
Inducing Energetic Switching Using Klotho Improves Vascular Smooth Muscle Cell Phenotype


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
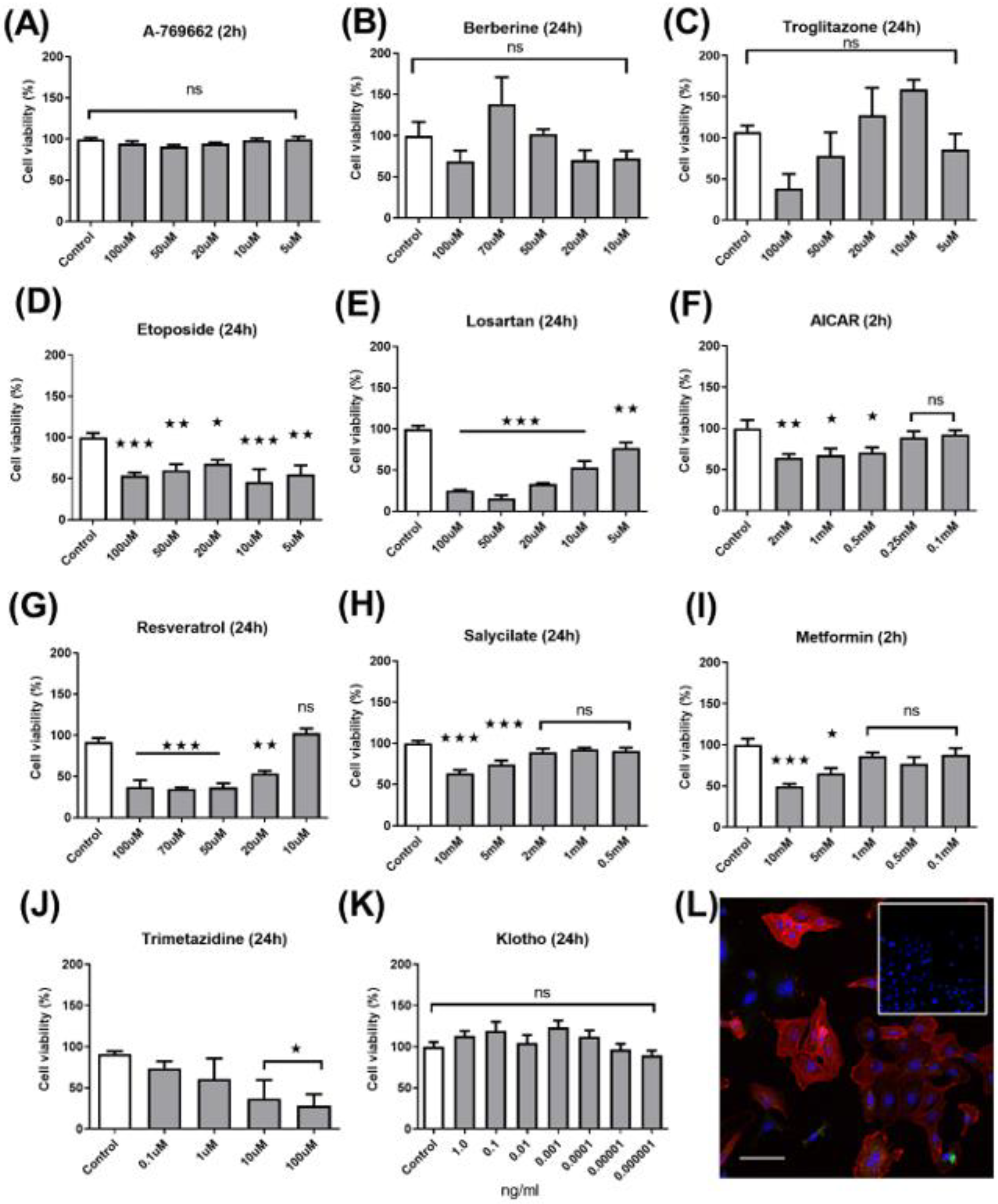
2.1. Dose–Response Cytotoxicity Assay
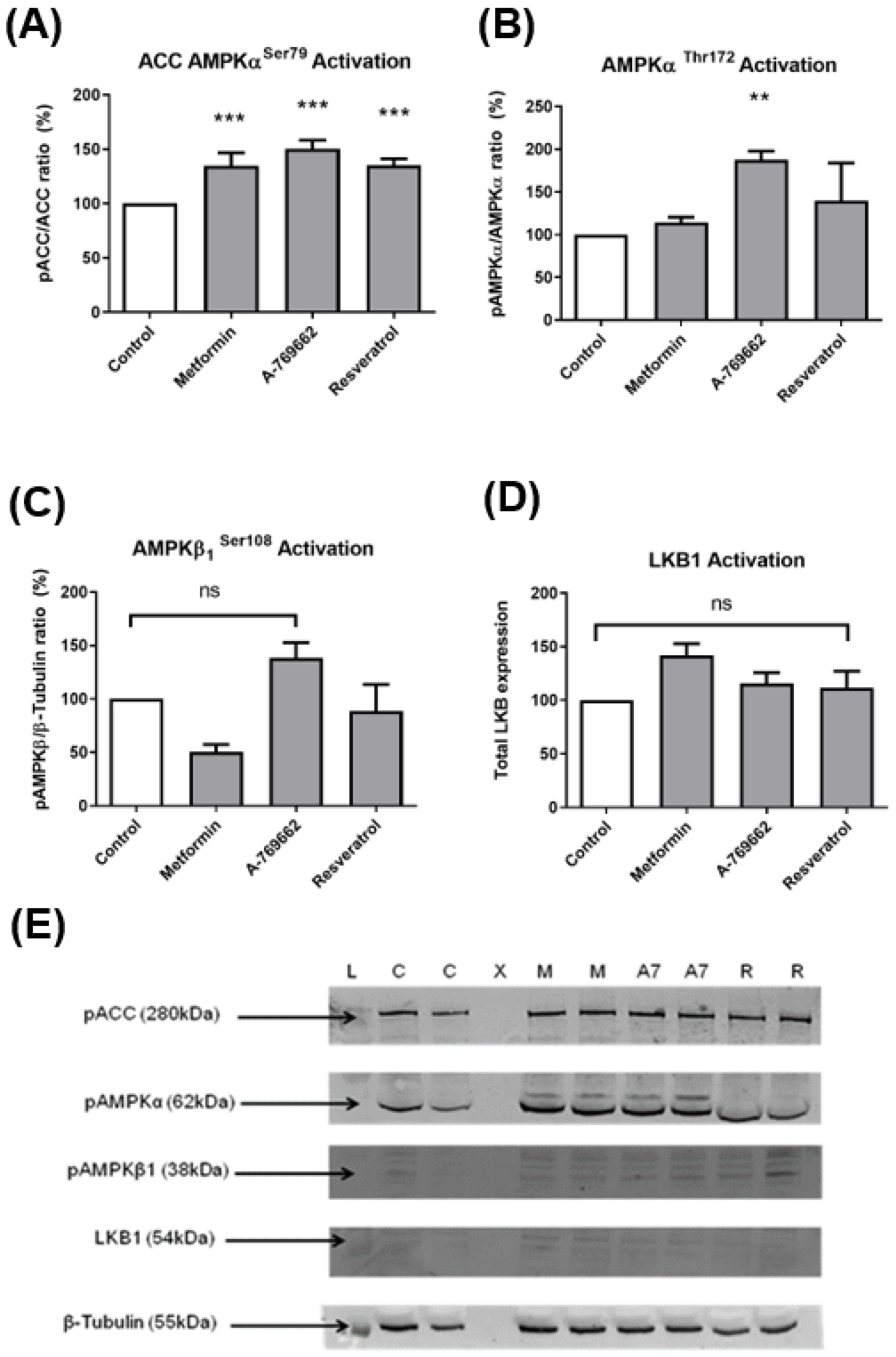
2.2. Protein Western Blot Analysis
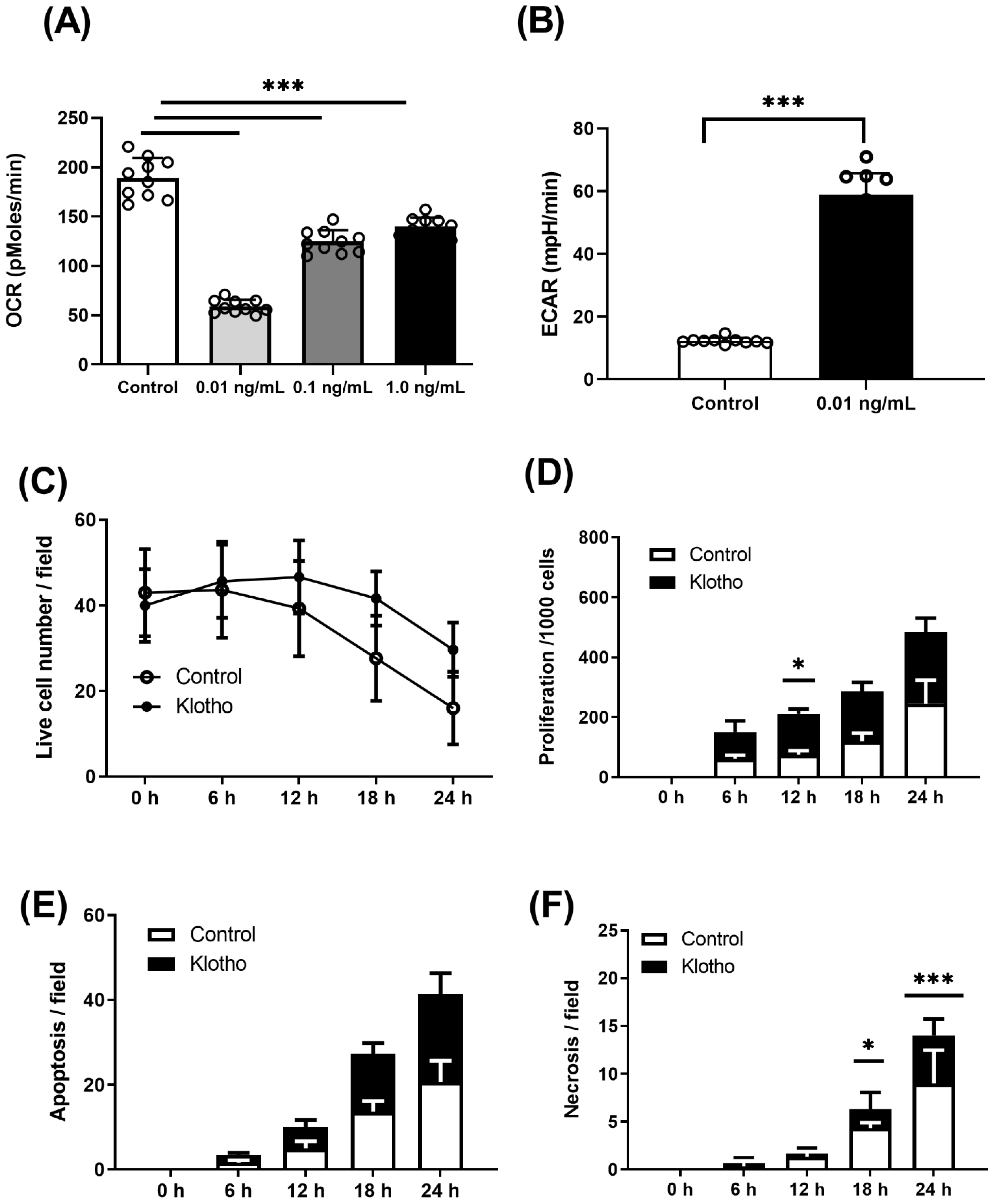
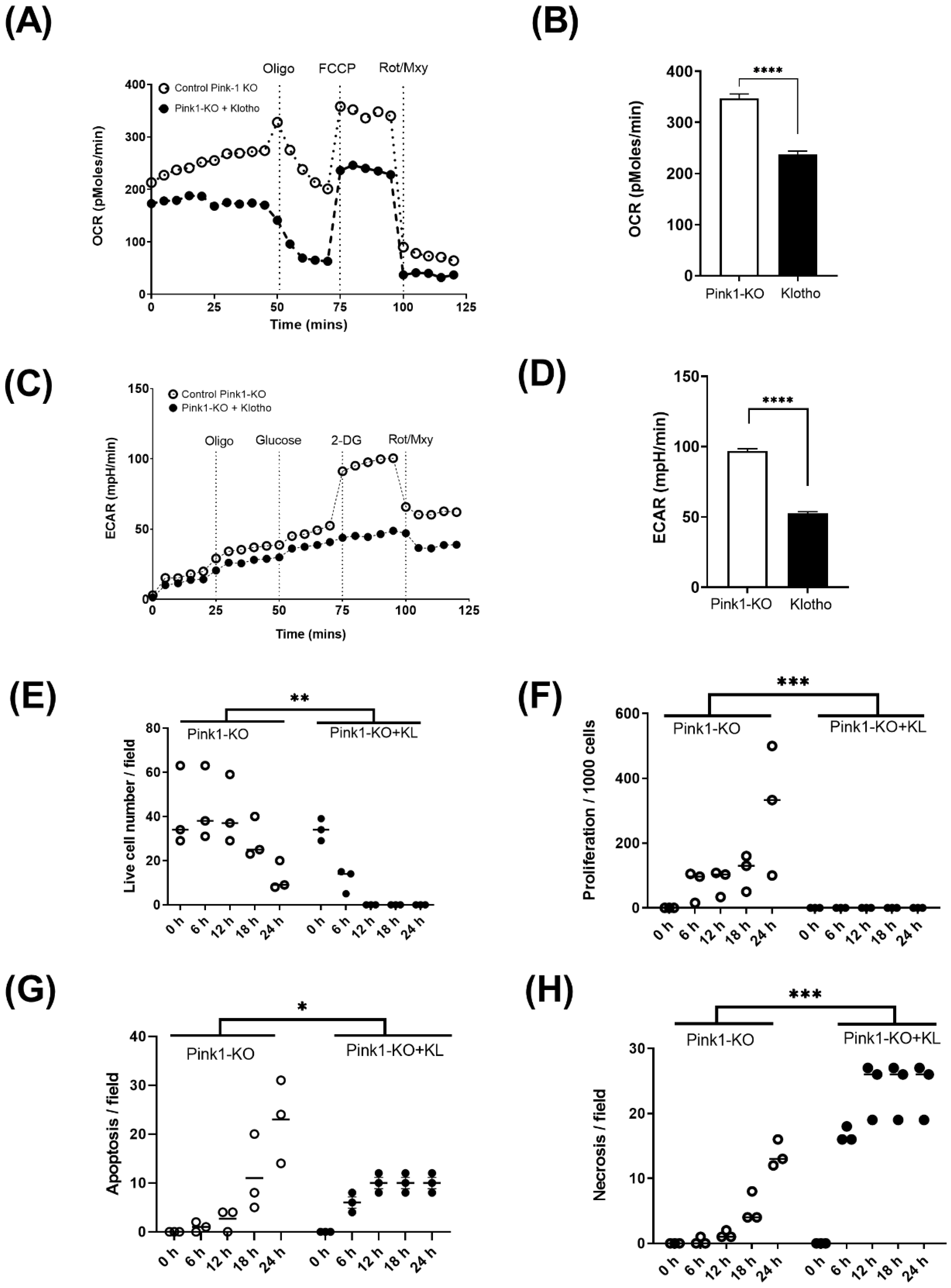
2.3. Energetic Analysis
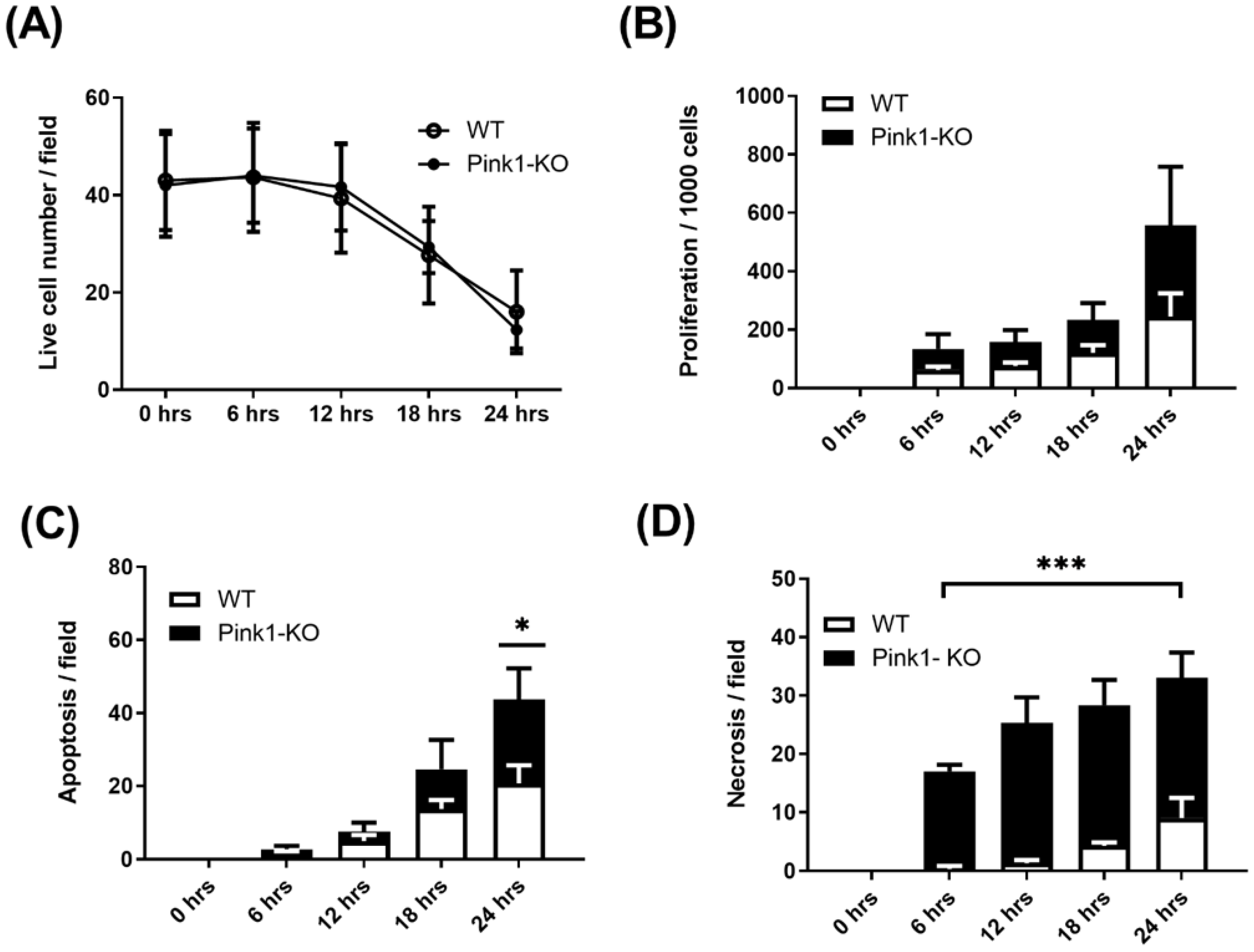
2.4. Live-Cell Imaging
3. Discussion
4. Materials and Methods
4.1. Generation of VSMC Wild-Type and Pink1 Cell Lines
4.2. MTT Viability Assay
4.3. Oxygen Respirometry
4.4. Live-Cell Scoring Analysis
4.5. Western Blotting
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Docherty, C.K.; Carswell, A.; Friel, E.; Mercer, J.R. Impaired mitochondrial respiration in human carotid plaque atherosclerosis: A potential role for Pink1 in vascular smooth muscle cell energetics. Atherosclerosis 2018, 268, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Chiriaco, M.; Georgiopoulos, G.; Duranti, E.; Antonioli, L.; Puxeddu, I.; Nannipieri, M.; Rosada, J.; Blandizzi, C.; Taddei, S.; Virdis, A.; et al. Inflammation and Vascular Ageing: From Telomeres to Novel Emerging Mechanisms. High Blood Press. Cardiovasc. Prev. 2019, 26, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Uryga, A.K.; Grootaert, M.O.J.; Garrido, A.M.; Oc, S.; Foote, K.; Chappell, J.; Finigan, A.; Rossiello, F.; d’Adda di Fagagna, F.; Aravani, D.; et al. Telomere damage promotes vascular smooth muscle cell senescence and immune cell recruitment after vessel injury. Commun. Biol. 2021, 4, 611. [Google Scholar] [CrossRef]
