
Multitalented Synthetic Antimicrobial Peptides and Their Antibacterial, Antifungal and Antiviral Mechanisms

 ,
,  ,
,  and
and 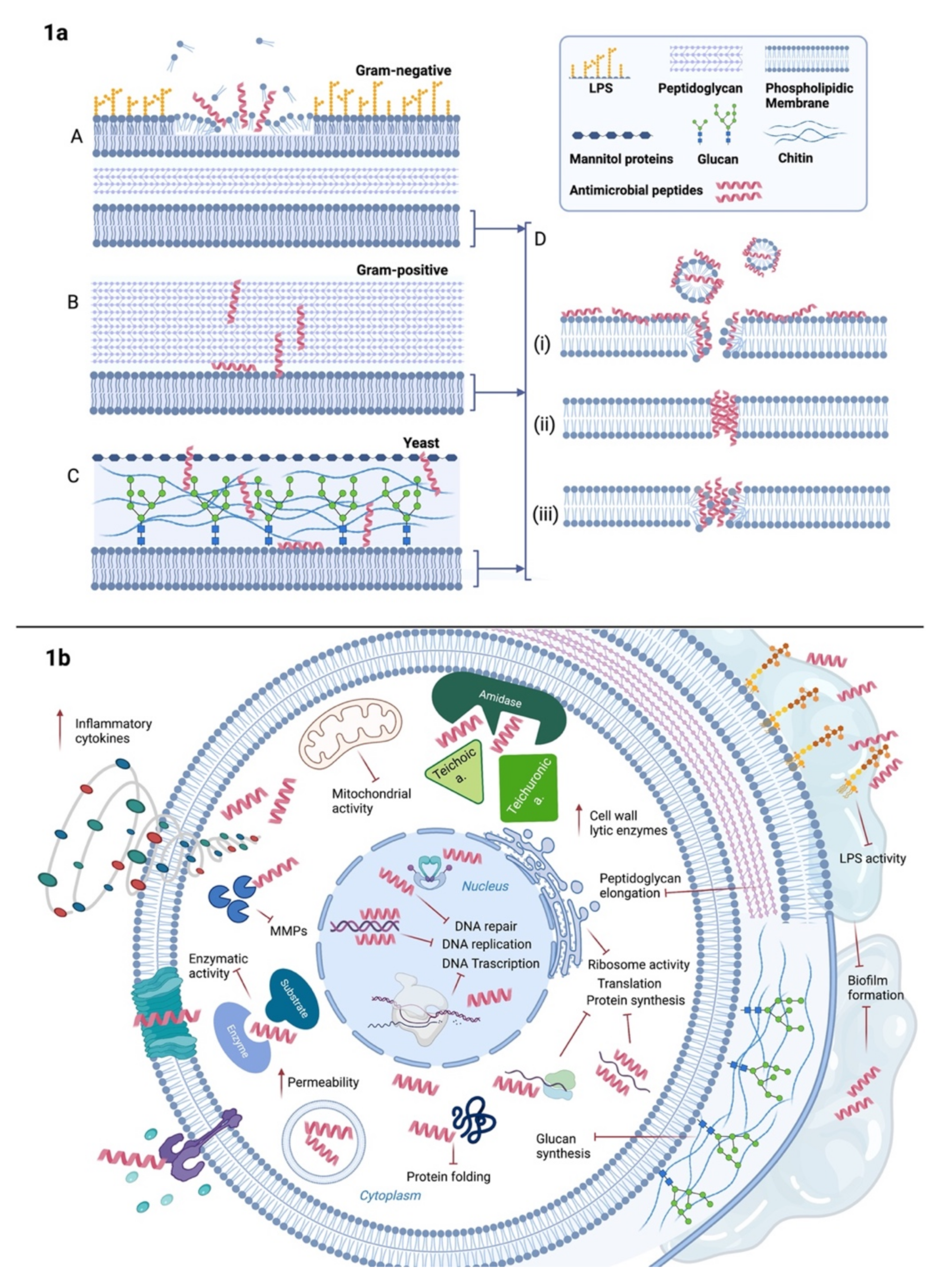{kind=link}
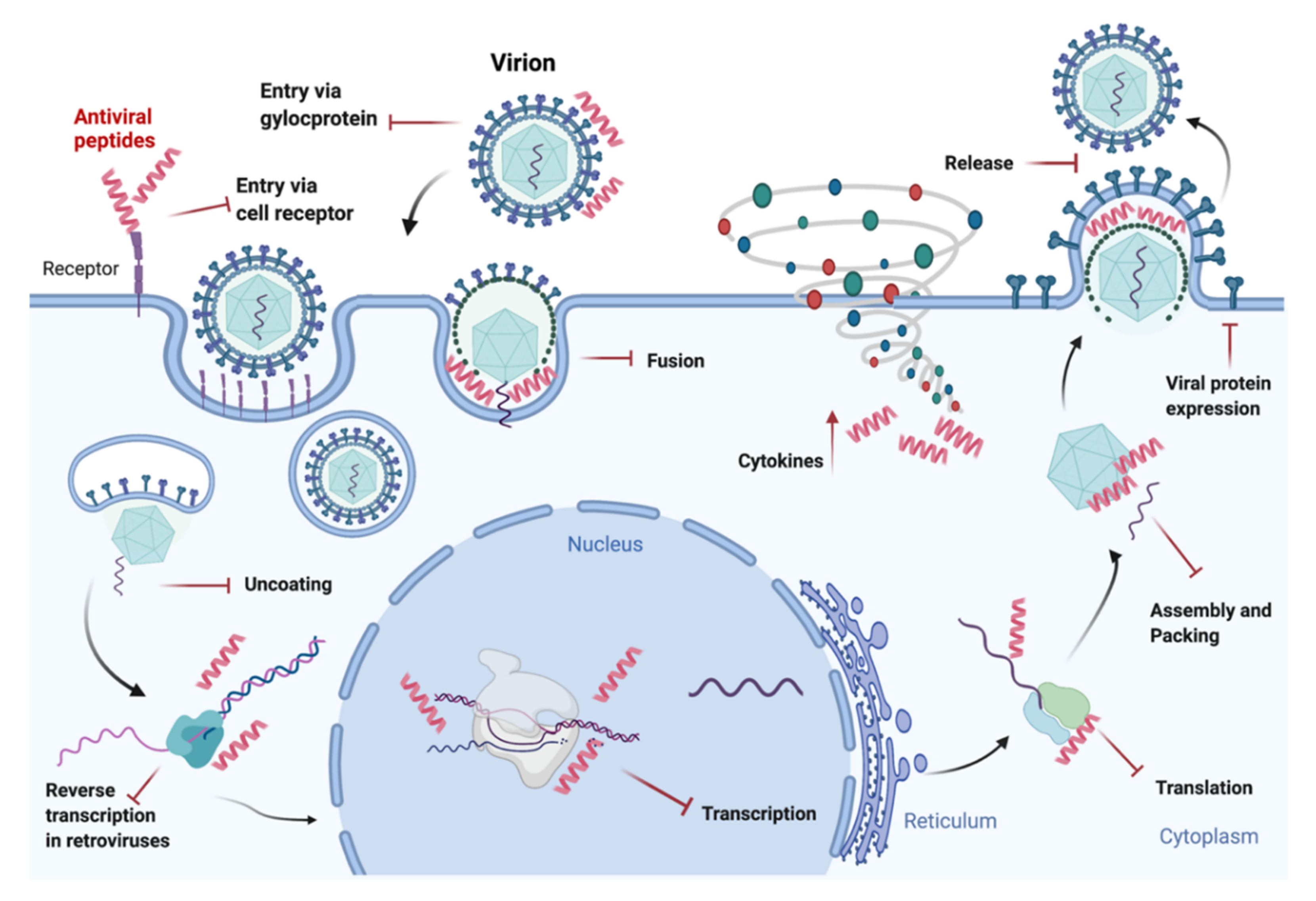{kind=link}
Abstract
:1. Introduction
2. Synthetic Antimicrobial Peptides
3. Antibacterial Peptides and Their Mechanism of Action
4. Antifungal Peptides
5. Antiviral Peptides
6. AMPs—Goods vs. Bads, and the Long Way towards Clinical Application
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fleming, A.; Allison, V.D. Observations on a Bacteriolytic Substance (“Lysozyme”) Found in Secretions and Tissues. Br. J. Exp. Pathol. 1922, 3, 252–260. [Google Scholar]
- Garrod, L.P. Alexander Fleming. A Dedication on the 50th Anniversary of the Discovery of Penicillin. Br. J. Exp. Pathol. 1979, 60, 1–2. [Google Scholar] [PubMed]
- Dubos, R.J. Studies on a Bactericidal Agent Extracted from a Soil Bacillus: I. Preparation of the Agent. Its Activity in Vitro. J. Exp. Med. 1939, 70, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakatsuji, T.; Gallo, R.L. Antimicrobial Peptides: Old Molecules with New Ideas. J. Investig. Dermatol. 2012, 132, 887–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Smet, K.; Contreras, R. Human Antimicrobial Peptides: Defensins, Cathelicidins and Histatins. Biotechnol. Lett. 2005, 27, 1337–1347. [Google Scholar] [CrossRef]
- Diamond, G.; Beckloff, N.; Weinberg, A.; Kisich, K.O. The Roles of Antimicrobial Peptides in Innate Host Defense. Curr. Pharm. Des. 2009, 15, 2377–2392. [Google Scholar] [CrossRef] [Green Version]
- Van der Does, A.M.; Hiemstra, P.S.; Mookherjee, N. Antimicrobial Host Defence Peptides: Immunomodulatory Functions and Translational Prospects. In Antimicrobial Peptides: Basics for Clinical Application; Matsuzaki, K., Ed.; Advances in Experimental Medicine and Biology; Springer: Singapore, 2019; pp. 149–171. ISBN 9789811335884. [Google Scholar]
- Boparai, J.K.; Sharma, P.K. Mini Review on Antimicrobial Peptides, Sources, Mechanism and Recent Applications. Protein Pept. Lett. 2020, 27, 4–16. [Google Scholar] [CrossRef]
- Geitani, R.; Moubareck, C.A.; Xu, Z.; Karam Sarkis, D.; Touqui, L. Expression and Roles of Antimicrobial Peptides in Innate Defense of Airway Mucosa: Potential Implication in Cystic Fibrosis. Front. Immunol. 2020, 11, 1198. [Google Scholar] [CrossRef]
