [Google Scholar] [CrossRef] [Green Version]
- Dyson, H.J.; Wright, P.E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 2005, 6, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Li, R. Juxtamembrane contribution to transmembrane signaling. Biopolymers 2015, 104, 317–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uversky, V.N. Intrinsic Disorder-based Protein Interactions and their Modulators. Curr. Pharm. Des. 2013, 19, 4191–4213. [Google Scholar] [CrossRef]
- Van Deventer, S.J.; Dunlock, V.-M.E.; Van Spriel, A.B. Molecular interactions shaping the tetraspanin web. Biochem. Soc. Trans. 2017, 45, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Haining, E.J.; Yang, J.; Bailey, R.L.; Khan, K.; Collier, R.; Tsai, S.; Watson, S.P.; Frampton, J.; Garcia, P.; Tomlinson, M.G. The TspanC8 Subgroup of Tetraspanins Interacts with A Disintegrin and Metalloprotease 10 (ADAM10) and Regulates Its Maturation and Cell Surface Expression. J. Biol. Chem. 2012, 287, 39753–39765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jouannet, S.; Saint-Pol, J.; Fernandez, L.; Nguyen, V.; Charrin, S.; Boucheix, C.; Brou, C.; Milhiet, P.-E.; Rubinstein, E. TspanC8 tetraspanins differentially regulate the cleavage of ADAM10 substrates, Notch activation and ADAM10 membrane compartmentalization. Cell Mol. Life Sci. 2016, 73, 1895–1915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noy, P.J.; Yang, J.; Reyat, J.S.; Matthews, A.L.; Charlton, A.E.; Furmston, J.; Rogers, D.A.; Rainger, G.E.; Tomlinson, M.G. TspanC8 Tetraspanins and A Disintegrin and Metalloprotease 10 (ADAM10) Interact via Their Extracellular Regions: Evidence for distinct binding mechanisms for different TspanC8 proteins. J. Biol. Chem. 2016, 291, 3145–3157. [Google Scholar] [CrossRef] [Green Version]
- Prox, J.; Willenbrock, M.; Weber, S.; Lehmann, T.; Schmidt-Arras, D.; Schwanbeck, R.; Saftig, P.; Schwake, M. Tetraspanin15 regulates cellular trafficking and activity of the ectodomain sheddase ADAM10. Cell. Mol. Life Sci. 2012, 69, 2919–2932. [Google Scholar] [CrossRef]
- Seipold, L.; Altmeppen, H.; Koudelka, T.; Tholey, A.; Kasparek, P.; Sedlacek, R.; Schweizer, M.; Bär, J.; Mikhaylova, M.; Glatzel, M.; et al. In vivo regulation of the A disintegrin and metalloproteinase 10 (ADAM10) by the tetraspanin. Cell Mol. Life Sci. 2018, 75, 3251–3267. [Google Scholar] [CrossRef]
- Marcello, E.; Gardoni, F.; Di Luca, M.; Pérez-Otaño, I. An Arginine Stretch Limits ADAM10 Exit from the Endoplasmic Reticulum. J. Biol. Chem. 2010, 285, 10376–10384. [Google Scholar] [CrossRef] [Green Version]
- Maretzky, T.; Evers, A.; Le Gall, S.; Alabi, R.O.; Speck, N.; Reiss, K.; Blobel, C.P. The Cytoplasmic Domain of A Disintegrin and Metalloproteinase 10 (ADAM10) Regulates Its Constitutive Activity but Is Dispensable for Stimulated ADAM10-dependent Shedding. J. Biol. Chem. 2015, 290, 7416–7425. [Google Scholar] [CrossRef] [Green Version]
- Saraceno, C.; Marcello, E.; Di Marino, D.; Borroni, B.; Claeysen, S.; Perroy, J.; Padovani, A.; Tramontano, A.; Gardoni, F.; Di Luca, M. SAP97-mediated ADAM10 trafficking from Golgi outposts depends on PKC phosphorylation. Cell Death Dis. 2014, 5, e1547. [Google Scholar] [CrossRef] [Green Version]
- Matthews, A.L.; Noy, P.J.; Reyat, J.S.; Tomlinson, M.G. Regulation of A disintegrin and metalloproteinase (ADAM) family sheddases ADAM10 and ADAM17: The emerging role of tetraspanins and rhomboids. Platelets 2017, 28, 333–341. [Google Scholar] [CrossRef] [Green Version]
- Dornier, E.; Coumailleau, F.; Ottavi, J.-F.; Moretti, J.; Boucheix, C.; Mauduit, P.; Schweisguth, F.; Rubinstein, E. TspanC8 tetraspanins regulate ADAM10/Kuzbanian trafficking and promote Notch activation in flies and mammals. J. Cell Biol. 2012, 199, 481–496. [Google Scholar] [CrossRef] [Green Version]