- Chappell, J.; Harman, J.L.; Narasimhan, V.M.; Yu, H.; Foote, K.; Simons, B.D.; Bennett, M.R.; Jorgensen, H.F. Extensive Proliferation of a Subset of Differentiated, yet Plastic, Medial Vascular Smooth Muscle Cells Contributes to Neointimal Formation in Mouse Injury and Atherosclerosis Models. Circ. Res. 2016, 119, 1313–1323. [Google Scholar] [CrossRef] [PubMed]
- Berezin AE, B.A. Impaired function of fibroblast growth factor 23/Klotho protein axis in prediabetes and diabetes mellitus: Promising predictor of cardiovascular risk. Diabetes Metab. Syndr. 2019, 13, 2549–2556. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Hwang, K.H.; Park, K.S.; Kong, I.D.; Cha, S.K. Biological Role of Anti-aging Protein Klotho. J. Lifestyle Med. 2015, 5, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Kurosu, H.; Yamamoto, M.; Clark, J.D.; Pastor, J.V.; Nandi, A.; Gurnani, P.; McGuinness, O.P.; Chikuda, H.; Yamaguchi, M.; Kawaguchi, H.; et al. Suppression of aging in mice by the hormone Klotho. Science 2005, 309, 1829–1833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olejnik, A.; Franczak, A.; Krzywonos-Zawadzka, A.; Kaluzna-Oleksy, M.; Bil-Lula, I. The Biological Role of Klotho Protein in the Development of Cardiovascular Diseases. BioMed Res. Int. 2018, 2018, 5171945. [Google Scholar] [CrossRef] [Green Version]
- Lim, K.; Groen, A.; Molostvov, G.; Lu, T.; Lilley, K.S.; Snead, D.; James, S.; Wilkinson, I.B.; Ting, S.; Hsiao, L.L.; et al. alpha-Klotho Expression in Human Tissues. J. Clin. Endocrinol. Metab. 2015, 100, E1308–E1318. [Google Scholar] [CrossRef] [Green Version]