- Wang, G. Antimicrobial Peptides: Discovery, Design and Novel Therapeutic Strategies, 2nd ed.; CABI: Wallingford, UK, 2017; ISBN 978-1-78639-039-4. [Google Scholar]
- Huan, Y.; Kong, Q.; Mou, H.; Yi, H. Antimicrobial Peptides: Classification, Design, Application and Research Progress in Multiple Fields. Front. Microbiol. 2020, 11, 2559. [Google Scholar] [CrossRef]
- Harris, F.; Dennison, S.R.; Phoenix, D.A. Anionic Antimicrobial Peptides from Eukaryotic Organisms. Curr. Protein Pept. Sci. 2009, 10, 585–606. [Google Scholar] [CrossRef]
- López Cascales, J.J.; Zenak, S.; García de la Torre, J.; Lezama, O.G.; Garro, A.; Enriz, R.D. Small Cationic Peptides: Influence of Charge on Their Antimicrobial Activity. ACS Omega 2018, 3, 5390–5398. [Google Scholar] [CrossRef]
- Hancock, R.E.W.; Scott, M.G. The Role of Antimicrobial Peptides in Animal Defenses. Proc. Natl. Acad. Sci. USA 2000, 97, 8856–8861. [Google Scholar] [CrossRef] [Green Version]
- Mishra, A.K.; Choi, J.; Moon, E.; Baek, K.-H. Tryptophan-Rich and Proline-Rich Antimicrobial Peptides. Molecules 2018, 23, 815. [Google Scholar] [CrossRef] [Green Version]
- The Vast Structural Diversity of Antimicrobial Peptides|Elsevier Enhanced Reader. Available online: https://reader.elsevier.com/reader/sd/pii/S0165614719300896?token=AC26A2A3A45C3A5FDA55579937D526F726B0848E72EAAF15FBC5BC9E4C015C21A91A1FDE71DE83D781B6A91FB50B08CE&originRegion=eu-west-1&originCreation=20211028065806 (accessed on 28 October 2021).
- Friedman, D.Z.P.; Schwartz, I.S. Emerging Fungal Infections: New Patients, New Patterns, and New Pathogens. J. Fungi 2019, 5, 67. [Google Scholar] [CrossRef] [Green Version]
- Lockhart, S.R.; Guarner, J. Emerging and Reemerging Fungal Infections. Semin. Diagn. Pathol. 2019, 36, 177–181. [Google Scholar] [CrossRef]
- Lima, P.G.; Oliveira, J.T.A.; Amaral, J.L.; Freitas, C.D.T.; Souza, P.F.N. Synthetic Antimicrobial Peptides: Characteristics, Design, and Potential as Alternative Molecules to Overcome Microbial Resistance. Life Sci. 2021, 278, 119647. [Google Scholar] [CrossRef]
- CDC Antibiotic-Resistant Germs: New Threats. Available online: https://www.cdc.gov/drugresistance/biggest-threats.html (accessed on 27 October 2021).
- WHO Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int (accessed on 11 October 2021).
