
Novel KCND3 Variant Underlying Nonprogressive Congenital Ataxia or SCA19/22 Disrupt KV4.3 Protein Expression and K+ Currents with Variable Effects on Channel Properties

 , , , , , ,
, , , , , ,
Abstract
:1. Introduction
2. Results
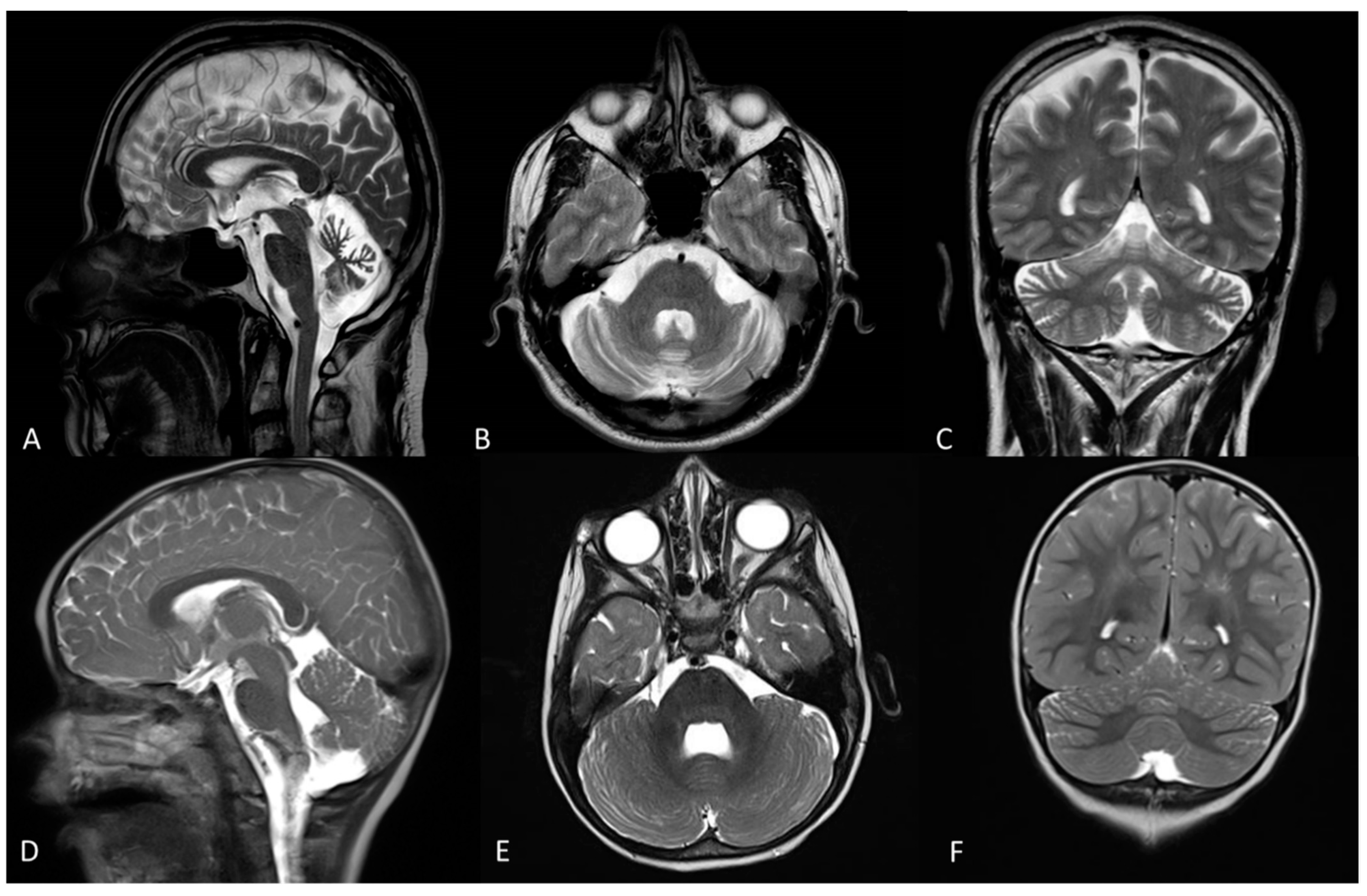
2.1. Clinical Description
2.2. Genetic Data
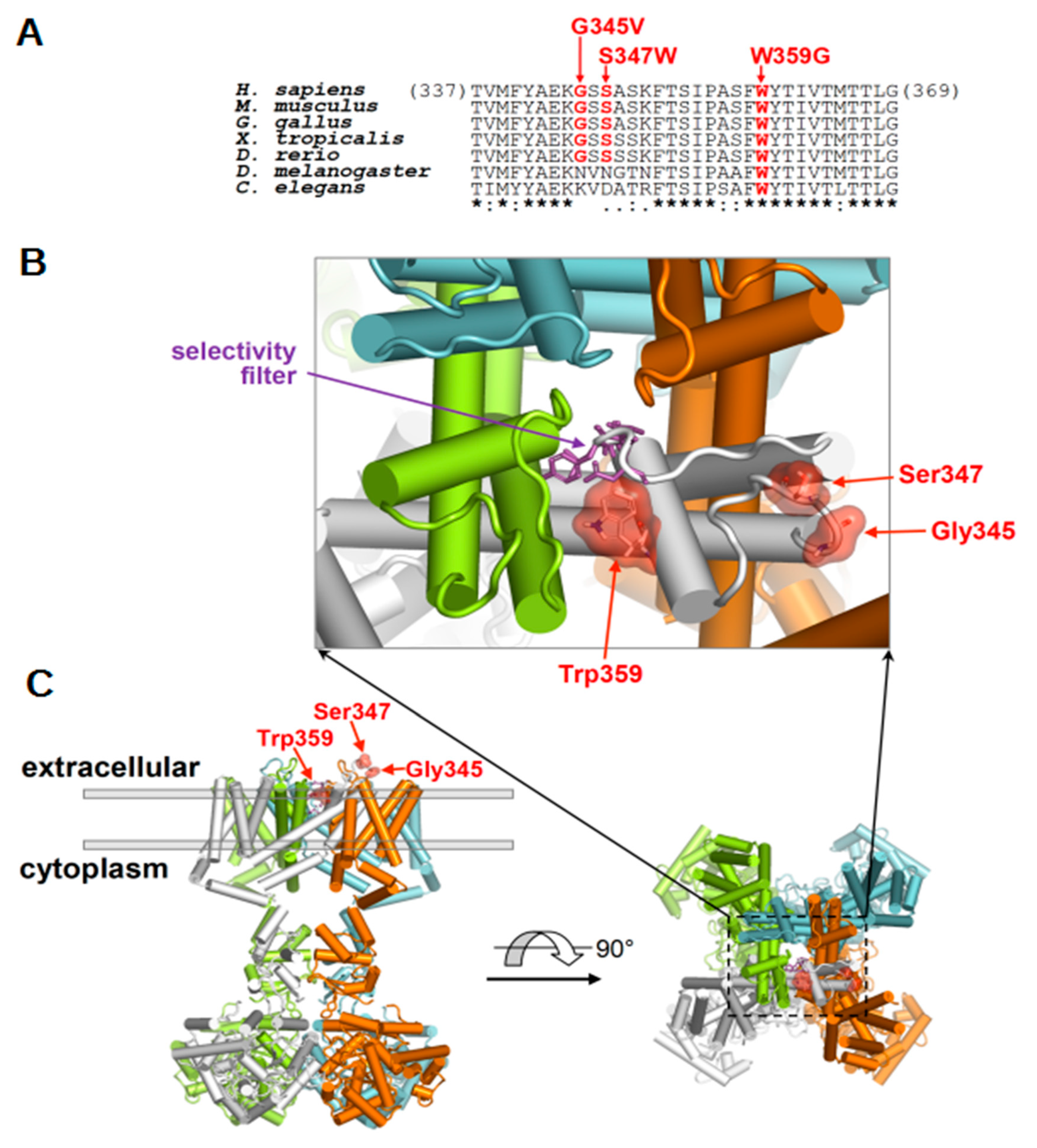
2.3. Molecular Localization of Disease-Associated KV4.3 Variants
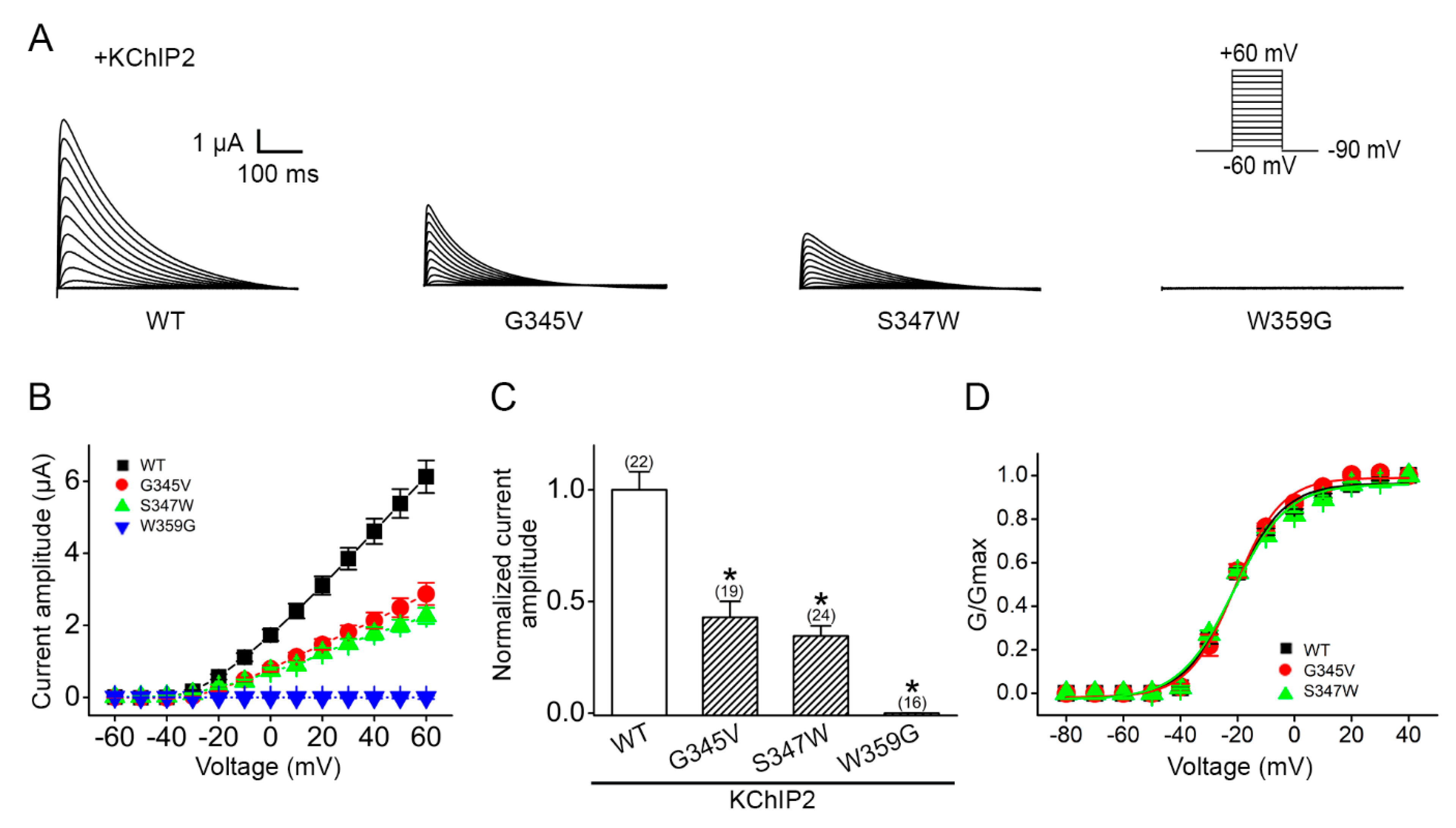
2.4. Altered Voltage Dependent Current Amplitudes of Disease-Associated KV4.3 Mutants
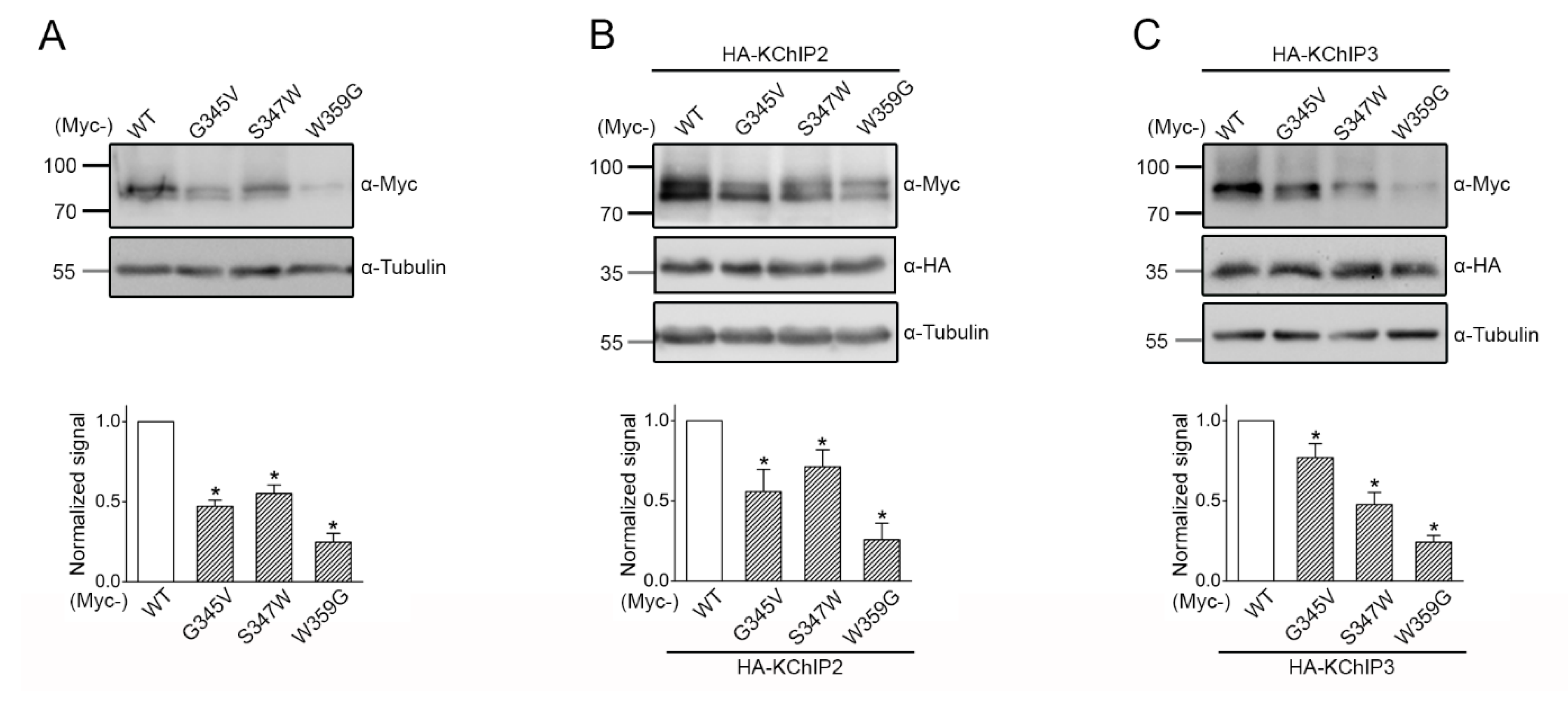
2.5. Reduced Protein Expression of the Disease-Associated KV4.3 Mutants
3. Discussion
4. Patients and Methods
4.1. Genetic Testing
4.2. Protein Homology Modeling
4.3. Expression Plasmids
4.4. Electrophysiology
4.5. Cell Culture and Transfection
4.6. Immunoblotting
4.7. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Duarri, A.; Jezierska, J.; Fokkens, M.; Meijer, M.; Schelhaas, H.J.; den Dunnen, W.F.; van Dijk, F.; Verschuuren-Bemelmans, C.; Hageman, G.; van de Vlies, P.; et al. Mutations in potassium channel KCND3 cause spinocerebellar ataxia type 19. Ann. Neurol. 2012, 72, 870–880. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.C.; Durr, A.; Majczenko, K.; Huang, Y.H.; Liu, Y.C.; Lien, C.C.; Tsai, P.C.; Ichikawa, Y.; Goto, J.; Monin, M.L.; et al. Mutations in KCND3 cause spinocerebellar ataxia type 22. Ann. Neurol. 2012, 72, 859–869. [Google Scholar] [CrossRef]