- Mazucanti, C.H.; Kawamoto, E.M.; Mattson, M.P.; Scavone, C.; Camandola, S. Activity-dependent neuronal Klotho enhances astrocytic aerobic glycolysis. J. Cereb. Blood Flow Metab. 2019, 39, 1544–1556. [Google Scholar] [CrossRef]
- Wolf, I.; Levanon-Cohen, S.; Bose, S.; Ligumsky, H.; Sredni, B.; Kanety, H.; Kuro-o, M.; Karlan, B.; Kaufman, B.; Koeffler, H.P.; et al. Klotho: A tumor suppressor and a modulator of the IGF-1 and FGF pathways in human breast cancer. Oncogene 2008, 27, 7094–7105. [Google Scholar] [CrossRef] [PubMed]
- Imura, A.; Tsuji, Y.; Murata, M.; Maeda, R.; Kubota, K.; Iwano, A.; Obuse, C.; Togashi, K.; Tominaga, M.; Kita, N.; et al. alpha-Klotho as a regulator of calcium homeostasis. Science 2007, 316, 1615–1618. [Google Scholar] [CrossRef] [PubMed]
- Donate-Correa, J.; Martin-Nunez, E.; Delgado, N.P.; de Fuentes, M.M.; Arduan, A.O.; Mora-Fernandez, C.; Navarro Gonzalez, J.F. Implications of Fibroblast growth factor/Klotho system in glucose metabolism and diabetes. Cytokine Growth Factor Rev. 2016, 28, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sun, Z. Current understanding of klotho. Ageing Res. Rev. 2009, 8, 43–51. [Google Scholar] [CrossRef] [Green Version]
- Docherty, C.K.; Bresciani, J.; Carswell, A.; Chanderseka, A.; Friel, E.; Stasi, M.; Mercer, J.R. An Inducible and Vascular Smooth Muscle Cell-Specific Pink1 Knockout Induces Mitochondrial Energetic Dysfunction during Atherogenesis. Int. J. Mol. Sci. 2021, 22, 9993. [Google Scholar] [CrossRef]
- Avin, K.G.; Coen, P.M.; Huang, W.; Stolz, D.B.; Sowa, G.A.; Dube, J.J.; Goodpaster, B.H.; O’Doherty, R.M.; Ambrosio, F. Skeletal muscle as a regulator of the longevity protein, Klotho. Front. Physiol. 2014, 5, 189. [Google Scholar] [CrossRef]