- Maretzky, T.; McIlwain, D.R.; Issuree, P.D.A.; Li, X.; Malapeira, J.; Amin, S.; Lang, P.A.; Mak, T.W.; Blobel, C.P. iRhom2 controls the substrate selectivity of stimulated ADAM17-dependent ectodomain shedding. Proc. Natl. Acad. Sci. USA 2013, 110, 11433–11438. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Maretzky, T.; Weskamp, G.; Monette, S.; Qing, X.; Issuree, P.D.A.; Crawford, H.C.; McIlwain, D.R.; Mak, T.W.; Salmon, J.E.; et al. iRhoms 1 and 2 are essential upstream regulators of ADAM17-dependent EGFR signaling. Proc. Natl. Acad. Sci. USA 2015, 112, 6080–6085. [Google Scholar] [CrossRef] [Green Version]
- Tang, B.; Li, X.; Maretzky, T.; Perez-Aguilar, J.M.; McIlwain, D.; Xie, Y.; Zheng, Y.; Mak, T.W.; Weinstein, H.; Blobel, C.P. Substrate-selective protein ectodomain shedding by ADAM17 and iRhom2 depends on their juxtamembrane and transmembrane domains. FASEB J. 2020, 34, 4956–4969. [Google Scholar] [CrossRef] [Green Version]
- Le Gall, S.M.; Maretzky, T.; Issuree, P.D.A.; Niu, X.-D.; Reiss, K.; Saftig, P.; Khokha, R.; Lundell, D.; Blobel, C.P. ADAM17 is regulated by a rapid and reversible mechanism that controls access to its catalytic site. J. Cell Sci. 2010, 123, 3913–3922. [Google Scholar] [CrossRef] [Green Version]
- Álvarez-Fernández, S.M.; Barbariga, M.; Cannizzaro, L.; Cannistraci, C.V.; Hurley, L.; Zanardi, A.; Conti, A.; Sanvito, F.; Innocenzi, A.; Pecorelli, N.; et al. Serological immune response against ADAM10 pro-domain is associated with favourable prognosis in stage III colorectal cancer patients. Oncotarget 2016, 7, 80059–80076. [Google Scholar] [CrossRef] [Green Version]
- Wong, E.; Maretzky, T.; Peleg, Y.; Blobel, C.P.; Sagi, I. The Functional Maturation of a Disintegrin and Metalloproteinase (ADAM) 9, 10, and 17 Requires Processing at a Newly Identified Proprotein Convertase (PC) Cleavage Site. J. Biol. Chem. 2015, 290, 12135–12146. [Google Scholar] [CrossRef] [Green Version]
- Wolfsberg, T.G.; Straight, P.D.; Gerena, R.L.; Huovila, A.-P.J.; Primàkoff, P.; Myles, D.G.; White, J.M. ADAM, a Widely Distributed and Developmentally Regulated Gene Family Encoding Membrane Proteins with ADisintegrin and Metalloprotease Domain. Dev. Biol. 1995, 169, 378–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolfsberg, T.G.; PrimakoffJ, P.; Myles, D.G.; White, J.M. ADAM, a novel family of membrane proteins containing A Disintegrin And Metalloprotease domain: Multipotential functions in cell-cell and cell-matrix interactions. J. Cell Biol. 1995, 131, 275–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merilahti, J.A.; Elenius, K. Gamma-secretase-dependent signaling of receptor tyrosine kinases. Oncogene 2019, 38, 151–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCarthy, A.J.; Coleman-Vaughan, C.; McCarthy, J.V. Regulated intramembrane proteolysis: Emergent role in cell signalling pathways. Biochem. Soc. Trans. 2017, 45, 1185–1202. [Google Scholar] [CrossRef] [PubMed]
- Urban, S. A guide to the rhomboid protein superfamily in development and disease. Semin. Cell Dev. Biol. 2016, 60, 1–4. [Google Scholar] [CrossRef]
- Adrain, C.; Strisovsky, K.; Zettl, M.; Hu, L.; Lemberg, M.K.; Freeman, M. Mammalian EGF receptor activation by the rhomboid protease RHBDL2. EMBO Rep. 2011, 12, 421–427. [Google Scholar] [CrossRef] [Green Version]
- Merilahti, J.A.M.; Ojala, V.K.; Knittle, A.M.; Pulliainen, A.T.; Elenius, K. Genome-wide screen of gamma-secretase–mediated intramembrane cleavage of receptor tyrosine kinases. Mol. Biol. Cell 2017, 28, 3123–3131. [Google Scholar] [CrossRef]
