
Integration of Genomic Profiling and Organoid Development in Precision Oncology

Abstract
:1. Introduction
2. Single-Cell Sequencing-Matched Cancer Treatment
2.1. Ovarian Cancer
2.2. Breast Cancer
3. Advanced Genome Profiling Combined with the Use of Cancer Organoids
3.1. Ovarian Cancer
3.2. Breast Cancer
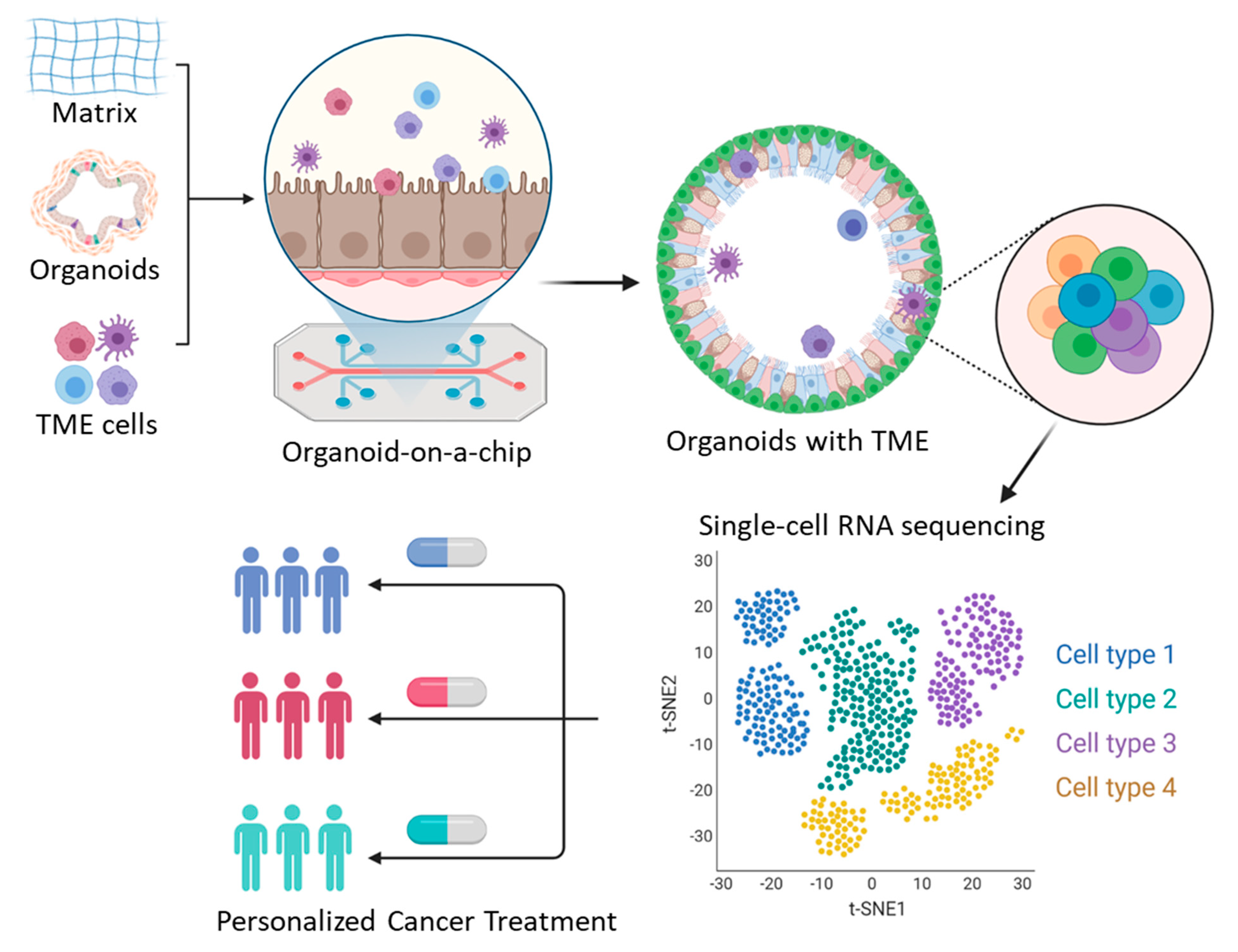
4. Organoids on a Chip
5. Conclusions

{kind=link}
{kind=link}
| Application | Functional Study | Reference | |
|---|---|---|---|
| Ovarian cancer | Basic research | Transcriptome expression profiles of individual cells; intratumoral heterogeneity within ovarian cancer and ascites (fibroblast, T cell, B cell, macrophages, dendritic cells) | [40,41,42,43,110] |
| Druggable target, translational research | Individual gene expression of immune cells in HGSOC; contribution of JAK/STAT signaling in inflammatory programming; drug screening with cucurbitacin I in vitro and in vivo; identification of grade and origin specific cell populations | [44,47,48] | |
| Cancer stem cell | Comparison of gene expression profiles between ovarian cancer and embryonic tissues; cell population expressing PEG10 modulates ovarian cancer stemness and drug resistance | [49] | |
| Drug resistance | Identification of chemo-resistant cell population in HGSOC; the cells express CD44, MYD88, and ALDH1 | [53] | |
| Omentum (ovarian cancer) | Druggable target | High T cell infiltration in the omentum in ovarian cancer patients; increase of antitumor response; providing therapeutic targeting | [52] |
| Breast cancer | Basic research | Single-cell transcriptome profiling of individual cells; clonal evolution; genomic evolution in TNBC; characterization of heterogeneous tumor cells with stromal and immune cells (T cell, B cell, macrophages, CAFs); intratumoral heterogeneity within breast cancer | [64,65,66,67,68] |
| Advanced scRNA-seq research | Nanogrid single-nuclear RNA sequencing; heterogenous phenotypic profiles of breast cancer related to angiogenesis, cell proliferation, and cancer stemness | [69] | |
| Translational research | scRNA-seq analysis using 40 TNBC patients with neoadjuvant anti-PD1; CCR2+ or MMP9+ macrophage and dendritic cells increased T cell expansion; providing therapeutic targeting for synergistic effect with anti-PD1 | [80] | |
| Cancer stem cell, Drug resistance Translational research | CAF-induced Hedgehog ligand promotes chemo-resistant and cancer stem cell population in TNBC; chemotherapy-induced transcriptional reprogramming of resistant signatures; smoothened inhibitors (SMOi) sensitize tumors with docetaxel in vivo; providing a therapeutic target in TNBC | [82,83] |
| Source | Development Efficiency | Features and Use | Reference | |
|---|---|---|---|---|
| Ovarian cancer | 56 organoid lines derived from 32 patients | Medium (~65%) | Maintaining CNVs, recurrent mutations and tumor heterogeneity; long-term expansion; providing drug screening platform; in vivo tumorigenicity; sensitive to platinum-based therapy | [91] |
| 33 organoid lines derived from 22 HGSOC patients | High (80–90%) | Maintaining DNA repair gene mutational status in HGSOC; providing DNA repair profiling and a rapid functional platform for therapeutic sensitivity testing | [92] | |
| 14 organoid lines derived from 3 HGSOC, 1 clear cell, 3endometrioid patients | High (~80%) | Replicating the mutational landscape of the primary tumors; maintaining similar CNVs and BRCA1 pathogenic variant; sensitivity to PARP inhibitor, olaparib, and platinum drugs | [93] | |
| 14 organoid lines derived from 21 gynecologic tumors | High (~95%) | Retaining features of histology and mutations of original tumors; retention of intra-tumoral heterogeneity; only 1 organoid model has in vivo tumorigenicity; drug response assay using organoid-derived spheroids | [90] | |
| Breast cancer | >100 organoid lines derived from >150 patients | High (>80%) | Matching the histopathology, hormone receptor status, and HER2 status of the parental tumor; generic variations retained after long-term expansion; providing in vitro drug screens; sensitive to drugs (e.g., afatinib and pictilisib) blocking the HER signaling pathway | [94] |