- Kim, J.E.; Choi, H.C. Losartan Inhibits Vascular Smooth Muscle Cell Proliferation through Activation of AMP-Activated Protein Kinase. Korean J. Physiol. Pharmacol. 2010, 14, 299–304. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Zhang, M.; Liang, B.; Shirwany, N.; Zhu, Y.; Zou, M.H. Activation of AMP-activated protein kinase is required for berberine-induced reduction of atherosclerosis in mice: The role of uncoupling protein 2. PLoS ONE 2011, 6, e25436. [Google Scholar] [CrossRef] [Green Version]
- Fullerton, M.D.; Ford, R.J.; McGregor, C.P.; LeBlond, N.D.; Snider, S.A.; Stypa, S.A.; Day, E.A.; Lhotak, S.; Schertzer, J.D.; Austin, R.C.; et al. Salicylate improves macrophage cholesterol homeostasis via activation of Ampk. J. Lipid Res. 2015, 56, 1025–1033. [Google Scholar] [CrossRef] [Green Version]
- Hawley, S.A.; Fullerton, M.D.; Ross, F.A.; Schertzer, J.D.; Chevtzoff, C.; Walker, K.J.; Peggie, M.W.; Zibrova, D.; Green, K.A.; Mustard, K.J.; et al. The ancient drug salicylate directly activates AMP-activated protein kinase. Science 2012, 336, 918–922. [Google Scholar] [CrossRef] [Green Version]
- Goirand, F.; Solar, M.; Athea, Y.; Viollet, B.; Mateo, P.; Fortin, D.; Leclerc, J.; Hoerter, J.; Ventura-Clapier, R.; Garnier, A. Activation of AMP kinase alpha1 subunit induces aortic vasorelaxation in mice. J. Physiol. 2007, 581, 1163–1171. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Connors, K.E.; Yang, D.Q. AICAR induces phosphorylation of AMPK in an ATM-dependent, LKB1-independent manner. Mol. Cell. Biochem. 2007, 306, 239–245. [Google Scholar] [CrossRef]