- Hartmann, D.; De Strooper, B.; Serneels, L.; Craessaerts, K.; Herreman, A.; Annaert, W.; Umans, L.; Lübke, T.; Illert, A.L.; Von Figura, K.; et al. The disintegrin/metalloprotease ADAM 10 is essential for Notch signalling but not for alpha-secretase activity in fibroblasts. Hum. Mol. Genet. 2002, 11, 2615–2624. [Google Scholar] [CrossRef] [Green Version]
- Qiu, H.; Tang, X.; Ma, J.; Shaverdashvili, K.; Zhang, K.; Bedogni, B. Notch1 Autoactivation via Transcriptional Regulation of Furin, Which Sustains Notch1 Signaling by Processing Notch1-Activating Proteases ADAM10 and Membrane Type 1 Matrix Metalloproteinase. Mol. Cell Biol. 2015, 35, 3622–3632. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Kesti, T.; Uğurlu, H.; Baur, A.S.; Fagerlund, R.; Saksela, K. Tyrosine phosphorylation directs TACE into extracellular vesicles via unconventional secretion. Traffic Cph. Den. 2019, 20, 202–212. [Google Scholar] [CrossRef] [Green Version]
- Singh, B.; Modica-Napolitano, J.S.; Singh, K.K. Defining the momiome: Promiscuous information transfer by mobile mitochondria and the mitochondrial genome. Semin. Cancer Biol. 2017, 47, 1–17. [Google Scholar] [CrossRef]
- Tousseyn, T.; Thathiah, A.; Jorissen, E.; Raemaekers, T.; Konietzko, U.; Reiss, K.; Maes, E.; Snellinx, A.; Serneels, L.; Nyabi, O.; et al. ADAM10, the Rate-limiting Protease of Regulated Intramembrane Proteolysis of Notch and Other Proteins, Is Processed by ADAMS-9, ADAMS-15, and the γ-Secretase. J. Biol. Chem. 2009, 284, 11738–11747. [Google Scholar] [CrossRef] [Green Version]
- Langemeyer, L.; Fröhlich, F.; Ungermann, C. Rab GTPase Function in Endosome and Lysosome Biogenesis. Trends Cell Biol. 2018, 28, 957–970. [Google Scholar] [CrossRef]
- Sun, L.; Li, X.; Shi, Y. Structural biology of intramembrane proteases: Mechanistic insights from rhomboid and S2P to γ-secretase. Curr. Opin. Struct. Biol. 2016, 37, 97–107. [Google Scholar] [CrossRef]
- Burton, J.C.; Grimsey, N.J. Ubiquitination as a Key Regulator of Endosomal Signaling by GPCRs. Front. Cell Dev. Biol. 2019, 7, 43. [Google Scholar] [CrossRef] [Green Version]
- Barbieri, E.; Di Fiore, P.P.; Sigismund, S. Endocytic control of signaling at the plasma membrane. Curr. Opin. Cell Biol. 2016, 39, 21–27. [Google Scholar] [CrossRef] [Green Version]
- Cormier, K.W.; Woodgett, J.R. Recent advances in understanding the cellular roles of GSK-3. F1000 Res. 2017, 6, 167. [Google Scholar] [CrossRef] [Green Version]
- De Robertis, E.M.; Ploper, D. Sperm Motility Requires Wnt/GSK3 Stabilization of Proteins. Dev. Cell 2015, 35, 401–402. [Google Scholar] [CrossRef] [Green Version]
- Stoica, R.; De Vos, K.J.; Paillusson, S.; Mueller, S.; Sancho, R.M.; Lau, K.-F.; Vizcay-Barrena, G.; Lin, W.-L.; Xu, Y.-F.; Lewis, J.; et al. ER–mitochondria associations are regulated by the VAPB–PTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43. Nat. Commun. 2014, 5, 3996. [Google Scholar] [CrossRef] [Green Version]
- Couchie, D.; Vaisman, B.; Abderrazak, A.; Mahmood, D.F.D.; Hamza, M.M.; Canesi, F.; Diderot, V.; El Hadri, K.; Nègre-Salvayre, A.E.; Le Page, A.; et al. Human Plasma Thioredoxin-80 Increases with Age and in ApoE −/− Mice Induces Inflammation, Angiogenesis, and Atherosclerosis. Circulation 2017, 136, 464–475. [Google Scholar] [CrossRef]
- Horvath, G.L.; Schrum, J.E.; De Nardo, C.M.; Latz, E. Intracellular sensing of microbes and danger signals by the inflammasomes. Immunol. Rev. 2011, 243, 119–135. [Google Scholar] [CrossRef]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Inflammaging: Disturbed interplay between autophagy and inflammasomes. Aging 2012, 4, 166–175. [Google Scholar] [CrossRef] [Green Version]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A.; Ojala, J.; Haapasalo, A.; Soininen, H.; Hiltunen, M. Impaired autophagy and APP processing in Alzheimer’s disease: The potential role of Beclin 1 interactome. Prog. Neurobiol. 2013, 106-107, 33–54. [Google Scholar] [CrossRef]