| 45 biobanked breast organoid cultures | Medium (55–70%) in most subtypes; Low (~40%) in TNBC | Organoids covering all major breast cancer subtypes; providing genetically edited normal breast organoids using CRISPR–Cas9; providing in vitro and in vivo drug screening platform | [97] | |
| 99 organoids derived from 132 samples | Medium (~75%) | Recapitulating the histopathologic and genetic characteristics of parental tumors; in vitro drug sensitivity screening; sensitive to microtubule-targeting drugs | [98] |
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dieras, V.; Han, H.S.; Kaufman, B.; Wildiers, H.; Friedlander, M.; Ayoub, J.P.; Puhalla, S.L.; Bondarenko, I.; Campone, M.; Jakobsen, E.H.; et al. Veliparib with carboplatin and paclitaxel in BRCA-mutated advanced breast cancer (BROCADE3): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2020, 21, 1269–1282. [Google Scholar] [CrossRef]
- Xie, W.; Sun, H.; Li, X.; Lin, F.; Wang, Z.; Wang, X. Ovarian cancer: Epigenetics, drug resistance, and progression. Cancer Cell Int. 2021, 21, 434. [Google Scholar] [CrossRef]
- Ford, D.; Easton, D.F.; Bishop, D.T.; Narod, S.A.; Goldgar, D.E. Risks of cancer in BRCA1-mutation carriers. Breast Cancer Linkage Consortium. Lancet 1994, 343, 692–695. [Google Scholar] [CrossRef]
- Van’t Veer, L.J.; Bernards, R. Enabling personalized cancer medicine through analysis of gene-expression patterns. Nature 2008, 452, 564–570. [Google Scholar] [CrossRef]
- Shin, S.H.; Bode, A.M.; Dong, Z. Precision medicine: The foundation of future cancer therapeutics. NPJ Precis. Oncol. 2017, 1, 12. [Google Scholar] [CrossRef]
- The Lancet. 20 years of precision medicine in oncology. Lancet 2021, 397, 1781. [Google Scholar] [CrossRef]
- Malone, E.R.; Oliva, M.; Sabatini, P.J.B.; Stockley, T.L.; Siu, L.L. Molecular profiling for precision cancer therapies. Genome Med. 2020, 12, 8. [Google Scholar] [CrossRef] [Green Version]
- Garofalo, A.; Sholl, L.; Reardon, B.; Taylor-Weiner, A.; Amin-Mansour, A.; Miao, D.; Liu, D.; Oliver, N.; MacConaill, L.; Ducar, M.; et al. The impact of tumor profiling approaches and genomic data strategies for cancer precision medicine. Genome Med. 2016, 8, 79. [Google Scholar] [CrossRef] [Green Version]
- Shyr, D.; Liu, Q. Next generation sequencing in cancer research and clinical application. Biol. Proced. Online 2013, 15, 4. [Google Scholar] [CrossRef] [Green Version]
- Morash, M.; Mitchell, H.; Beltran, H.; Elemento, O.; Pathak, J. The Role of Next-Generation Sequencing in Precision Medicine: A Review of Outcomes in Oncology. J. Pers. Med. 2018, 8, 30. [Google Scholar] [CrossRef] [Green Version]
- Cheng, F.; Zhao, J.; Zhao, Z. Advances in computational approaches for prioritizing driver mutations and significantly mutated genes in cancer genomes. Brief. Bioinform. 2016, 17, 642–656. [Google Scholar] [CrossRef] [Green Version]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef]
- El-Sayes, N.; Vito, A.; Mossman, K. Tumor Heterogeneity: A Great Barrier in the Age of Cancer Immunotherapy. Cancers 2021, 13, 806. [Google Scholar] [CrossRef] [PubMed]
- Grun, D.; van Oudenaarden, A. Design and Analysis of Single-Cell Sequencing Experiments. Cell 2015, 163, 799–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Lareau, C.; Andreani, T.; Vinyard, M.E.; Garcia, S.P.; Clement, K.; Andrade-Navarro, M.A.; Buenrostro, J.D.; Pinello, L. Assessment of computational methods for the analysis of single-cell ATAC-seq data. Genome Biol. 2019, 20, 241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navin, N.; Kendall, J.; Troge, J.; Andrews, P.; Rodgers, L.; McIndoo, J.; Cook, K.; Stepansky, A.; Levy, D.; Esposito, D.; et al. Tumour evolution inferred by single-cell sequencing. Nature 2011, 472, 90–94. [Google Scholar] [CrossRef] [Green Version]
- Rotem, A.; Ram, O.; Shoresh, N.; Sperling, R.A.; Goren, A.; Weitz, D.A.; Bernstein, B.E. Single-cell ChIP-seq reveals cell subpopulations defined by chromatin state. Nat. Biotechnol. 2015, 33, 1165–1172. [Google Scholar] [CrossRef]