- Treebak, J.T.; Birk, J.B.; Hansen, B.F.; Olsen, G.S.; Wojtaszewski, J.F. A-769662 activates AMPK beta1-containing complexes but induces glucose uptake through a PI3-kinase-dependent pathway in mouse skeletal muscle. Am. J. Physiol.-Cell Physiol. 2009, 297, C1041–C1052. [Google Scholar] [CrossRef] [Green Version]
- Goransson, O.; McBride, A.; Hawley, S.A.; Ross, F.A.; Shpiro, N.; Foretz, M.; Viollet, B.; Hardie, D.G.; Sakamoto, K. Mechanism of action of A-769662, a valuable tool for activation of AMP-activated protein kinase. J. Biol. Chem. 2007, 282, 32549–32560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.S.; Kim, W.S.; Kim, K.H.; Yoon, M.J.; Cho, H.J.; Shen, Y.; Ye, J.M.; Lee, C.H.; Oh, W.K.; Kim, C.T.; et al. Berberine, a natural plant product, activates AMP-activated protein kinase with beneficial metabolic effects in diabetic and insulin-resistant states. Diabetes 2006, 55, 2256–2264. [Google Scholar] [CrossRef] [Green Version]
- Fu, X.; Wan, S.; Lyu, Y.L.; Liu, L.F.; Qi, H. Etoposide induces ATM-dependent mitochondrial biogenesis through AMPK activation. PLoS ONE 2008, 3, e2009. [Google Scholar] [CrossRef]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Price, N.L.; Gomes, A.P.; Ling, A.J.; Duarte, F.V.; Martin-Montalvo, A.; North, B.J.; Agarwal, B.; Ye, L.; Ramadori, G.; Teodoro, J.S.; et al. SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab. 2012, 15, 675–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, S.; Du, Y.; Peng, Q.; Fan, X.; Li, J.; Chen, M. Trimetazidine Protects Against Atherosclerosis by Changing Energy Charge and Oxidative Stress. Med. Sci. Monit. 2018, 24, 8459–8468. [Google Scholar] [CrossRef] [PubMed]