- Le Gall, S.M.; Bobé, P.; Reiss, K.; Horiuchi, K.; Niu, X.-D.; Lundell, D.; Gibb, D.R.; Conrad, D.; Saftig, P.; Blobel, C.P. ADAMs 10 and 17 Represent Differentially Regulated Components of a General Shedding Machinery for Membrane Proteins Such as Transforming Growth Factor α, L-Selectin, and Tumor Necrosis Factor α. Mol. Biol. Cell 2009, 20, 1785–1794. [Google Scholar] [CrossRef] [Green Version]
- Bleibaum, F.; Sommer, A.; Veit, M.; Rabe, B.; Andrä, J.; Kunzelmann, K.; Nehls, C.; Correa, W.; Gutsmann, T.; Grötzinger, J.; et al. ADAM10 sheddase activation is controlled by cell membrane asymmetry. J. Mol. Cell Biol. 2019, 11, 979–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartmann, M.; Parra, L.M.; Ruschel, A.; Lindner, C.; Morrison, H.; Herrlich, A.; Herrlich, P. Inside-out Regulation of Ectodomain Cleavage of Cluster-of-Differentiation-44 (CD44) and of Neuregulin-1 Requires Substrate Dimerization. J. Biol. Chem. 2015, 290, 17041–17054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cochet, M.; Donneger, R.; Cassier, E.; Gaven, F.; Lichtenthaler, S.F.; Marin, P.; Bockaert, J.; Dumuis, A.; Claeysen, S. 5-HT4Receptors Constitutively Promote the Non-Amyloidogenic Pathway of APP Cleavage and Interact with ADAM10. ACS Chem. Neurosci. 2012, 4, 130–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilardaga, J.-P.; Jean-Alphonse, F.G.; Gardella, T.J. Endosomal generation of cAMP in GPCR signaling. Nat. Chem. Biol. 2014, 10, 700–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irannejad, R.; Pessino, V.; Mika, D.; Huang, B.; Wedegaertner, P.B.; Conti, M.; Von Zastrow, M. Functional selectivity of GPCR-directed drug action through location bias. Nat. Chem. Biol. 2017, 13, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Hundhausen, C.; Misztela, D.; Berkhout, T.A.; Broadway, N.; Saftig, P.; Reiss, K.; Hartmann, D.; Fahrenholz, F.; Postina, R.; Matthews, V.; et al. The disintegrin-like metalloproteinase ADAM10 is involved in constitutive cleavage of CX3CL1 (fractalkine) and regulates CX3CL1-mediated cell-cell adhesion. Blood 2003, 102, 1186–1195. [Google Scholar] [CrossRef] [Green Version]
- Hundhausen, C.; Schulte, A.; Schulz, B.; Andrzejewski, M.G.; Schwarz, N.; Von Hundelshausen, P.; Winter, U.; Paliga, K.; Reiss, K.; Saftig, P.; et al. Regulated shedding of transmembrane chemokines by the disintegrin and metalloproteinase 10 facilitates detachment of adherent leukocytes. J. Immunol. 2007, 178, 8064–8072. [Google Scholar] [CrossRef] [Green Version]
- Horion, J.; Gloire, G.; El Mjiyad, N.; Quivy, V.; Vermeulen, L.; Berghe, W.V.; Haegeman, G.; Van Lint, C.; Piette, J.; Habraken, Y. Histone Deacetylase Inhibitor Trichostatin a Sustains Sodium Pervanadate-induced NF-kappaB Activation by Delaying IkappaBalpha mRNA Resynthesis: Comparison with tumor necrosis factor alpha. J. Biol. Chem. 2007, 282, 15383–15393. [Google Scholar] [CrossRef] [Green Version]
- Groth, E.; Pruessmeyer, J.; Babendreyer, A.; Schumacher, J.; Pasqualon, T.; Dreymueller, D.; Higashiyama, S.; Lorenzen, I.; Grötzinger, J.; Cataldo, D.; et al. Stimulated release and functional activity of surface expressed metalloproteinase ADAM17 in exosomes. Biochim. Biophys. Acta 2016, 1863, 2795–2808. [Google Scholar] [CrossRef]
- Gutwein, P.; Mechtersheimer, S.; Riedle, S.; Stoeck, A.; Gast, D.; Joumaa, S.; Zentgraf, H.; Fogel, M.; Altevogt, D.P. ADAM10-mediated cleavage of L1 adhesion molecule at the cell surface and in released membrane vesicles. FASEB J. 2003, 17, 292–294. [Google Scholar] [CrossRef]