- Tuveson, D.; Clevers, H. Cancer modeling meets human organoid technology. Science 2019, 364, 952–955. [Google Scholar] [CrossRef]
- Drost, J.; Clevers, H. Organoids in cancer research. Nat. Rev. Cancer 2018, 18, 407–418. [Google Scholar] [CrossRef]
- Fan, H.; Demirci, U.; Chen, P. Emerging organoid models: Leaping forward in cancer research. J. Hematol. Oncol. 2019, 12, 142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Tang, P.; Cai, S.; Peng, J.; Hua, G. Organoid based personalized medicine: From bench to bedside. Cell Regen. 2020, 9, 21. [Google Scholar] [CrossRef]
- Park, S.E.; Georgescu, A.; Huh, D. Organoids-on-a-chip. Science 2019, 364, 960–965. [Google Scholar] [CrossRef]
- Wrzeszczynski, K.O.; Felice, V.; Abhyankar, A.; Kozon, L.; Geiger, H.; Manaa, D.; London, F.; Robinson, D.; Fang, X.; Lin, D.; et al. Analytical Validation of Clinical Whole-Genome and Transcriptome Sequencing of Patient-Derived Tumors for Reporting Targetable Variants in Cancer. J. Mol. Diagn. 2018, 20, 822–835. [Google Scholar] [CrossRef] [Green Version]
- Eberwine, J.; Sul, J.Y.; Bartfai, T.; Kim, J. The promise of single-cell sequencing. Nat. Methods 2014, 11, 25–27. [Google Scholar] [CrossRef] [PubMed]
- Saliba, A.E.; Westermann, A.J.; Gorski, S.A.; Vogel, J. Single-cell RNA-seq: Advances and future challenges. Nucleic Acids Res. 2014, 42, 8845–8860. [Google Scholar] [CrossRef]
- Tirosh, I.; Venteicher, A.S.; Hebert, C.; Escalante, L.E.; Patel, A.P.; Yizhak, K.; Fisher, J.M.; Rodman, C.; Mount, C.; Filbin, M.G.; et al. Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature 2016, 539, 309–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seow, J.J.W.; Wong, R.M.M.; Pai, R.; Sharma, A. Single-Cell RNA Sequencing for Precision Oncology: Current State-of-Art. J. Indian Inst. Sci. 2020, 100, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Shih Ie, M.; Kurman, R.J. Ovarian tumorigenesis: A proposed model based on morphological and molecular genetic analysis. Am. J. Pathol. 2004, 164, 1511–1518. [Google Scholar] [CrossRef]
- Olivier, R.I.; van Beurden, M.; Lubsen, M.A.; Rookus, M.A.; Mooij, T.M.; van de Vijver, M.J.; van’t Veer, L.J. Clinical outcome of prophylactic oophorectomy in BRCA1/BRCA2 mutation carriers and events during follow-up. Br. J. Cancer 2004, 90, 1492–1497. [Google Scholar] [CrossRef]
- Alsop, K.; Fereday, S.; Meldrum, C.; deFazio, A.; Emmanuel, C.; George, J.; Dobrovic, A.; Birrer, M.J.; Webb, P.M.; Stewart, C.; et al. BRCA mutation frequency and patterns of treatment response in BRCA mutation-positive women with ovarian cancer: A report from the Australian Ovarian Cancer Study Group. J. Clin. Oncol. 2012, 30, 2654–2663. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Yang, D.; Sun, Y.; Shmulevich, I.; Xue, F.; Sood, A.K.; Zhang, W. Differing clinical impact of BRCA1 and BRCA2 mutations in serous ovarian cancer. Pharmacogenomics 2012, 13, 1523–1535. [Google Scholar] [CrossRef] [Green Version]
- Endris, V.; Stenzinger, A.; Pfarr, N.; Penzel, R.; Mobs, M.; Lenze, D.; Darb-Esfahani, S.; Hummel, M.; Sabine Merkelbach, B.; Jung, A.; et al. NGS-based BRCA1/2 mutation testing of high-grade serous ovarian cancer tissue: Results and conclusions of the first international round robin trial. Virchows Arch. 2016, 468, 697–705. [Google Scholar] [CrossRef] [PubMed]
- Murakami, R.; Matsumura, N.; Mandai, M.; Yoshihara, K.; Tanabe, H.; Nakai, H.; Yamanoi, K.; Abiko, K.; Yoshioka, Y.; Hamanishi, J.; et al. Establishment of a Novel Histopathological Classification of High-Grade Serous Ovarian Carcinoma Correlated with Prognostically Distinct Gene Expression Subtypes. Am. J. Pathol. 2016, 186, 1103–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyarskikh, U.A.; Gulyaeva, L.F.; Avdalyan, A.M.; Kechin, A.A.; Khrapov, E.A.; Lazareva, D.G.; Kushlinskii, N.E.; Melkonyan, A.; Arakelyan, A.; Filipenko, M.L. Spectrum of TP53 Mutations in BRCA1/2 Associated High-Grade Serous Ovarian Cancer. Front. Oncol. 2020, 10, 1103. [Google Scholar] [CrossRef] [PubMed]