- Chrusciel, P.; Rysz, J.; Banach, M. Defining the role of trimetazidine in the treatment of cardiovascular disorders: Some insights on its role in heart failure and peripheral artery disease. Drugs 2014, 74, 971–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marzilli, M.; Vinereanu, D.; Lopaschuk, G.; Chen, Y.; Dalal, J.J.; Danchin, N.; Etriby, E.; Ferrari, R.; Gowdak, L.H.; Lopatin, Y.; et al. Trimetazidine in cardiovascular medicine. Int. J. Cardiol. 2019, 293, 39–44. [Google Scholar] [CrossRef] [PubMed]
- LeBrasseur, N.K.; Kelly, M.; Tsao, T.S.; Farmer, S.R.; Saha, A.K.; Ruderman, N.B.; Tomas, E. Thiazolidinediones can rapidly activate AMP-activated protein kinase in mammalian tissues. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E175–E181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, E.; Calvert, P.A.; Mercer, J.R.; Harrison, J.; Baker, L.; Figg, N.L.; Kumar, S.; Wang, J.C.; Hurst, L.A.; Obaid, D.R.; et al. Mitochondrial DNA damage can promote atherosclerosis independently of reactive oxygen species through effects on smooth muscle cells and monocytes and correlates with higher-risk plaques in humans. Circulation 2013, 128, 702–712. [Google Scholar] [CrossRef] [Green Version]
- King, G.D.; Rosene, D.L.; Abraham, C.R. Promoter methylation and age-related downregulation of Klotho in rhesus monkey. Age 2012, 34, 1405–1419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahu, A.; Mamiya, H.; Shinde, S.N.; Cheikhi, A.; Winter, L.L.; Vo, N.V.; Stolz, D.; Roginskaya, V.; Tang, W.Y.; St Croix, C.; et al. Age-related declines in alpha-Klotho drive progenitor cell mitochondrial dysfunction and impaired muscle regeneration. Nat. Commun. 2018, 9, 4859. [Google Scholar] [CrossRef] [PubMed]