- Hancock, R.E. Cationic Peptides: Effectors in Innate Immunity and Novel Antimicrobials. Lancet Infect. Dis. 2001, 1, 156–164. [Google Scholar] [CrossRef]
- Sarkar, T.; Chetia, M.; Chatterjee, S. Antimicrobial Peptides and Proteins: From Nature’s Reservoir to the Laboratory and Beyond. Front. Chem. 2021, 9, 691532. [Google Scholar] [CrossRef]
- Giuliani, A.; Rinaldi, A. Beyond Natural Antimicrobial Peptides: Multimeric Peptides and Other Peptidomimetic Approaches. Cell. Mol. Life Sci. 2011, 68, 717. [Google Scholar] [CrossRef]
- Nordström, R.; Malmsten, M. Delivery Systems for Antimicrobial Peptides. Adv. Colloid Interface Sci. 2017, 242, 17–34. [Google Scholar] [CrossRef]
- Marr, A.K.; Gooderham, W.J.; Hancock, R.E. Antibacterial Peptides for Therapeutic Use: Obstacles and Realistic Outlook. Curr. Opin. Pharmacol. 2006, 6, 468–472. [Google Scholar] [CrossRef]
- Lei, J.; Sun, L.; Huang, S.; Zhu, C.; Li, P.; He, J.; Mackey, V.; Coy, D.H.; He, Q. The Antimicrobial Peptides and Their Potential Clinical Applications. Am. J. Transl. Res. 2019, 11, 3919–3931. [Google Scholar]
- Jaradat, D.M.M. Thirteen Decades of Peptide Synthesis: Key Developments in Solid Phase Peptide Synthesis and Amide Bond Formation Utilized in Peptide Ligation. Amino Acids 2018, 50, 39–68. [Google Scholar] [CrossRef]
- González-Henríquez, C.M.; Sarabia-Vallejos, M.A.; Rodríguez-Hernández, J. Strategies to Fabricate Polypeptide-Based Structures via Ring-Opening Polymerization of N-Carboxyanhydrides. Polymers 2017, 9, 551. [Google Scholar] [CrossRef] [Green Version]
- Cardoso, M.H.; Orozco, R.Q.; Rezende, S.B.; Rodrigues, G.; Oshiro, K.G.N.; Cândido, E.S.; Franco, O.L. Computer-Aided Design of Antimicrobial Peptides: Are We Generating Effective Drug Candidates? Front. Microbiol. 2020, 10, 3097. [Google Scholar] [CrossRef]
- Mohamed, M.F.; Abdelkhalek, A.; Seleem, M.N. Evaluation of Short Synthetic Antimicrobial Peptides for Treatment of Drug-Resistant and Intracellular Staphylococcus Aureus. Sci. Rep. 2016, 6, 29707. [Google Scholar] [CrossRef]
- Goodman, M.; Zapf, C.; Rew, Y. New Reagents, Reactions, and Peptidomimetics for Drug Design. Biopolymers 2001, 60, 229–245. [Google Scholar] [CrossRef]
- Chou, K.C. Prediction of Protein Cellular Attributes Using Pseudo-Amino Acid Composition. Proteins 2001, 43, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Adessi, C.; Soto, C. Converting a Peptide into a Drug: Strategies to Improve Stability and Bioavailability. Curr. Med. Chem. 2002, 9, 963–978. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Pramanik, A.; Bhattacharjya, S.; Balaram, P. Omega Amino Acids in Peptide Design: Incorporation into Helices. Biopolymers 1996, 39, 769–777. [Google Scholar] [CrossRef]
- Godballe, T.; Nilsson, L.L.; Petersen, P.D.; Jenssen, H. Antimicrobial β-Peptides and α-Peptoids. Chem. Biol. Drug Des. 2011, 77, 107–116. [Google Scholar] [CrossRef]
- Evans, B.J.; King, A.T.; Katsifis, A.; Matesic, L.; Jamie, J.F. Methods to Enhance the Metabolic Stability of Peptide-Based PET Radiopharmaceuticals. Molecules 2020, 25, 2314. [Google Scholar] [CrossRef]
- Gan, B.H.; Gaynord, J.; Rowe, S.M.; Deingruber, T.; Spring, D.R. The Multifaceted Nature of Antimicrobial Peptides: Current Synthetic Chemistry Approaches and Future Directions. Chem. Soc. Rev. 2021, 50, 7820–7880. [Google Scholar] [CrossRef]
- Falciani, C.; Lozzi, L.; Scali, S.; Brunetti, J.; Bracci, L.; Pini, A. Site-Specific Pegylation of an Antimicrobial Peptide Increases Resistance to Pseudomonas Aeruginosa Elastase. Amino Acids 2014, 46, 1403–1407. [Google Scholar] [CrossRef]
- Gong, Y.; Andina, D.; Nahar, S.; Leroux, J.-C.; Gauthier, M.A. Releasable and Traceless PEGylation of Arginine-Rich Antimicrobial Peptides. Chem. Sci. 2017, 8, 4082–4086. [Google Scholar] [CrossRef] [Green Version]
- Rounds, T.; Straus, S.K. Lipidation of Antimicrobial Peptides as a Design Strategy for Future Alternatives to Antibiotics. Int. J. Mol. Sci. 2020, 21, 9692. [Google Scholar] [CrossRef]
- Tang, J.; Chen, H.; He, Y.; Sheng, W.; Bai, Q.; Wang, H. Peptide-Guided Functionalization and Macrocyclization of Bioactive Peptidosulfonamides by Pd(II)-Catalyzed Late-Stage C-H Activation. Nat. Commun. 2018, 9, 3383. [Google Scholar] [CrossRef]
- Abbasi, E.; Aval, S.F.; Akbarzadeh, A.; Milani, M.; Nasrabadi, H.T.; Joo, S.W.; Hanifehpour, Y.; Nejati-Koshki, K.; Pashaei-Asl, R. Dendrimers: Synthesis, Applications, and Properties. Nanoscale Res. Lett. 2014, 9, 247. [Google Scholar] [CrossRef] [Green Version]
- Tam, J.P. Synthetic Peptide Vaccine Design: Synthesis and Properties of a High-Density Multiple Antigenic Peptide System. Proc. Natl. Acad. Sci. USA 1988, 85, 5409–5413. [Google Scholar] [CrossRef] [Green Version]