- Bayani, J.; Brenton, J.D.; Macgregor, P.F.; Beheshti, B.; Albert, M.; Nallainathan, D.; Karaskova, J.; Rosen, B.; Murphy, J.; Laframboise, S.; et al. Parallel analysis of sporadic primary ovarian carcinomas by spectral karyotyping, comparative genomic hybridization, and expression microarrays. Cancer Res. 2002, 62, 3466–3476. [Google Scholar] [PubMed]
- Kobel, M.; Huntsman, D.; Gilks, C.B. Critical molecular abnormalities in high-grade serous carcinoma of the ovary. Expert Rev. Mol. Med. 2008, 10, e22. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.A.; Etemadmoghadam, D.; Temple, J.; Lynch, A.G.; Riad, M.; Sharma, R.; Stewart, C.; Fereday, S.; Caldas, C.; Defazio, A.; et al. Driver mutations in TP53 are ubiquitous in high grade serous carcinoma of the ovary. J. Pathol. 2010, 221, 49–56. [Google Scholar] [CrossRef] [Green Version]
- Khalique, L.; Ayhan, A.; Weale, M.E.; Jacobs, I.J.; Ramus, S.J.; Gayther, S.A. Genetic intra-tumour heterogeneity in epithelial ovarian cancer and its implications for molecular diagnosis of tumours. J. Pathol. 2007, 211, 286–295. [Google Scholar] [CrossRef]
- Torres, L.; Ribeiro, F.R.; Pandis, N.; Andersen, J.A.; Heim, S.; Teixeira, M.R. Intratumor genomic heterogeneity in breast cancer with clonal divergence between primary carcinomas and lymph node metastases. Breast Cancer Res. Treat. 2007, 102, 143–155. [Google Scholar] [CrossRef]
- Winterhoff, B.J.; Maile, M.; Mitra, A.K.; Sebe, A.; Bazzaro, M.; Geller, M.A.; Abrahante, J.E.; Klein, M.; Hellweg, R.; Mullany, S.A.; et al. Single cell sequencing reveals heterogeneity within ovarian cancer epithelium and cancer associated stromal cells. Gynecol. Oncol. 2017, 144, 598–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef]
- Cai, D.L.; Jin, L.P. Immune Cell Population in Ovarian Tumor Microenvironment. J. Cancer 2017, 8, 2915–2923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, Q.; Li, J.; Zhang, Q.; Xu, F.; Xie, B.; Lu, H.; Wu, X.; Zhou, X. Single-cell transcriptomes reveal heterogeneity of high-grade serous ovarian carcinoma. Clin. Transl. Med. 2021, 11, e500. [Google Scholar] [CrossRef] [PubMed]
- Shih, A.J.; Menzin, A.; Whyte, J.; Lovecchio, J.; Liew, A.; Khalili, H.; Bhuiya, T.; Gregersen, P.K.; Lee, A.T. Identification of grade and origin specific cell populations in serous epithelial ovarian cancer by single cell RNA-seq. PLoS ONE 2018, 13, e0206785. [Google Scholar]
- Ahmed, N.; Stenvers, K.L. Getting to know ovarian cancer ascites: Opportunities for targeted therapy-based translational research. Front. Oncol. 2013, 3, 256. [Google Scholar] [CrossRef] [Green Version]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H., 2nd; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef] [Green Version]
- Izar, B.; Tirosh, I.; Stover, E.H.; Wakiro, I.; Cuoco, M.S.; Alter, I.; Rodman, C.; Leeson, R.; Su, M.J.; Shah, P.; et al. A single-cell landscape of high-grade serous ovarian cancer. Nat. Med. 2020, 26, 1271–1279. [Google Scholar] [CrossRef]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehar, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Zhao, H.; Gao, Y.; Miao, J.; Chen, S.; Li, J.; Li, Z.; Yin, C.; Yue, W. Single-cell RNA-seq highlights a specific carcinoembryonic cluster in ovarian cancer. Cell Death Dis. 2021, 12, 1082. [Google Scholar] [CrossRef]
- Stanton, S.E.; Disis, M.L. Clinical significance of tumor-infiltrating lymphocytes in breast cancer. J. Immunother. Cancer 2016, 4, 59. [Google Scholar] [CrossRef] [Green Version]
- Santoiemma, P.P.; Powell, D.J., Jr. Tumor infiltrating lymphocytes in ovarian cancer. Cancer Biol. Ther. 2015, 16, 807–820. [Google Scholar] [CrossRef]
- Olalekan, S.; Xie, B.; Back, R.; Eckart, H.; Basu, A. Characterizing the tumor microenvironment of metastatic ovarian cancer by single-cell transcriptomics. Cell Rep. 2021, 35, 109165. [Google Scholar] [CrossRef]
- Villani, A.C.; Satija, R.; Reynolds, G.; Sarkizova, S.; Shekhar, K.; Fletcher, J.; Griesbeck, M.; Butler, A.; Zheng, S.; Lazo, S.; et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science 2017, 356, eaah4573. [Google Scholar] [CrossRef] [Green Version]