- Glasl, L.; Kloos, K.; Giesert, F.; Roethig, A.; Di Benedetto, B.; Kuhn, R.; Zhang, J.; Hafen, U.; Zerle, J.; Hofmann, A.; et al. Pink1-deficiency in mice impairs gait, olfaction and serotonergic innervation of the olfactory bulb. Exp. Neurol. 2012, 235, 214–227. [Google Scholar] [CrossRef]
- Seidler, E.; Wohlrab, F. Suitability of monotetrazolium salt MTT derivatives for histochemical dehydrogenase demonstration. Acta Histochem. 1973, 46, 202–208. [Google Scholar] [PubMed]
- Kavurma, M.M.; Figg, N.; Bennett, M.R.; Mercer, J.; Khachigian, L.M.; Littlewood, T.D. Oxidative stress regulates IGF1R expression in vascular smooth-muscle cells via p53 and HDAC recruitment. Biochem. J. 2007, 407, 79–87. [Google Scholar] [CrossRef] [Green Version]
- Mercer, J.; Figg, N.; Stoneman, V.; Braganza, D.; Bennett, M.R. Endogenous p53 protects vascular smooth muscle cells from apoptosis and reduces atherosclerosis in ApoE knockout mice. Circ. Res. 2005, 96, 667–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercer, J.R.; Cheng, K.K.; Figg, N.; Gorenne, I.; Mahmoudi, M.; Griffin, J.; Vidal-Puig, A.; Logan, A.; Murphy, M.P.; Bennett, M. DNA damage links mitochondrial dysfunction to atherosclerosis and the metabolic syndrome. Circ. Res. 2010, 107, 1021–1031. [Google Scholar] [CrossRef] [PubMed]
- Mercer, J.R.; Yu, E.; Figg, N.; Cheng, K.K.; Prime, T.A.; Griffin, J.L.; Masoodi, M.; Vidal-Puig, A.; Murphy, M.P.; Bennett, M.R. The mitochondria-targeted antioxidant MitoQ decreases features of the metabolic syndrome in ATM+/−/ApoE−/− mice. Free Radic. Biol. Med. 2012, 52, 841–849. [Google Scholar] [CrossRef] [PubMed]