- Tam, J.P.; Spetzler, J.C. Synthesis and Application of Peptide Dendrimers as Protein Mimetics. Curr. Protoc. Immunol. 2001, 9, 96. [Google Scholar] [CrossRef]
- Bracci, L.; Falciani, C.; Lelli, B.; Lozzi, L.; Runci, Y.; Pini, A.; De Montis, M.G.; Tagliamonte, A.; Neri, P. Synthetic Peptides in the Form of Dendrimers Become Resistant to Protease Activity. J. Biol. Chem. 2003, 278, 46590–46595. [Google Scholar] [CrossRef] [Green Version]
- Falciani, C.; Lozzi, L.; Pini, A.; Corti, F.; Fabbrini, M.; Bernini, A.; Lelli, B.; Niccolai, N.; Bracci, L. Molecular Basis of Branched Peptides Resistance to Enzyme Proteolysis. Chem. Biol. Drug Des. 2007, 69, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Wadhwani, P.; Reichert, J.; Bürck, J.; Ulrich, A.S. Antimicrobial and Cell-Penetrating Peptides Induce Lipid Vesicle Fusion by Folding and Aggregation. Eur. Biophys. J. 2012, 41, 177–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, N.; Chou, S.; Li, J.; Xue, C.; Li, X.; Cheng, B.; Shan, A.; Xu, L. Short Symmetric-End Antimicrobial Peptides Centered on β-Turn Amino Acids Unit Improve Selectivity and Stability. Front. Microbiol. 2018, 9, 2832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benfield, A.H.; Henriques, S.T. Mode-of-Action of Antimicrobial Peptides: Membrane Disruption vs. Intracellular Mechanisms. Front. Med. Technol. 2020, 2, 20. [Google Scholar] [CrossRef]
- Mwangi, J.; Hao, X.; Lai, R.; Zhang, Z.-Y. Antimicrobial Peptides: New Hope in the War against Multidrug Resistance. Zool. Res. 2019, 40, 488–505. [Google Scholar] [CrossRef]
- Scocchi, M.; Mardirossian, M.; Runti, G.; Benincasa, M. Non-Membrane Permeabilizing Modes of Action of Antimicrobial Peptides on Bacteria. Curr. Top. Med. Chem. 2016, 16, 76–88. [Google Scholar] [CrossRef]
- Gabriel, G.J.; Madkour, A.E.; Dabkowski, J.M.; Nelson, C.F.; Nüsslein, K.; Tew, G.N. Synthetic Mimic of Antimicrobial Peptide with Nonmembrane-Disrupting Antibacterial Properties. Biomacromolecules 2008, 9, 2980–2983. [Google Scholar] [CrossRef] [Green Version]
- Yeaman, M.R.; Yount, N.Y. Mechanisms of Antimicrobial Peptide Action and Resistance. Pharmacol. Rev. 2003, 55, 27–55. [Google Scholar] [CrossRef] [Green Version]
- McHenry, A.J.; Sciacca, M.F.M.; Brender, J.R.; Ramamoorthy, A. Does Cholesterol Suppress the Antimicrobial Peptide Induced Disruption of Lipid Raft Containing Membranes? Biochim. Biophys. Acta 2012, 1818, 3019–3024. [Google Scholar] [CrossRef] [Green Version]
- Silhavy, T.J.; Kahne, D.; Walker, S. The Bacterial Cell Envelope. Cold Spring Harb. Perspect. Biol. 2010, 2, a000414. [Google Scholar] [CrossRef]
- Malanovic, N.; Lohner, K. Antimicrobial Peptides Targeting Gram-Positive Bacteria. Pharmaceuticals 2016, 9, 59. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Koh, J.-J.; Liu, S.; Lakshminarayanan, R.; Verma, C.S.; Beuerman, R.W. Membrane Active Antimicrobial Peptides: Translating Mechanistic Insights to Design. Front. Neurosci. 2017, 11, 73. [Google Scholar] [CrossRef] [Green Version]
- Wimley, W.C. Describing the Mechanism of Antimicrobial Peptide Action with the Interfacial Activity Model. ACS Chem. Biol. 2010, 5, 905–917. [Google Scholar] [CrossRef] [Green Version]
- Brogden, K.A. Antimicrobial Peptides: Pore Formers or Metabolic Inhibitors in Bacteria? Nat. Rev. Microbiol. 2005, 3, 238–250. [Google Scholar] [CrossRef]
- Manzini, M.C.; Perez, K.R.; Riske, K.A.; Bozelli, J.C.; Santos, T.L.; da Silva, M.A.; Saraiva, G.K.V.; Politi, M.J.; Valente, A.P.; Almeida, F.C.L.; et al. Peptide:Lipid Ratio and Membrane Surface Charge Determine the Mechanism of Action of the Antimicrobial Peptide BP100. Conformational and Functional Studies. Biochim. Biophys. Acta 2014, 1838, 1985–1999. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Harroun, T.A.; Weiss, T.M.; Ding, L.; Huang, H.W. Barrel-Stave Model or Toroidal Model? A Case Study on Melittin Pores. Biophys. J. 2001, 81, 1475–1485. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Singh, D.; Kumari, P.; Malik, R.S.; Poonam, P.K.; Parang, K.; Tiwari, R.K. PEGylation and Cell-Penetrating Peptides: Glimpse into the Past and Prospects in the Future. Curr. Top. Med. Chem. 2020, 20, 337–348. [Google Scholar] [CrossRef]
- Sengupta, D.; Leontiadou, H.; Mark, A.E.; Marrink, S.-J. Toroidal Pores Formed by Antimicrobial Peptides Show Significant Disorder. Biochim. Biophys. Acta 2008, 1778, 2308–2317. [Google Scholar] [CrossRef] [Green Version]
- Tuerkova, A.; Kabelka, I.; Králová, T.; Sukeník, L.; Pokorná, Š.; Hof, M.; Vácha, R. Effect of Helical Kink in Antimicrobial Peptides on Membrane Pore Formation. eLife 2020, 9, e47946. [Google Scholar] [CrossRef]