- Higgins, M.J.; Baselga, J. Targeted therapies for breast cancer. J. Clin. Investig. 2011, 121, 3797–3803. [Google Scholar] [CrossRef]
- Arteaga, C.L.; Sliwkowski, M.X.; Osborne, C.K.; Perez, E.A.; Puglisi, F.; Gianni, L. Treatment of HER2-positive breast cancer: Current status and future perspectives. Nat. Rev. Clin. Oncol. 2011, 9, 16–32. [Google Scholar] [CrossRef] [PubMed]
- Hicks, D.G.; Kulkarni, S. HER2+ breast cancer: Review of biologic relevance and optimal use of diagnostic tools. Am. J. Clin. Pathol. 2008, 129, 263–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Xu, B. Targeted therapeutic options and future perspectives for HER2-positive breast cancer. Signal Transduct. Target. Ther. 2019, 4, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Yu, D. Mechanisms of ErbB2-mediated paclitaxel resistance and trastuzumab-mediated paclitaxel sensitization in ErbB2-overexpressing breast cancers. Semin. Oncol. 2001, 28 (Suppl. 16), 12–17. [Google Scholar] [CrossRef]
- Pohlmann, P.R.; Mayer, I.A.; Mernaugh, R. Resistance to Trastuzumab in Breast Cancer. Clin. Cancer Res. 2009, 15, 7479–7491. [Google Scholar] [CrossRef] [Green Version]
- Shattuck, D.L.; Miller, J.K.; Carraway, K.L., 3rd; Sweeney, C. Met receptor contributes to trastuzumab resistance of Her2-overexpressing breast cancer cells. Cancer Res. 2008, 68, 1471–1477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, C.; Zhao, H.; Chen, H.; Yao, Q. CXCR4 in breast cancer: Oncogenic role and therapeutic targeting. Drug Des. Dev. Ther. 2015, 9, 4953–4964. [Google Scholar]
- Yousefi, H.; Vatanmakanian, M.; Mahdiannasser, M.; Mashouri, L.; Alahari, N.V.; Monjezi, M.R.; Ilbeigi, S.; Alahari, S.K. Understanding the role of integrins in breast cancer invasion, metastasis, angiogenesis, and drug resistance. Oncogene 2021, 40, 1043–1063. [Google Scholar] [CrossRef]
- Chung, W.; Eum, H.H.; Lee, H.O.; Lee, K.M.; Lee, H.B.; Kim, K.T.; Ryu, H.S.; Kim, S.; Lee, J.E.; Park, Y.H.; et al. Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer. Nat. Commun. 2017, 8, 15081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Waters, J.; Leung, M.L.; Unruh, A.; Roh, W.; Shi, X.; Chen, K.; Scheet, P.; Vattathil, S.; Liang, H.; et al. Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature 2014, 512, 155–160. [Google Scholar] [CrossRef] [Green Version]
- Karaayvaz, M.; Cristea, S.; Gillespie, S.M.; Patel, A.P.; Mylvaganam, R.; Luo, C.C.; Specht, M.C.; Bernstein, B.E.; Michor, F.; Ellisen, L.W. Unravelling subclonal heterogeneity and aggressive disease states in TNBC through single-cell RNA-seq. Nat. Commun. 2018, 9, 3588. [Google Scholar] [CrossRef] [Green Version]
- Bartoschek, M.; Oskolkov, N.; Bocci, M.; Lovrot, J.; Larsson, C.; Sommarin, M.; Madsen, C.D.; Lindgren, D.; Pekar, G.; Karlsson, G.; et al. Spatially and functionally distinct subclasses of breast cancer-associated fibroblasts revealed by single cell RNA sequencing. Nat. Commun. 2018, 9, 5150. [Google Scholar] [CrossRef] [Green Version]
- Grosselin, K.; Durand, A.; Marsolier, J.; Poitou, A.; Marangoni, E.; Nemati, F.; Dahmani, A.; Lameiras, S.; Reyal, F.; Frenoy, O.; et al. High-throughput single-cell ChIP-seq identifies heterogeneity of chromatin states in breast cancer. Nat. Genet. 2019, 51, 1060–1066. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Kim, C.; Sei, E.; Foukakis, T.; Crosetto, N.; Chan, L.K.; Srinivasan, M.; Zhang, H.; Meric-Bernstam, F.; Navin, N. Nanogrid single-nucleus RNA sequencing reveals phenotypic diversity in breast cancer. Nat. Commun. 2017, 8, 228. [Google Scholar] [CrossRef] [PubMed]
- Von Minckwitz, G.; Jonat, W.; Fasching, P.; du Bois, A.; Kleeberg, U.; Luck, H.J.; Kettner, E.; Hilfrich, J.; Eiermann, W.; Torode, J.; et al. A multicentre phase II study on gefitinib in taxane- and anthracycline-pretreated metastatic breast cancer. Breast Cancer Res. Treat. 2005, 89, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J.; Albanell, J.; Ruiz, A.; Lluch, A.; Gascon, P.; Guillem, V.; Gonzalez, S.; Sauleda, S.; Marimon, I.; Tabernero, J.M.; et al. Phase II and tumor pharmacodynamic study of gefitinib in patients with advanced breast cancer. J. Clin. Oncol. 2005, 23, 5323–5333. [Google Scholar] [CrossRef]