- Zimmerman, B.; Kelly, B.; McMillan, B.J.; Seegar, T.C.; Dror, R.O.; Kruse, A.C.; Blacklow, S.C. Crystal Structure of a Full-Length Human Tetraspanin Reveals a Cholesterol-Binding Pocket. Cell 2016, 167, 1041–1051.e11. [Google Scholar] [CrossRef] [Green Version]
- Austin, C.D.; Shields, D. Formation of nascent secretory vesicles from the trans-Golgi network of endocrine cells is inhibited by tyrosine kinase and phosphatase inhibitors. J. Cell Biol. 1996, 135, 1471–1483. [Google Scholar] [CrossRef]
- Kleino, I.; Järviluoma, A.; Hepojoki, J.; Huovila, A.P.; Saksela, K. Preferred SH3 Domain Partners of ADAM Metalloproteases Include Shared and ADAM-Specific SH3 Interactions. PLoS ONE 2015, 10, e0121301. [Google Scholar] [CrossRef] [Green Version]
- Nussinov, R.; Tsai, C.-J.; Ma, B. The Underappreciated Role of Allostery in the Cellular Network. Annu. Rev. Biophys. 2013, 42, 169–189. [Google Scholar] [CrossRef]
- Arduise, C.; Abache, T.; Li, L.; Billard, M.; Chabanon, A.; Ludwig, A.; Mauduit, P.; Boucheix, C.; Rubinstein, E.; Le Naour, F. Tetraspanins Regulate ADAM10-Mediated Cleavage of TNF-α and Epidermal Growth Factor. J. Immunol. 2008, 181, 7002–7013. [Google Scholar] [CrossRef] [Green Version]
- Shiuan, E.; Chen, J. Eph Receptor Tyrosine Kinases in Tumor Immunity. Cancer Res. 2016, 76, 6452–6457. [Google Scholar] [CrossRef] [Green Version]
- Janes, P.W.; Saha, N.; Barton, W.A.; Kolev, M.V.; Wimmer-Kleikamp, S.H.; Nievergall, E.; Blobel, C.P.; Himanen, J.-P.; Lackmann, M.; Nikolov, D.B. Adam Meets Eph: An ADAM Substrate Recognition Module Acts as a Molecular Switch for Ephrin Cleavage In trans. Cell 2005, 123, 291–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solanas, G.; Cortina, C.; Sevillano, M.; Batlle, E. Cleavage of E-cadherin by ADAM10 mediates epithelial cell sorting downstream of EphB signalling. Nat. Cell Biol. 2011, 13, 1100–1107. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.J.; Hwang, Y.-S.; Mood, K.; Cho, H.-J.; Lee, H.-S.; Winterbottom, E.; Cousin, H.; Daar, I.O. EphrinB2 Affects Apical Constriction in Xenopus Embryos and is Regulated by ADAM10 and Flotillin-1. Nat. Commun. 2014, 5, 3516. [Google Scholar] [CrossRef] [Green Version]
- Greene, A.C.; Lord, S.J.; Tian, A.; Rhodes, C.; Kai, H.; Groves, J.T. Spatial Organization of EphA2 at the Cell-Cell Interface Modulates Trans-Endocytosis of EphrinA1. Biophys. J. 2014, 106, 2196–2205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.; Wang, Y.; Forbes, K.; Vignali, K.M.; Heale, B.S.; Saftig, P.; Hartmann, D.; A Black, R.; Rossi, J.J.; Blobel, C.P.; et al. Metalloproteases regulate T-cell proliferation and effector function via LAG-3. EMBO J. 2007, 26, 494–504. [Google Scholar] [CrossRef] [Green Version]
- Uche, U.U.; Piccirillo, A.R.; Kataoka, S.; Grebinoski, S.J.; D’Cruz, L.M.; Kane, L.P. PIK3IP1/TrIP restricts activation of T cells through inhibition of PI3K/Akt. J. Exp. Med. 2018, 215, 3165–3179. [Google Scholar] [CrossRef] [Green Version]
- Jia, H.P.; Look, D.C.; Tan, P.; Shi, L.; Hickey, M.; Gakhar, L.; Chappell, M.C.; Wohlford-Lenane, C.; McCray, P.B. Ectodomain shedding of angiotensin converting enzyme 2 in human airway epithelia. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 297, L84–L96. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Sriramula, S.; Xia, H.; Moreno-Walton, L.; Culicchia, F.; Domenig, O.; Poglitsch, M.; Lazartigues, E. Clinical Relevance and Role of Neuronal AT1 Receptors in ADAM17-Mediated ACE2 Shedding in Neurogenic Hypertension. Circ. Res. 2017, 121, 43–55. [Google Scholar] [CrossRef]