- Clark, S.; Jowitt, T.A.; Harris, L.K.; Knight, C.G.; Dobson, C.B. The Lexicon of Antimicrobial Peptides: A Complete Set of Arginine and Tryptophan Sequences. Commun. Biol. 2021, 4, 605. [Google Scholar] [CrossRef] [PubMed]
- Moravej, H.; Moravej, Z.; Yazdanparast, M.; Heiat, M.; Mirhosseini, A.; Moosazadeh Moghaddam, M.; Mirnejad, R. Antimicrobial Peptides: Features, Action, and Their Resistance Mechanisms in Bacteria. Microb. Drug Resist. 2018, 24, 747–767. [Google Scholar] [CrossRef] [PubMed]
- Le, C.-F.; Fang, C.-M.; Sekaran, S.D. Intracellular Targeting Mechanisms by Antimicrobial Peptides. Antimicrob. Agents Chemother. 2017, 61, e02340–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, E.Y.; Lee, M.W.; Wong, G.C.L. Modulation of Toll-like Receptor Signaling by Antimicrobial Peptides. Semin. Cell Dev. Biol. 2019, 88, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Ryu, M.; Park, J.; Yeom, J.-H.; Joo, M.; Lee, K. Rediscovery of Antimicrobial Peptides as Therapeutic Agents. J. Microbiol. 2021, 59, 113–123. [Google Scholar] [CrossRef]
- Madani, F.; Lindberg, S.; Langel, Ü.; Futaki, S.; Gräslund, A. Mechanisms of Cellular Uptake of Cell-Penetrating Peptides. J. Biophys. 2011, 2011, 414729. [Google Scholar] [CrossRef] [Green Version]
- Tang, Z.; Ma, Q.; Chen, X.; Chen, T.; Ying, Y.; Xi, X.; Wang, L.; Ma, C.; Shaw, C.; Zhou, M. Recent Advances and Challenges in Nanodelivery Systems for Antimicrobial Peptides (AMPs). Antibiotics 2021, 10, 990. [Google Scholar] [CrossRef]
- Birkemo, G.A.; Lüders, T.; Andersen, Ø.; Nes, I.F.; Nissen-Meyer, J. Hipposin, a Histone-Derived Antimicrobial Peptide in Atlantic Halibut (Hippoglossus Hippoglossus L.). Biochim. Biophys. Acta 2003, 1646, 207–215. [Google Scholar] [CrossRef]
- Marchand, C.; Krajewski, K.; Lee, H.-F.; Antony, S.; Johnson, A.A.; Amin, R.; Roller, P.; Kvaratskhelia, M.; Pommier, Y. Covalent Binding of the Natural Antimicrobial Peptide Indolicidin to DNA Abasic Sites. Nucleic Acids Res. 2006, 34, 5157–5165. [Google Scholar] [CrossRef]
- Braffman, N.R.; Piscotta, F.J.; Hauver, J.; Campbell, E.A.; Link, A.J.; Darst, S.A. Structural Mechanism of Transcription Inhibition by Lasso Peptides Microcin J25 and Capistruin. Proc. Natl. Acad. Sci. USA 2019, 116, 1273–1278. [Google Scholar] [CrossRef] [Green Version]
- Heddle, J.G.; Blance, S.J.; Zamble, D.B.; Hollfelder, F.; Miller, D.A.; Wentzell, L.M.; Walsh, C.T.; Maxwell, A. The Antibiotic Microcin B17 Is a DNA Gyrase Poison: Characterisation of the Mode of Inhibition11Edited by J. Karn. J. Mol. Biol. 2001, 307, 1223–1234. [Google Scholar] [CrossRef] [Green Version]
- Polikanov, Y.S.; Aleksashin, N.A.; Beckert, B.; Wilson, D.N. The Mechanisms of Action of Ribosome-Targeting Peptide Antibiotics. Front. Mol. Biosci. 2018, 5, 48. [Google Scholar] [CrossRef] [Green Version]
- Friedrich, C.L.; Rozek, A.; Patrzykat, A.; Hancock, R.E.W. Structure and Mechanism of Action of an Indolicidin Peptide Derivative with Improved Activity against Gram-Positive Bacteria*. J. Biol. Chem. 2001, 276, 24015–24022. [Google Scholar] [CrossRef] [Green Version]
- Patrzykat, A.; Friedrich, C.L.; Zhang, L.; Mendoza, V.; Hancock, R.E.W. Sublethal Concentrations of Pleurocidin-Derived Antimicrobial Peptides Inhibit Macromolecular Synthesis in Escherichia Coli. Antimicrob. Agents Chemother. 2002, 46, 605–614. [Google Scholar] [CrossRef] [Green Version]
- Ho, Y.-H.; Shah, P.; Chen, Y.-W.; Chen, C.-S. Systematic Analysis of Intracellular-Targeting Antimicrobial Peptides, Bactenecin 7, Hybrid of Pleurocidin and Dermaseptin, Proline–Arginine-Rich Peptide, and Lactoferricin B, by Using Escherichia Coli Proteome Microarrays*. Mol. Cell. Proteom. 2016, 15, 1837–1847. [Google Scholar] [CrossRef] [Green Version]
- Florin, T.; Maracci, C.; Graf, M.; Karki, P.; Klepacki, D.; Berninghausen, O.; Beckmann, R.; Vázquez-Laslop, N.; Wilson, D.N.; Rodnina, M.V.; et al. An Antimicrobial Peptide That Inhibits Translation by Trapping Release Factors on the Ribosome. Nat. Struct. Mol. Biol. 2017, 24, 752–757. [Google Scholar] [CrossRef]
- El-Mowafi, S.A.; Sineva, E.; Alumasa, J.N.; Nicoloff, H.; Tomsho, J.W.; Ades, S.E.; Keiler, K.C. Identification of Inhibitors of a Bacterial Sigma Factor Using a New High-Throughput Screening Assay. Antimicrob. Agents Chemother. 2015, 59, 193–205. [Google Scholar] [CrossRef] [Green Version]
- Van Eijk, E.; Wittekoek, B.; Kuijper, E.J.; Smits, W.K. DNA Replication Proteins as Potential Targets for Antimicrobials in Drug-Resistant Bacterial Pathogens. J. Antimicrob. Chemother. 2017, 72, 1275–1284. [Google Scholar] [CrossRef]