- Carey, L.A.; Rugo, H.S.; Marcom, P.K.; Mayer, E.L.; Esteva, F.J.; Ma, C.X.; Liu, M.C.; Storniolo, A.M.; Rimawi, M.F.; Forero-Torres, A.; et al. TBCRC 001: Randomized phase II study of cetuximab in combination with carboplatin in stage IV triple-negative breast cancer. J. Clin. Oncol. 2012, 30, 2615–2623. [Google Scholar] [CrossRef] [Green Version]
- Chung, C.H.; Mirakhur, B.; Chan, E.; Le, Q.T.; Berlin, J.; Morse, M.; Murphy, B.A.; Satinover, S.M.; Hosen, J.; Mauro, D.; et al. Cetuximab-induced anaphylaxis and IgE specific for galactose-alpha-1,3-galactose. N. Engl. J. Med. 2008, 358, 1109–1117. [Google Scholar] [CrossRef] [Green Version]
- Montagut, C.; Dalmases, A.; Bellosillo, B.; Crespo, M.; Pairet, S.; Iglesias, M.; Salido, M.; Gallen, M.; Marsters, S.; Tsai, S.P.; et al. Identification of a mutation in the extracellular domain of the Epidermal Growth Factor Receptor conferring cetuximab resistance in colorectal cancer. Nat. Med. 2012, 18, 221–223. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.T.; Chen, J.S.; Liao, W.Y.; Ho, C.C.; Hsu, C.L.; Yang, C.Y.; Chen, K.Y.; Lee, J.H.; Lin, Z.Z.; Shih, J.Y.; et al. Clinical outcomes and secondary epidermal growth factor receptor (EGFR) T790M mutation among first-line gefitinib, erlotinib and afatinib-treated non-small cell lung cancer patients with activating EGFR mutations. Int. J. Cancer 2019, 144, 2887–2896. [Google Scholar] [CrossRef]
- Savage, P.; Blanchet-Cohen, A.; Revil, T.; Badescu, D.; Saleh, S.M.I.; Wang, Y.C.; Zuo, D.; Liu, L.; Bertos, N.R.; Munoz-Ramos, V.; et al. A Targetable EGFR-Dependent Tumor-Initiating Program in Breast Cancer. Cell Rep. 2017, 21, 1140–1149. [Google Scholar] [CrossRef] [Green Version]
- Loi, S.; Sirtaine, N.; Piette, F.; Salgado, R.; Viale, G.; Van Eenoo, F.; Rouas, G.; Francis, P.; Crown, J.P.; Hitre, E.; et al. Prognostic and predictive value of tumor-infiltrating lymphocytes in a phase III randomized adjuvant breast cancer trial in node-positive breast cancer comparing the addition of docetaxel to doxorubicin with doxorubicin-based chemotherapy: BIG 02-98. J. Clin. Oncol. 2013, 31, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Denkert, C.; von Minckwitz, G.; Darb-Esfahani, S.; Lederer, B.; Heppner, B.I.; Weber, K.E.; Budczies, J.; Huober, J.; Klauschen, F.; Furlanetto, J.; et al. Tumour-infiltrating lymphocytes and prognosis in different subtypes of breast cancer: A pooled analysis of 3771 patients treated with neoadjuvant therapy. Lancet Oncol. 2018, 19, 40–50. [Google Scholar] [CrossRef]
- Bravaccini, S.; Ravaioli, S.; Tumedei, M.M.; Rocca, A.; Palleschi, M.; Puccetti, M.; Parrella, P.; Altini, M.; Maltoni, R. Atezolizumab Plus Nab-paclitaxel in PD-L1-Positive TNBC-Letter. Clin. Cancer Res. 2020, 26, 3892–3893. [Google Scholar] [CrossRef] [PubMed]
- Bassez, A.; Vos, H.; Van Dyck, L.; Floris, G.; Arijs, I.; Desmedt, C.; Boeckx, B.; Vanden Bempt, M.; Nevelsteen, I.; Lambein, K.; et al. A single-cell map of intratumoral changes during anti-PD1 treatment of patients with breast cancer. Nat. Med. 2021, 27, 820–832. [Google Scholar] [CrossRef] [PubMed]
- Scioli, M.G.; Storti, G.; D’Amico, F.; Gentile, P.; Fabbri, G.; Cervelli, V.; Orlandi, A. The Role of Breast Cancer Stem Cells as a Prognostic Marker and a Target to Improve the Efficacy of Breast Cancer Therapy. Cancers 2019, 11, 1021. [Google Scholar] [CrossRef] [Green Version]
- Cazet, A.S.; Hui, M.N.; Elsworth, B.L.; Wu, S.Z.; Roden, D.; Chan, C.L.; Skhinas, J.N.; Collot, R.; Yang, J.; Harvey, K.; et al. Targeting stromal remodeling and cancer stem cell plasticity overcomes chemoresistance in triple negative breast cancer. Nat. Commun. 2018, 9, 2897. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.; Gao, R.; Sei, E.; Brandt, R.; Hartman, J.; Hatschek, T.; Crosetto, N.; Foukakis, T.; Navin, N.E. Chemoresistance Evolution in Triple-Negative Breast Cancer Delineated by Single-Cell Sequencing. Cell 2018, 173, 879–893.e13. [Google Scholar] [CrossRef] [Green Version]