- Docherty, C.K.; Salt, I.P.; Mercer, J.R. Lin28A induces energetic switching to glycolytic metabolism in human embryonic kidney cells. Stem Cell Res. Ther. 2016, 7, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Requejo-Aguilar, R.; Lopez-Fabuel, I.; Fernandez, E.; Martins, L.M.; Almeida, A.; Bolanos, J.P. PINK1 deficiency sustains cell proliferation by reprogramming glucose metabolism through HIF1. Nat. Commun. 2014, 5, 4514. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Docherty, C.K.; Strembitska, A.; Baker, C.P.; Schmidt, F.F.; Reay, K.; Mercer, J.R. Inducing Energetic Switching Using Klotho Improves Vascular Smooth Muscle Cell Phenotype. Int. J. Mol. Sci. 2022, 23, 217. https://doi.org/10.3390/ijms23010217
Docherty CK, Strembitska A, Baker CP, Schmidt FF, Reay K, Mercer JR. Inducing Energetic Switching Using Klotho Improves Vascular Smooth Muscle Cell Phenotype. International Journal of Molecular Sciences. 2022; 23(1):217. https://doi.org/10.3390/ijms23010217
Chicago/Turabian StyleDocherty, Craig K., Anastasiya Strembitska, Christa P. Baker, Fiona F. Schmidt, Kieran Reay, and John R. Mercer. 2022. "Inducing Energetic Switching Using Klotho Improves Vascular Smooth Muscle Cell Phenotype" International Journal of Molecular Sciences 23, no. 1: 217. https://doi.org/10.3390/ijms23010217
APA StyleDocherty, C. K., Strembitska, A., Baker, C. P., Schmidt, F. F., Reay, K., & Mercer, J. R. (2022). Inducing Energetic Switching Using Klotho Improves Vascular Smooth Muscle Cell Phenotype. International Journal of Molecular Sciences, 23(1), 217. https://doi.org/10.3390/ijms23010217