- Knappe, D.; Goldbach, T.; Hatfield, M.P.D.; Palermo, N.Y.; Weinert, S.; Sträter, N.; Hoffmann, R.; Lovas, S. Proline-Rich Antimicrobial Peptides Optimized for Binding to Escherichia Coli Chaperone DnaK. Protein Pept. Lett. 2016, 23, 1061–1071. [Google Scholar] [CrossRef]
- Bhowmick, M.; Tokmina-Roszyk, D.; Onwuha-Ekpete, L.; Harmon, K.; Robichaud, T.; Fuerst, R.; Stawikowska, R.; Steffensen, B.; Roush, W.; Wong, H.R.; et al. Second Generation Triple-Helical Peptide Inhibitors of Matrix Metalloproteinases. J. Med. Chem. 2017, 60, 3814–3827. [Google Scholar] [CrossRef]
- Tay, C.X.; Quah, S.Y.; Lui, J.N.; Yu, V.S.H.; Tan, K.S. Matrix Metalloproteinase Inhibitor as an Antimicrobial Agent to Eradicate Enterococcus Faecalis Biofilm. J. Endod. 2015, 41, 858–863. [Google Scholar] [CrossRef]
- Rahman, F.; Nguyen, T.-M.; Adekoya, O.A.; Campestre, C.; Tortorella, P.; Sylte, I.; Winberg, J.-O. Inhibition of Bacterial and Human Zinc-Metalloproteases by Bisphosphonate- and Catechol-Containing Compounds. J. Enzyme Inhib. Med. Chem. 2021, 36, 819–830. [Google Scholar] [CrossRef]
- Salomón, R.A.; Farías, R.N. Microcin 25, a Novel Antimicrobial Peptide Produced by Escherichia Coli. J. Bacteriol. 1992, 174, 7428–7435. [Google Scholar] [CrossRef] [Green Version]
- Chileveru, H.R.; Lim, S.A.; Chairatana, P.; Wommack, A.J.; Chiang, I.-L.; Nolan, E.M. Visualizing Attack of Escherichia Coli by the Antimicrobial Peptide Human Defensin 5. Biochemistry 2015, 54, 1767–1777. [Google Scholar] [CrossRef] [Green Version]
- Scherer, K.M.; Spille, J.-H.; Sahl, H.-G.; Grein, F.; Kubitscheck, U. The Lantibiotic Nisin Induces Lipid II Aggregation, Causing Membrane Instability and Vesicle Budding. Biophys. J. 2015, 108, 1114–1124. [Google Scholar] [CrossRef] [Green Version]
- Wiedemann, I.; Böttiger, T.; Bonelli, R.R.; Schneider, T.; Sahl, H.-G.; Martínez, B. Lipid II-Based Antimicrobial Activity of the Lantibiotic Plantaricin C. Appl. Environ. Microbiol. 2006, 72, 2809–2814. [Google Scholar] [CrossRef] [Green Version]
- Brötz, H.; Bierbaum, G.; Reynolds, P.E.; Sahl, H.-G. The Lantibiotic Mersacidin Inhibits Peptidoglycan Biosynthesis at the Level of Transglycosylation. Eur. J. Biochem. 1997, 246, 193–199. [Google Scholar] [CrossRef]
- Bruschi, M.; Pirri, G.; Giuliani, A.; Nicoletto, S.F.; Baster, I.; Scorciapino, M.A.; Casu, M.; Rinaldi, A.C. Synthesis, Characterization, Antimicrobial Activity and LPS-Interaction Properties of SB041, a Novel Dendrimeric Peptide with Antimicrobial Properties. Peptides 2010, 31, 1459–1467. [Google Scholar] [CrossRef]
- Costa-Orlandi, C.B.; Sardi, J.C.O.; Pitangui, N.S.; de Oliveira, H.C.; Scorzoni, L.; Galeane, M.C.; Medina-Alarcón, K.P.; Melo, W.C.M.A.; Marcelino, M.Y.; Braz, J.D.; et al. Fungal Biofilms and Polymicrobial Diseases. J. Fungi 2017, 3, 22. [Google Scholar] [CrossRef]
- Das, B.; Bhadra, R.K. (P)PpGpp Metabolism and Antimicrobial Resistance in Bacterial Pathogens. Front. Microbiol. 2020, 11, 2415. [Google Scholar] [CrossRef]
- Luo, Y.; Song, Y. Mechanism of Antimicrobial Peptides: Antimicrobial, Anti-Inflammatory and Antibiofilm Activities. Int. J. Mol. Sci. 2021, 22, 11401. [Google Scholar] [CrossRef] [PubMed]
- Hancock, R.E.W.; Alford, M.A.; Haney, E.F. Antibiofilm Activity of Host Defence Peptides: Complexity Provides Opportunities. Nat. Rev. Microbiol. 2021, 19, 786–797. [Google Scholar] [CrossRef] [PubMed]
- Dostert, M.; Belanger, C.R.; Hancock, R.E.W. Design and Assessment of Anti-Biofilm Peptides: Steps toward Clinical Application. J. Innate Immun. 2019, 11, 193–204. [Google Scholar] [CrossRef]
- Luong, H.X.; Thanh, T.T.; Tran, T.H. Antimicrobial Peptides—Advances in Development of Therapeutic Applications. Life Sci. 2020, 260, 118407. [Google Scholar] [CrossRef] [PubMed]
- Dostert, M.; Trimble, M.; Hancock, R.E. Antibiofilm Peptides: Overcoming Biofilm-Related Treatment Failure. RSC Adv. 2021, 11, 2718–2728. [Google Scholar] [CrossRef]
- Buda De Cesare, G.; Cristy, S.A.; Garsin, D.A.; Lorenz, M.C. Antimicrobial Peptides: A New Frontier in Antifungal Therapy. mBio 2020, 11, e02123–20. [Google Scholar] [CrossRef]
- Firacative, C. Invasive Fungal Disease in Humans: Are We Aware of the Real Impact? Mem. Inst. Oswaldo Cruz 2020, 115, e200430. [Google Scholar] [CrossRef]
- Brown, G.D.; Denning, D.W.; Gow, N.A.R.; Levitz, S.M.; Netea, M.G.; White, T.C. Hidden Killers: Human Fungal Infections. Sci. Transl. Med. 2012, 4, 165rv13. [Google Scholar] [CrossRef] [Green Version]
- Candida Auris|Candida Auris|Fungal Diseases|CDC. Available online: https://www.cdc.gov/fungal/candida-auris/index.html (accessed on 31 October 2021).