- Smets, K.; Duarri, A.; Deconinck, T.; Ceulemans, B.; van de Warrenburg, B.P.; Zuchner, S.; Gonzalez, M.A.; Schule, R.; Synofzik, M.; Van der Aa, N.; et al. First de novo KCND3 mutation causes severe Kv4.3 channel dysfunction leading to early onset cerebellar ataxia, intellectual disability, oral apraxia and epilepsy. BMC Med. Genet. 2015, 16, 51. [Google Scholar] [CrossRef] [PubMed]
- Carrasco-Marina, M.L.; Quijada-Fraile, P.; Fernandez-Marmiesse, A.; Gutierrez-Cruz, N.; Martin-Del Valle, F. De novo sporadic mutation in the KCND3 gene in a patient with early onset chronic ataxia. Rev. Neurol. 2019, 68, 398–399. [Google Scholar] [PubMed]
- Serodio, P.; Vega-Saenz De Miera, E.; Rudy, B. Cloning of a novel component of A-type K+ channels operating at subthreshold potentials with unique expression in heart and brain. J. Neurophysiol. 1996, 75, 2174–2179. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.H.; Huang, H.Y.; Tsaur, M.L. Contrasting expression of Kv4.3, an A-type K+ channel, in migrating Purkinje cells and other post-migratory cerebellar neurons. Eur. J. Neurosci. 2003, 18, 601–612. [Google Scholar] [CrossRef]
- Li-Smerin, Y.; Hackos, D.H.; Swartz, K.J. Alpha-helical structural elements within the voltage-sensing domains of a K+ channel. J. Gen. Physiol. 2000, 115, 33–50. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yan, Y.; Liu, Q.; Huang, Y.; Shen, Y.; Chen, L.; Chen, Y.; Yang, Q.; Hao, Q.; Wang, K.; et al. Structural basis for modulation of Kv4 K+ channels by auxiliary KChIP subunits. Nat. Neurosci. 2007, 10, 32–39. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Takimoto, K.; Yang, E.K.; Conforti, L. Palmitoylation of KChIP splicing variants is required for efficient cell surface expression of Kv4.3 channels. J. Biol. Chem. 2002, 277, 26904–26911. [Google Scholar] [CrossRef] [PubMed]
- An, W.F.; Bowlby, M.R.; Betty, M.; Cao, J.; Ling, H.P.; Mendoza, G.; Hinson, J.W.; Mattsson, K.I.; Strassle, B.W.; Trimmer, J.S.; et al. Modulation of A-type potassium channels by a family of calcium sensors. Nature 2000, 403, 553–556. [Google Scholar] [CrossRef] [PubMed]
- Jerng, H.H.; Pfaffinger, P.J. Modulatory mechanisms and multiple functions of somatodendritic A-type K (+) channel auxiliary subunits. Front. Cell. Neurosci. 2014, 8, 82. [Google Scholar] [CrossRef] [PubMed]
- Klockgether, T.; Mariotti, C.; Paulson, H.L. Spinocerebellar ataxia. Nat. Rev. Dis. Prim. 2019, 5, 24. [Google Scholar] [CrossRef] [PubMed]
- Bertini, E.; Zanni, G.; Boltshauser, E. Nonprogressive congenital ataxias. Handb. Clin. Neurol. 2018, 155, 91–103. [Google Scholar] [PubMed]
- Pollini, L.; Galosi, S.; Tolve, M.; Caputi, C.; Carducci, C.; Angeloni, A.; Leuzzi, V. KCND3-Related Neurological Disorders: From Old to Emerging Clinical Phenotypes. Int. J. Mol. Sci. 2020, 21, 5802. [Google Scholar] [CrossRef]