- Shah, J.; Rouaud, F.; Guerrera, D.; Vasileva, E.; Popov, L.M.; Kelley, W.L.; Rubinstein, E.; Carette, J.E.; Amieva, M.R.; Citi, S. A Dock-and-Lock Mechanism Clusters ADAM10 at Cell-Cell Junctions to Promote α-Toxin Cytotoxicity. Cell Rep. 2018, 25, 2132–2147.e7. [Google Scholar] [CrossRef] [Green Version]
- Britton, G.J.; Ambler, R.; Clark, D.J.; Hill, E.V.; Tunbridge, H.M.; E McNally, K.; Burton, B.R.; Butterweck, P.; Sabatos-Peyton, C.; A Hampton-O’Neil, L.; et al. PKCθ links proximal T cell and Notch signaling through localized regulation of the actin cytoskeleton. eLife 2017, 6, 2896. [Google Scholar] [CrossRef] [Green Version]
- Defourny, J.; Peuckert, C.; Kullander, K.; Malgrange, B. EphA4-ADAM10 Interplay Patterns the Cochlear Sensory Epithelium through Local Disruption of Adherens Junctions. iScience 2019, 11, 246–257. [Google Scholar] [CrossRef] [Green Version]
- Millichip, M.I.; Dallas, D.J.; Wu, E.; Dale, S.; McKie, N. The Metallo-Disintegrin ADAM10 (MADM) from Bovine Kidney Has Type IV Collagenase Activity in Vitro. Biochem. Biophys. Res. Commun. 1998, 245, 594–598. [Google Scholar] [CrossRef]
- Grötzinger, J.; Lorenzen, I.; Düsterhöft, S. Molecular insights into the multilayered regulation of ADAM17: The role of the extracellular region. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 2088–2095. [Google Scholar] [CrossRef]
- Stoeck, A.; Keller, S.; Riedle, S.; Sanderson, M.P.; Runz, S.; Le Naour, F.; Gutwein, P.; Ludwig, A.; Rubinstein, E.; Altevogt, P. A role for exosomes in the constitutive and stimulus-induced ectodomain cleavage of L1 and CD44. Biochem. J. 2006, 393, 609–618. [Google Scholar] [CrossRef]
- Andrzejewski, M.G.; Koelsch, A.; Kögel, T.; Dreymueller, D.; Schwarz, N.; Ludwig, A. Distinct role of the intracellular C-terminus for subcellular expression, shedding and function of the murine transmembrane chemokine CX3CL1. Biochem. Biophys. Res. Commun. 2010, 395, 178–184. [Google Scholar] [CrossRef]
- Mathews, J.A.; Gibb, D.R.; Chen, B.-H.; Scherle, P.; Conrad, D.H. CD23 Sheddase A Disintegrin and Metalloproteinase 10 (ADAM10) Is Also Required for CD23 Sorting into B Cell-derived Exosomes. J. Biol. Chem. 2010, 285, 37531–37541. [Google Scholar] [CrossRef] [Green Version]
- Gibb, D.R.; Saleem, S.J.; Chaimowitz, N.S.; Mathews, J.; Conrad, D.H. The emergence of ADAM10 as a regulator of lymphocyte development and autoimmunity. Mol. Immunol. 2011, 48, 1319–1327. [Google Scholar] [CrossRef] [Green Version]
- Evans, S.F.; Irmady, K.; Ostrow, K.; Kim, T.; Nykjaer, A.; Saftig, P.; Blobel, C.; Hempstead, B.L. Neuronal Brain-derived Neurotrophic Factor Is Synthesized in Excess, with Levels Regulated by Sortilin-mediated Trafficking and Lysosomal Degradation. J. Biol. Chem. 2011, 286, 29556–29567. [Google Scholar] [CrossRef] [Green Version]
- Ebsen, H.; Lettau, M.; Kabelitz, D.; Janssen, O. Subcellular localization and activation of ADAM proteases in the context of FasL shedding in T lymphocytes. Mol. Immunol. 2015, 65, 416–428. [Google Scholar] [CrossRef]
- Lee, J.; Dieckmann, N.M.; Edgar, J.R.; Griffiths, G.M.; Siegel, R.M. Fas Ligand localizes to intraluminal vesicles within NK cell cytolytic granules and is enriched at the immune synapse. Immun. Inflamm. Dis. 2018, 6, 312–321. [Google Scholar] [CrossRef]
- Kowal, J.; Arras, G.; Colombo, M.; Jouve, M.; Morath, J.P.; Primdal-Bengtson, B.; Dingli, F.; Loew, D.; Tkach, M.; Théry, C. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl. Acad. Sci. USA 2016, 113, E968–E977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalra, H.; Drummen, G.P.C.; Mathivanan, S. Focus on Extracellular Vesicles: Introducing the Next Small Big Thing. Int. J. Mol. Sci. 2016, 17, 170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chastagner, P.; Rubinstein, E.; Brou, C. Ligand-activated Notch undergoes DTX4-mediated ubiquitylation and bilateral endocytosis before ADAM10 processing. Sci. Signal. 2017, 10, eaag2989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinbuck, M.P.; Arakcheeva, K.; Winandy, S. Novel TCR-Mediated Mechanisms of Notch Activation and Signaling. J. Immunol. 2018, 200, 997–1007. [Google Scholar] [CrossRef] [Green Version]