- Aranda, F.J.; Teruel, J.A.; Ortiz, A. Further Aspects on the Hemolytic Activity of the Antibiotic Lipopeptide Iturin A. Biochim. Biophys. Acta 2005, 1713, 51–56. [Google Scholar] [CrossRef] [Green Version]
- Matejuk, A.; Leng, Q.; Begum, M.D.; Woodle, M.C.; Scaria, P.; Chou, S.-T.; Mixson, A.J. Peptide-Based Antifungal Therapies against Emerging Infections. Drugs Future 2010, 35, 197. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, K.N.; Hirota, K.; Kono, K.; Takeda, S.; Sakamuru, S.; Xia, M.; Huang, R.; Austin, C.P.; Witt, K.L.; Tice, R.R. Characterization of Environmental Chemicals with Potential for DNA Damage Using Isogenic DNA Repair-Deficient Chicken DT40 Cell Lines. Environ. Mol. Mutagenesis 2011, 52, 547–561. [Google Scholar] [CrossRef]
- Li, T.; Li, L.; Du, F.; Sun, L.; Shi, J.; Long, M.; Chen, Z. Activity and Mechanism of Action of Antifungal Peptides from Microorganisms: A Review. Molecules 2021, 26, 3438. [Google Scholar] [CrossRef]
- Hayes, B.M.E.; Bleackley, M.R.; Wiltshire, J.L.; Anderson, M.A.; Traven, A.; van der Weerden, N.L. Identification and Mechanism of Action of the Plant Defensin NaD1 as a New Member of the Antifungal Drug Arsenal against Candida albicans. Antimicrob. Agents Chemother. 2013, 57, 3667–3675. [Google Scholar] [CrossRef] [Green Version]
- Han, Q.; Wu, F.; Wang, X.; Qi, H.; Shi, L.; Ren, A.; Liu, Q.; Zhao, M.; Tang, C. The Bacterial Lipopeptide Iturins Induce Verticillium Dahliae Cell Death by Affecting Fungal Signalling Pathways and Mediate Plant Defence Responses Involved in Pathogen-Associated Molecular Pattern-Triggered Immunity. Environ. Microbiol. 2015, 17, 1166–1188. [Google Scholar] [CrossRef]
- Edgerton, M.; Koshlukova, S.E.; Araujo, M.W.B.; Patel, R.C.; Dong, J.; Bruenn, J.A. Salivary Histatin 5 and Human Neutrophil Defensin 1 Kill Candida Albicans via Shared Pathways. Antimicrob. Agents Chemother. 2000, 44, 3310–3316. [Google Scholar] [CrossRef] [Green Version]
- Trudel, J.; Grenier, J.; Potvin, C.; Asselin, A. Several Thaumatin-Like Proteins Bind to β-1,3-Glucans. Plant Physiol. 1998, 118, 1431–1438. [Google Scholar] [CrossRef] [Green Version]
- Thevissen, K.; Warnecke, D.C.; François, I.E.J.A.; Leipelt, M.; Heinz, E.; Ott, C.; Zähringer, U.; Thomma, B.P.H.J.; Ferket, K.K.A.; Cammue, B.P.A. Defensins from Insects and Plants Interact with Fungal Glucosylceramides. J. Biol. Chem. 2004, 279, 3900–3905. [Google Scholar] [CrossRef] [Green Version]
- Aeed, P.A.; Young, C.L.; Nagiec, M.M.; Elhammer, A.P. Inhibition of Inositol Phosphorylceramide Synthase by the Cyclic Peptide Aureobasidin A. Antimicrob. Agents Chemother. 2009, 53, 496–504. [Google Scholar] [CrossRef] [Green Version]
- Walsh, T.J.; Giri, N. Pradimicins: A Novel Class of Broad-Spectrum Antifungal Compounds. Eur. J. Clin. Microbiol. Infect. Dis. 1997, 16, 93–97. [Google Scholar] [CrossRef]
- Liu, Y.; Lu, J.; Sun, J.; Zhu, X.; Zhou, L.; Lu, Z.; Lu, Y. C16-Fengycin A Affect the Growth of Candida albicans by Destroying Its Cell Wall and Accumulating Reactive Oxygen Species. Appl. Microbiol. Biotechnol. 2019, 103, 8963–8975. [Google Scholar] [CrossRef]
- Guzmán-de-Peña, D.L.; Correa-González, A.M.; Valdés-Santiago, L.; León-Ramírez, C.G.; Valdés-Rodríguez, S. In Vitro Effect of Recombinant Amaranth Cystatin (AhCPI) on Spore Germination, Mycelial Growth, Stress Response and Cellular Integrity of Aspergillus niger and Aspergillus parasiticus. Mycology 2015, 6, 168–175. [Google Scholar] [CrossRef]
- Li, X.S.; Reddy, M.S.; Baev, D.; Edgerton, M. Candida Albicans Ssa1/2p Is the Cell Envelope Binding Protein for Human Salivary Histatin 5. J. Biol. Chem. 2003, 278, 28553–28561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puri, S.; Edgerton, M. How Does It Kill?: Understanding the Candidacidal Mechanism of Salivary Histatin 5. Eukaryot. Cell 2014, 13, 958–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, I.; Ohsumi, K.; Takeda, S.; Katsumata, K.; Matsumoto, S.; Akamatsu, S.; Mitori, H.; Nakai, T. ASP2397 Is a Novel Natural Compound That Exhibits Rapid and Potent Fungicidal Activity against Aspergillus Species through a Specific Transporter. Antimicrob. Agents Chemother. 2019, 63, e02689-18. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Chen, F.; Chen, H.-Y.; Peng, H.; Hao, H.; Wang, K.-J. A Novel Antimicrobial Peptide Scyreprocin from Mud Crab Scylla Paramamosain Showing Potent Antifungal and Anti-Biofilm Activity. Front. Microbiol. 2020, 11, 1589. [Google Scholar] [CrossRef]
- Hacioglu, M.; Oyardi, O.; Bozkurt-Guzel, C.; Savage, P.B. Antibiofilm Activities of Ceragenins and Antimicrobial Peptides against Fungal-Bacterial Mono and Multispecies Biofilms. J. Antibiot. 2020, 73, 455–462. [Google Scholar] [CrossRef]
- Singulani, J.d.L.; Oliveira, L.T.; Ramos, M.D.; Fregonezi, N.F.; Gomes, P.C.; Galeane, M.C.; Palma, M.S.; Fusco Almeida, A.M.; Mendes Giannini, M.J.S. The Antimicrobial Peptide MK58911-NH2 Acts on Planktonic, Biofilm, and Intramacrophage Cells of Cryptococcus Neoformans. Antimicrob. Agents Chemother. 2021, 65, e00904-20. [Google Scholar] [CrossRef]
- Graham, C.E.; Cruz, M.R.; Garsin, D.A.; Lorenz, M.C. Enterococcus Faecalis Bacteriocin EntV Inhibits Hyphal Morphogenesis, Biofilm Formation, and Virulence of Candida Albicans. Proc. Natl. Acad. Sci. USA 2017, 114, 4507–4512. [Google Scholar] [CrossRef] [Green Version]
- Roscetto, E.; Contursi, P.; Vollaro, A.; Fusco, S.; Notomista, E.; Catania, M.R. Antifungal and Anti-Biofilm Activity of the First Cryptic Antimicrobial Peptide from an Archaeal Protein against Candida Spp. Clinical Isolates. Sci. Rep. 2018, 8, 17570. [Google Scholar] [CrossRef]