- Schott, A.F.; Landis, M.D.; Dontu, G.; Griffith, K.A.; Layman, R.M.; Krop, I.; Paskett, L.A.; Wong, H.; Dobrolecki, L.E.; Lewis, M.T.; et al. Preclinical and clinical studies of gamma secretase inhibitors with docetaxel on human breast tumors. Clin. Cancer Res. 2013, 19, 1512–1524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parson, W.; Kirchebner, R.; Muhlmann, R.; Renner, K.; Kofler, A.; Schmidt, S.; Kofler, R. Cancer cell line identification by short tandem repeat profiling: Power and limitations. FASEB J. 2005, 19, 434–436. [Google Scholar] [CrossRef] [PubMed]
- Rockwell, S. In Vivo-In Vitro tumour cell lines: Characteristics and limitations as models for human cancer. Br. J. Cancer Suppl. 1980, 4, 118–122. [Google Scholar] [PubMed]
- Darmanis, S.; Sloan, S.A.; Croote, D.; Mignardi, M.; Chernikova, S.; Samghababi, P.; Zhang, Y.; Neff, N.; Kowarsky, M.; Caneda, C.; et al. Single-Cell RNA-Seq Analysis of Infiltrating Neoplastic Cells at the Migrating Front of Human Glioblastoma. Cell Rep. 2017, 21, 1399–1410. [Google Scholar] [CrossRef] [Green Version]
- Krieger, T.G.; Tirier, S.M.; Park, J.; Jechow, K.; Eisemann, T.; Peterziel, H.; Angel, P.; Eils, R.; Conrad, C. Modeling glioblastoma invasion using human brain organoids and single-cell transcriptomics. Neuro-Oncol. 2020, 22, 1138–1149. [Google Scholar] [CrossRef] [Green Version]
- Norkin, M.; Ordonez-Moran, P.; Huelsken, J. High-content, targeted RNA-seq screening in organoids for drug discovery in colorectal cancer. Cell Rep. 2021, 35, 109026. [Google Scholar] [CrossRef]
- Maru, Y.; Tanaka, N.; Itami, M.; Hippo, Y. Efficient use of patient-derived organoids as a preclinical model for gynecologic tumors. Gynecol. Oncol. 2019, 154, 189–198. [Google Scholar] [CrossRef]
- Kopper, O.; de Witte, C.J.; Lohmussaar, K.; Valle-Inclan, J.E.; Hami, N.; Kester, L.; Balgobind, A.V.; Korving, J.; Proost, N.; Begthel, H.; et al. An organoid platform for ovarian cancer captures intra- and interpatient heterogeneity. Nat. Med. 2019, 25, 838–849. [Google Scholar] [CrossRef]
- Hill, S.J.; Decker, B.; Roberts, E.A.; Horowitz, N.S.; Muto, M.G.; Worley, M.J., Jr.; Feltmate, C.M.; Nucci, M.R.; Swisher, E.M.; Nguyen, H.; et al. Prediction of DNA Repair Inhibitor Response in Short-Term Patient-Derived Ovarian Cancer Organoids. Cancer Discov. 2018, 8, 1404–1421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nanki, Y.; Chiyoda, T.; Hirasawa, A.; Ookubo, A.; Itoh, M.; Ueno, M.; Akahane, T.; Kameyama, K.; Yamagami, W.; Kataoka, F.; et al. Patient-derived ovarian cancer organoids capture the genomic profiles of primary tumours applicable for drug sensitivity and resistance testing. Sci. Rep. 2020, 10, 12581. [Google Scholar] [CrossRef]
- Sachs, N.; de Ligt, J.; Kopper, O.; Gogola, E.; Bounova, G.; Weeber, F.; Balgobind, A.V.; Wind, K.; Gracanin, A.; Begthel, H.; et al. A Living Biobank of Breast Cancer Organoids Captures Disease Heterogeneity. Cell 2018, 172, 373–386.e10. [Google Scholar] [CrossRef] [Green Version]
- Goldhammer, N.; Kim, J.; Timmermans-Wielenga, V.; Petersen, O.W. Characterization of organoid cultured human breast cancer. Breast Cancer Res. 2019, 21, 141. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, V.; Huycke, T.R.; Phong, K.T.; Gartner, Z.J. Organoid models for mammary gland dynamics and breast cancer. Curr. Opin. Cell Biol. 2020, 66, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Dekkers, J.F.; van Vliet, E.J.; Sachs, N.; Rosenbluth, J.M.; Kopper, O.; Rebel, H.G.; Wehrens, E.J.; Piani, C.; Visvader, J.E.; Verissimo, C.S.; et al. Long-term culture, genetic manipulation and xenotransplantation of human normal and breast cancer organoids. Nat. Protoc. 2021, 16, 1936–1965. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Zhang, X.; Ding, R.; Yang, L.; Lyu, X.; Zeng, J.; Lei, J.H.; Wang, L.; Bi, J.; Shao, N.; et al. Patient-Derived Organoids Can Guide Personalized-Therapies for Patients with Advanced Breast Cancer. Adv. Sci. 2021, 8, e2101176. [Google Scholar] [CrossRef]