- Tosetti, F.; Venè, R.; Camodeca, C.; Nuti, E.; Rossello, A.; D’Arrigo, C.; Galante, D.; Ferrari, N.; Poggi, A.; Zocchi, M.R. Specific ADAM10 inhibitors localize in exosome-like vesicles released by Hodgkin lymphoma and stromal cells and prevent sheddase activity carried to bystander cells. Oncoimmunology 2018, 7, e1421889. [Google Scholar] [CrossRef] [Green Version]
- Linford, A.; Yoshimura, S.-I.; Bastos, R.N.; Langemeyer, L.; Gerondopoulos, A.; Rigden, D.J.; Barr, F.A. Rab14 and Its Exchange Factor FAM116 Link Endocytic Recycling and Adherens Junction Stability in Migrating Cells. Dev. Cell 2012, 22, 952–966. [Google Scholar] [CrossRef] [Green Version]
- Ashiru, O.; Boutet, P.; Fernández-Messina, L.; Agüera-González, S.; Skepper, J.N.; Valés-Gómez, M.; Reyburn, H.T. Natural Killer Cell Cytotoxicity Is Suppressed by Exposure to the Human NKG2D Ligand MICA*008 That Is Shed by Tumor Cells in Exosomes. Cancer Res. 2010, 70, 481–489. [Google Scholar] [CrossRef] [Green Version]
- Hansen, H.P.; Engels, H.-M.; Dams, M.; Leme, A.F.P.; Pauletti, B.A.; Simhadri, V.L.; Dürkop, H.; Reiners, K.S.; Barnert, S.; Engert, A.; et al. Protrusion-guided extracellular vesicles mediate CD30 trans-signalling in the microenvironment of Hodgkin’s lymphoma. J. Pathol. 2014, 232, 405–414. [Google Scholar] [CrossRef]
- Ebsen, H.; Lettau, M.; Kabelitz, D.; Janssen, O. Identification of SH3 Domain Proteins Interacting with the Cytoplasmic Tail of the A Disintegrin and Metalloprotease 10 (ADAM10). PLoS ONE 2014, 9, e102899. [Google Scholar] [CrossRef] [Green Version]
- Marcello, E.; Gardoni, F.; Mauceri, D.; Romorini, S.; Jeromin, A.; Epis, R.; Borroni, B.; Cattabeni, F.; Sala, C.; Padovani, A.; et al. Synapse-Associated Protein-97 Mediates α-Secretase ADAM10 Trafficking and Promotes Its Activity. J. Neurosci. 2007, 27, 1682–1691. [Google Scholar] [CrossRef] [Green Version]
- Wunderle, L.; Knopf, J.D.; Kühnle, N.; Morlé, A.; Hehn, B.; Adrain, C.; Strisovsky, K.; Freeman, M.; Lemberg, M.K. Rhomboid intramembrane protease RHBDL4 triggers ER-export and non-canonical secretion of membrane-anchored TGFα. Sci. Rep. 2016, 6, 27342. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.; Shirakabe, K.; Werb, Z. The metalloprotease Kuzbanian (ADAM10) mediates the transactivation of EGF receptor by G protein–coupled receptors. J. Cell Biol. 2002, 158, 221–226. [Google Scholar] [CrossRef] [Green Version]
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schiöth, H.B.; Gloriam, D.E. Trends in GPCR drug discovery: New agents, targets and indications. Nat. Rev. Drug Discov. 2017, 16, 829–842. [Google Scholar] [CrossRef]
- Perez-Hernandez, D.; Gutiérrez-Vázquez, C.; Jorge, I.; López-Martín, S.; Ursa, A.; Sánchez-Madrid, F.; Vázquez, J.; Yáñez-Mó, M. The Intracellular Interactome of Tetraspanin-enriched Microdomains Reveals Their Function as Sorting Machineries toward Exosomes. J. Biol. Chem. 2013, 288, 11649–11661. [Google Scholar] [CrossRef] [Green Version]
- Ghadially, H.; Brown, L.; Lloyd, C.; Lewis, L.; Lewis, A.; Dillon, J.; Sainson, R.; Jovanovic, J.; Tigue, N.J.; Bannister, D.; et al. MHC class I chain-related protein A and B (MICA and MICB) are predominantly expressed intracellularly in tumour and normal tissue. Br. J. Cancer 2017, 116, 1208–1217. [Google Scholar] [CrossRef] [Green Version]