- Hoffman, D.A.; Magee, J.C.; Colbert, C.M.; Johnston, D. K+ channel regulation of signal propagation in dendrites of hippocampal pyramidal neurons. Nature 1997, 387, 869–875. [Google Scholar] [CrossRef]
- Nadal, M.S.; Amarillo, Y.; Vega-Saenz de Miera, E.; Rudy, B. Evidence for the presence of a novel Kv4-mediated A-type K+ channel-modifying factor. J. Physiol. 2001, 537 Pt 3, 801–809. [Google Scholar] [CrossRef]
- Hsiao, C.T.; Fu, S.J.; Liu, Y.T.; Lu, Y.H.; Zhong, C.Y.; Tang, C.Y.; Soong, B.W.; Jeng, C.J. Novel SCA19/22-associated KCND3 mutations disrupt human K(V) 4.3 protein biosynthesis and channel gating. Hum. Mutat. 2019, 40, 2088–2107. [Google Scholar] [CrossRef] [PubMed]
- Huin, V.; Strubi-Vuillaume, I.; Dujardin, K.; Brion, M.; Delliaux, M.; Dellacherie, D.; Cuvellier, J.C.; Cuisset, J.M.; Riquet, A.; Moreau, C.; et al. Expanding the phenotype of SCA19/22: Parkinsonism, cognitive impairment and epilepsy. Parkinsonism Relat. Disord. 2017, 45, 85–89. [Google Scholar] [CrossRef]
- Kurihara, M.; Ishiura, H.; Sasaki, T.; Otsuka, J.; Hayashi, T.; Terao, Y.; Matsukawa, T.; Mitsui, J.; Kaneko, J.; Nishiyama, K.; et al. Novel De Novo KCND3 Mutation in a Japanese Patient with Intellectual Disability, Cerebellar Ataxia, Myoclonus, and Dystonia. Cerebellum 2018, 17, 237–242. [Google Scholar] [CrossRef]