- Habedanck, R.; Stierhof, Y.D.; Wilkinson, C.J.; Nigg, E.A. The Polo kinase Plk4 functions in centriole duplication. Nat. Cell Biol. 2005, 7, 1140–1146. [Google Scholar] [CrossRef]
- Garvey, D.R.; Chhabra, G.; Ndiaye, M.A.; Ahmad, N. Role of Polo-Like Kinase 4 (PLK4) in Epithelial Cancers and Recent Progress in its Small Molecule Targeting for Cancer Management. Mol. Cancer Ther. 2021, 20, 632–640. [Google Scholar] [CrossRef]
- Denu, R.A.; Zasadil, L.M.; Kanugh, C.; Laffin, J.; Weaver, B.A.; Burkard, M.E. Centrosome amplification induces high grade features and is prognostic of worse outcomes in breast cancer. BMC Cancer 2016, 16, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neal, J.T.; Li, X.; Zhu, J.; Giangarra, V.; Grzeskowiak, C.L.; Ju, J.; Liu, I.H.; Chiou, S.H.; Salahudeen, A.A.; Smith, A.R.; et al. Organoid Modeling of the Tumor Immune Microenvironment. Cell 2018, 175, 1972–1988.e16. [Google Scholar] [CrossRef] [Green Version]
- Zumwalde, N.A.; Haag, J.D.; Sharma, D.; Mirrielees, J.A.; Wilke, L.G.; Gould, M.N.; Gumperz, J.E. Analysis of Immune Cells from Human Mammary Ductal Epithelial Organoids Reveals Vdelta2+ T Cells That Efficiently Target Breast Carcinoma Cells in the Presence of Bisphosphonate. Cancer Prev. Res. 2016, 9, 305–316. [Google Scholar] [CrossRef] [Green Version]
- Homan, K.A.; Gupta, N.; Kroll, K.T.; Kolesky, D.B.; Skylar-Scott, M.; Miyoshi, T.; Mau, D.; Valerius, M.T.; Ferrante, T.; Bonventre, J.V.; et al. Flow-enhanced vascularization and maturation of kidney organoids in vitro. Nat. Methods 2019, 16, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Takebe, T.; Sekine, K.; Enomura, M.; Koike, H.; Kimura, M.; Ogaeri, T.; Zhang, R.R.; Ueno, Y.; Zheng, Y.W.; Koike, N.; et al. Vascularized and functional human liver from an iPSC-derived organ bud transplant. Nature 2013, 499, 481–484. [Google Scholar] [CrossRef]
- Cakir, B.; Xiang, Y.; Tanaka, Y.; Kural, M.H.; Parent, M.; Kang, Y.J.; Chapeton, K.; Patterson, B.; Yuan, Y.; He, C.S.; et al. Engineering of human brain organoids with a functional vascular-like system. Nat. Methods 2019, 16, 1169–1175. [Google Scholar] [CrossRef]
- Wang, H.F.; Ran, R.; Liu, Y.; Hui, Y.; Zeng, B.; Chen, D.; Weitz, D.A.; Zhao, C.X. Tumor-Vasculature-on-a-Chip for Investigating Nanoparticle Extravasation and Tumor Accumulation. ACS Nano 2018, 12, 11600–11609. [Google Scholar] [CrossRef]
- Ayuso, J.M.; Truttschel, R.; Gong, M.M.; Humayun, M.; Virumbrales-Munoz, M.; Vitek, R.; Felder, M.; Gillies, S.D.; Sondel, P.; Wisinski, K.B.; et al. Evaluating natural killer cell cytotoxicity against solid tumors using a microfluidic model. Oncoimmunology 2019, 8, 1553477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freeman, F.E.; Pitacco, P.; van Dommelen, L.H.A.; Nulty, J.; Browe, D.C.; Shin, J.Y.; Alsberg, E.; Kelly, D.J. 3D bioprinting spatiotemporally defined patterns of growth factors to tightly control tissue regeneration. Sci. Adv. 2020, 6, eabb5093. [Google Scholar] [CrossRef]
- Schelker, M.; Feau, S.; Du, J.; Ranu, N.; Klipp, E.; MacBeath, G.; Schoeberl, B.; Raue, A. Estimation of immune cell content in tumour tissue using single-cell RNA-seq data. Nat. Commun. 2017, 8, 2032. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoon, H.; Lee, S. Integration of Genomic Profiling and Organoid Development in Precision Oncology. Int. J. Mol. Sci. 2022, 23, 216. https://doi.org/10.3390/ijms23010216
Yoon H, Lee S. Integration of Genomic Profiling and Organoid Development in Precision Oncology. International Journal of Molecular Sciences. 2022; 23(1):216. https://doi.org/10.3390/ijms23010216
Chicago/Turabian StyleYoon, Hyunho, and Sanghoon Lee. 2022. "Integration of Genomic Profiling and Organoid Development in Precision Oncology" International Journal of Molecular Sciences 23, no. 1: 216. https://doi.org/10.3390/ijms23010216
APA StyleYoon, H., & Lee, S. (2022). Integration of Genomic Profiling and Organoid Development in Precision Oncology. International Journal of Molecular Sciences, 23(1), 216. https://doi.org/10.3390/ijms23010216