- Zocchi, M.R.; Catellani, S.; Canevali, P.; Tavella, S.; Garuti, A.; Villaggio, B.; Zunino, A.; Gobbi, M.; Fraternali-Orcioni, G.; Kunkl, A.; et al. High ERp5/ADAM10 expression in lymph node microenvironment and impaired NKG2D ligands recognition in Hodgkin lymphomas. Blood 2012, 119, 1479–1489. [Google Scholar] [CrossRef]
- Hansen, H.P.; Trad, A.; Dams, M.; Zigrino, P.; Moss, M.; Tator, M.; Schön, G.; Grenzi, P.C.; Bachurski, D.; Aquino, B.; et al. CD30 on extracellular vesicles from malignant Hodgkin cells supports damaging of CD30 ligand-expressing bystander cells with Brentuximab-Vedotin, in vitro. Oncotarget 2016, 7, 30523–30535. [Google Scholar] [CrossRef] [Green Version]
- Cvjetkovic, A.; Jang, S.C.; Konečná, B.; Höög, J.L.; Sihlbom, C.; Lässer, C.; Lötvall, J. Detailed Analysis of Protein Topology of Extracellular Vesicles–Evidence of Unconventional Membrane Protein Orientation. Sci. Rep. 2016, 6, 36338. [Google Scholar] [CrossRef] [Green Version]
- Piffoux, M.; Nicolás-Boluda, A.; Mulens-Arias, V.; Richard, S.; Rahmi, G.; Gazeau, F.; Wilhelm, C.; Silva, A.K. Extracellular vesicles for personalized medicine: The input of physically triggered production, loading and theranostic properties. Adv. Drug Deliv. Rev. 2019, 138, 247–258. [Google Scholar] [CrossRef]
- Kedersha, N.; Ivanov, P.; Anderson, P. Stress granules and cell signaling: More than just a passing phase? Trends Biochem. Sci. 2013, 38, 494–506. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| ADAM10 Target | Site | ADAM10/17 Interacting Proteins/Small Signalling-Accessory Molecules | Docking/Locking/Motor Proteins | Ref. |
|---|---|---|---|---|
| Notch | cell membrane endocytic/trans-endocytotic vesicles | E3 ubiquitin ligase DTX4, dynamin | dynamin, actin, epsin | [103] |
| cell membrane endocytic vesicles | γ-secretase, MT1-MMP, furin | [49] | ||
| endocytic vesicles | TCR, DAG-PKC | [104] | ||
| FasL | lysosome, lipid rafts | TCR, Src | [99] | |
| TNFα | tetraspanin web | MEK/ERK | Tspan (CD81,CD9) | [79] |
| CD23 | endosomes | Ca2+ | clathrin | [96] |
| CD44 | endosomes, exosomes | homodimerization, Ca2+ | [48,94,97] | |
| L1 | endosomes exosomes | Ca++, cholesterol depletion, pervanadate, phorbol esters, Src | [74,94,97] | |
| E-cadherin | adherens junctions | EphA4, EphB2, EphB, GPI-anchored EphB1 | [82,91,106] | |
| S.aureusα-toxin | adherens junctions | PLEKHA7 | Tspan33, PDZD11, afadin | [85] |
| EphA1 | trans-endocytic vesicles | EphA2 | clathrin, dynamin, | [84] |
| EphA5 | cell-cell interface | EphA3 (in trans), RTK | [81] | |
| sortilin | endosomes | BDNF, PKC, calmodulin, Ca2+ | [98] | |
| APP | membrane endosomes | γ-secretase presenilin | Tspan3 | [29] |
| MBP, collagen IV | membrane, microsomes | furin (209RKKR cleavage) | [92] | |
| CD30, MICA008/ULPBs | exosomes | [105,107,108] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tosetti, F.; Alessio, M.; Poggi, A.; Zocchi, M.R. ADAM10 Site-Dependent Biology: Keeping Control of a Pervasive Protease. Int. J. Mol. Sci. 2021, 22, 4969. https://doi.org/10.3390/ijms22094969
Tosetti F, Alessio M, Poggi A, Zocchi MR. ADAM10 Site-Dependent Biology: Keeping Control of a Pervasive Protease. International Journal of Molecular Sciences. 2021; 22(9):4969. https://doi.org/10.3390/ijms22094969
Chicago/Turabian StyleTosetti, Francesca, Massimo Alessio, Alessandro Poggi, and Maria Raffaella Zocchi. 2021. "ADAM10 Site-Dependent Biology: Keeping Control of a Pervasive Protease" International Journal of Molecular Sciences 22, no. 9: 4969. https://doi.org/10.3390/ijms22094969
APA StyleTosetti, F., Alessio, M., Poggi, A., & Zocchi, M. R. (2021). ADAM10 Site-Dependent Biology: Keeping Control of a Pervasive Protease. International Journal of Molecular Sciences, 22(9), 4969. https://doi.org/10.3390/ijms22094969