- Paucar, M.; Ågren, R.; Li, T.; Lissmats, S.; Bergendal, Å.; Weinberg, J.; Nilsson, D.; Savichetva, I.; Sahlholm, K.; Nilsson, J.; et al. V374A KCND3 Pathogenic Variant Associated with Paroxysmal Ataxia Exacerbations. Neurol. Genet. 2021, 7, e546. [Google Scholar] [CrossRef] [PubMed]
- Duarri, A.; Nibbeling, E.; Fokkens, M.R.; Meijer, M.; Boddeke, E.; Lagrange, E.; Stevanin, G.; Brice, A.; Durr, A.; Verbeek, D.S. The L450F [Corrected] mutation in KCND3 brings spinocerebellar ataxia and Brugada syndrome closer together. Neurogenetics 2013, 14, 257–258. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.D.; Kim, J.S.; Kim, H.J.; Jung, I.; Jeong, S.H.; Lee, S.H.; Kim, D.U.; Kim, S.H.; Choi, S.Y.; Shin, J.H.; et al. Genetic Variants Associated with Episodic Ataxia in Korea. Sci. Rep. 2017, 7, 13855. [Google Scholar] [CrossRef] [PubMed]
- Coutelier, M.; Coarelli, G.; Monin, M.L.; Konop, J.; Davoine, C.S.; Tesson, C.; Valter, R.; Anheim, M.; Behin, A.; Castelnovo, G.; et al. A panel study on patients with dominant cerebellar ataxia highlights the frequency of channelopathies. Brain A J. Neurol. 2017, 140, 1579–1594. [Google Scholar] [CrossRef]
- Coutelier, M.; Hammer, M.B.; Stevanin, G.; Monin, M.L.; Davoine, C.S.; Mochel, F.; Labauge, P.; Ewenczyk, C.; Ding, J.; Gibbs, J.R.; et al. Efficacy of Exome-Targeted Capture Sequencing to Detect Mutations in Known Cerebellar Ataxia Genes. JAMA Neurol. 2018, 75, 591–599. [Google Scholar] [CrossRef]
- Hsieh, J.Y.; Ulrich, B.N.; Issa, F.A.; Lin, M.A.; Brown BPapazian, D.M. Infant and adult SCA13 mutations differentially affect Purkinje cell excitability, maturation, and viability in vivo. eLife 2020, 9, e573. [Google Scholar] [CrossRef] [PubMed]
- Krivov, G.G.; Shapovalov, M.V.; Dunbrack, R.L., Jr. Improved prediction of protein side-chain conformations with SCWRL4. Proteins 2009, 77, 778–795. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Family C (Lee et al. 2012) | 1 | 2 |
|---|---|---|---|
| Genetic variant | c.1034G > T | c.1040C > G | c.1075T > G |
| Protein | p.G345V | p.S347W | p.W359G |
| Inheritance | AD | n.d | de novo |
| Incomplete penetrance | Yes | - | - |
| Onset | 35–55 y | 45 y | 1.5 y |
| First symptom | Gait disorder | Gait disorder | Hypotonia, DD |
| Ataxia | Yes | Yes | Yes |
| Nystagmus | No | No | No |
| Dysarthria | Yes | Yes | Yes |
| Saccadic pursuit | Yes (1/3) | No | Yes |
| Cognitive delay | No | No | Yes |
| Developmental delay | No | No | Yes |
| Paroxysmal features | No | No | No |
| Movement disorders | No | No | No |
| Pyramidal signs | Yes (1/3) | No | No |
| Seizures Clinical course Brain MRI | No Slowly progressive CA | No Slowly progressive CA | No stable mild CA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zanni, G.; Hsiao, C.-T.; Fu, S.-J.; Tang, C.-Y.; Capuano, A.; Bosco, L.; Graziola, F.; Bellacchio, E.; Servidei, S.; Primiano, G.; et al. Novel KCND3 Variant Underlying Nonprogressive Congenital Ataxia or SCA19/22 Disrupt KV4.3 Protein Expression and K+ Currents with Variable Effects on Channel Properties. Int. J. Mol. Sci. 2021, 22, 4986. https://doi.org/10.3390/ijms22094986
Zanni G, Hsiao C-T, Fu S-J, Tang C-Y, Capuano A, Bosco L, Graziola F, Bellacchio E, Servidei S, Primiano G, et al. Novel KCND3 Variant Underlying Nonprogressive Congenital Ataxia or SCA19/22 Disrupt KV4.3 Protein Expression and K+ Currents with Variable Effects on Channel Properties. International Journal of Molecular Sciences. 2021; 22(9):4986. https://doi.org/10.3390/ijms22094986
Chicago/Turabian StyleZanni, Ginevra, Cheng-Tsung Hsiao, Ssu-Ju Fu, Chih-Yung Tang, Alessandro Capuano, Luca Bosco, Federica Graziola, Emanuele Bellacchio, Serenella Servidei, Guido Primiano, and et al. 2021. "Novel KCND3 Variant Underlying Nonprogressive Congenital Ataxia or SCA19/22 Disrupt KV4.3 Protein Expression and K+ Currents with Variable Effects on Channel Properties" International Journal of Molecular Sciences 22, no. 9: 4986. https://doi.org/10.3390/ijms22094986
APA StyleZanni, G., Hsiao, C. -T., Fu, S. -J., Tang, C. -Y., Capuano, A., Bosco, L., Graziola, F., Bellacchio, E., Servidei, S., Primiano, G., Soong, B. -W., & Jeng, C. -J. (2021). Novel KCND3 Variant Underlying Nonprogressive Congenital Ataxia or SCA19/22 Disrupt KV4.3 Protein Expression and K+ Currents with Variable Effects on Channel Properties. International Journal of Molecular Sciences, 22(9), 4986. https://doi.org/10.3390/ijms22094986