- Vilas Boas, L.C.P.; Campos, M.L.; Berlanda, R.L.A.; de Carvalho Neves, N.; Franco, O.L. Antiviral Peptides as Promising Therapeutic Drugs. Cell. Mol. Life Sci. 2019, 76, 3525–3542. [Google Scholar] [CrossRef]
- Qureshi, A.; Thakur, N.; Tandon, H.; Kumar, M. AVPdb: A Database of Experimentally Validated Antiviral Peptides Targeting Medically Important Viruses. Nucleic Acids Res. 2014, 42, D1147–D1153. [Google Scholar] [CrossRef] [Green Version]
- Vigant, F.; Santos, N.C.; Lee, B. Broad-Spectrum Antivirals against Viral Fusion. Nat. Rev. Microbiol. 2015, 13, 426–437. [Google Scholar] [CrossRef]
- Jenssen, H.; Hamill, P.; Hancock, R.E.W. Peptide Antimicrobial Agents. Clin. Microbiol. Rev. 2006, 19, 491–511. [Google Scholar] [CrossRef] [Green Version]
- Badani, H.; Garry, R.F.; Wimley, W.C. Peptide Entry Inhibitors of Enveloped Viruses: The Importance of Interfacial Hydrophobicity. Biochim. Biophys. Acta 2014, 1838, 2180–2197. [Google Scholar] [CrossRef] [Green Version]
- LaBonte, J.; Lebbos, J.; Kirkpatrick, P. Enfuvirtide. Nat. Rev. Drug Discov. 2003, 2, 345–346. [Google Scholar] [CrossRef]
- Eggink, D.; Bontjer, I.; de Taeye, S.W.; Langedijk, J.P.M.; Berkhout, B.; Sanders, R.W. HIV-1 Anchor Inhibitors and Membrane Fusion Inhibitors Target Distinct but Overlapping Steps in Virus Entry. J. Biol. Chem. 2019, 294, 5736–5746. [Google Scholar] [CrossRef] [Green Version]
- Lok, S.-M.; Costin, J.M.; Hrobowski, Y.M.; Hoffmann, A.R.; Rowe, D.K.; Kukkaro, P.; Holdaway, H.; Chipman, P.; Fontaine, K.A.; Holbrook, M.R.; et al. Release of Dengue Virus Genome Induced by a Peptide Inhibitor. PLoS ONE 2012, 7, e50995. [Google Scholar] [CrossRef]
- Cetina-Corona, A.; López-Sánchez, U.; Salinas-Trujano, J.; Méndez-Tenorio, A.; Barrón, B.L.; Torres-Flores, J. Peptides Derived from Glycoproteins H and B of Herpes Simplex Virus Type 1 and Herpes Simplex Virus Type 2 Are Capable of Blocking Herpetic Infection in Vitro. Intervirology 2016, 59, 235–242. [Google Scholar] [CrossRef]
- Han, D.P.; Penn-Nicholson, A.; Cho, M.W. Identification of Critical Determinants on ACE2 for SARS-CoV Entry and Development of a Potent Entry Inhibitor. Virology 2006, 350, 15–25. [Google Scholar] [CrossRef] [Green Version]
- Schütz, D.; Ruiz-Blanco, Y.B.; Münch, J.; Kirchhoff, F.; Sanchez-Garcia, E.; Müller, J.A. Peptide and Peptide-Based Inhibitors of SARS-CoV-2 Entry. Adv. Drug Deliv. Rev. 2020, 167, 47–65. [Google Scholar] [CrossRef]
- Zhao, H.; To, K.K.W.; Sze, K.-H.; Yung, T.T.-M.; Bian, M.; Lam, H.; Yeung, M.L.; Li, C.; Chu, H.; Yuen, K.-Y. A Broad-Spectrum Virus- and Host-Targeting Peptide against Respiratory Viruses Including Influenza Virus and SARS-CoV-2. Nat. Commun. 2020, 11, 4252. [Google Scholar] [CrossRef]
- Zhao, H.; To, K.K.W.; Lam, H.; Zhou, X.; Chan, J.F.-W.; Peng, Z.; Lee, A.C.Y.; Cai, J.; Chan, W.-M.; Ip, J.D.; et al. Cross-Linking Peptide and Repurposed Drugs Inhibit Both Entry Pathways of SARS-CoV-2. Nat. Commun. 2021, 12, 1517. [Google Scholar] [CrossRef]
- Wang, C.; Wang, S.; Li, D.; Zhao, X.; Han, S.; Wang, T.; Zhao, G.; Chen, Y.; Chen, F.; Zhao, J.; et al. Lectin-like Intestinal Defensin Inhibits 2019-NCoV Spike Binding to ACE2. BioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.-H.; Chen, C.; Jou, M.-L.; Lee, A.Y.-L.; Lin, Y.-C.; Yu, Y.-P.; Huang, W.-T.; Wu, S.-H. Structural and DNA-Binding Studies on the Bovine Antimicrobial Peptide, Indolicidin: Evidence for Multiple Conformations Involved in Binding to Membranes and DNA. Nucleic Acids Res. 2005, 33, 4053–4064. [Google Scholar] [CrossRef] [Green Version]
- Scott, M.G.; Davidson, D.J.; Gold, M.R.; Bowdish, D.; Hancock, R.E.W. The Human Antimicrobial Peptide LL-37 Is a Multifunctional Modulator of Innate Immune Responses. J. Immunol. 2002, 169, 3883–3891. [Google Scholar] [CrossRef] [Green Version]
- Wohlford-Lenane, C.L.; Meyerholz, D.K.; Perlman, S.; Zhou, H.; Tran, D.; Selsted, M.E.; McCray, P.B. Rhesus Theta-Defensin Prevents Death in a Mouse Model of Severe Acute Respiratory Syndrome Coronavirus Pulmonary Disease. J. Virol. 2009, 83, 11385–11390. [Google Scholar] [CrossRef] [Green Version]
- Mahendran, A.S.K.; Lim, Y.S.; Fang, C.-M.; Loh, H.-S.; Le, C.F. The Potential of Antiviral Peptides as COVID-19 Therapeutics. Front. Pharmacol. 2020, 11, 1475. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Ballatori, N. Endogenous Glutathione Conjugates: Occurrence and Biological Functions. Pharmacol. Rev. 1998, 50, 335–356. [Google Scholar] [PubMed]
- Lu, S.C. Glutathione Synthesis. Biochim. Biophys. Acta 2013, 1830, 3143–3153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraternale, A.; Paoletti, M.F.; Casabianca, A.; Nencioni, L.; Garaci, E.; Palamara, A.T.; Magnani, M. GSH and Analogs in Antiviral Therapy. Mol. Asp. Med. 2009, 30, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Polonikov, A. Endogenous Deficiency of Glutathione as the Most Likely Cause of Serious Manifestations and Death in COVID-19 Patients. ACS Infect. Dis. 2020, 6, 1558–1562. [Google Scholar] [CrossRef]
- Cacciatore, I.; Cornacchia, C.; Pinnen, F.; Mollica, A.; Di Stefano, A. Prodrug Approach for Increasing Cellular Glutathione Levels. Molecules 2010, 15, 1242–1264. [Google Scholar] [CrossRef] [Green Version]
- Ghezzi, P. Redox Regulation of Immunity and the Role of Small Molecular Weight Thiols. Redox Biol. 2021, 44, 102001. [Google Scholar] [CrossRef]
- Fraternale, A.; Zara, C.; De Angelis, M.; Nencioni, L.; Palamara, A.T.; Retini, M.; Di Mambro, T.; Magnani, M.; Crinelli, R. Intracellular Redox-Modulated Pathways as Targets for Effective Approaches in the Treatment of Viral Infection. Int. J. Mol. Sci. 2021, 22, 3603. [Google Scholar] [CrossRef]
- Checconi, P.; De Angelis, M.; Marcocci, M.E.; Fraternale, A.; Magnani, M.; Palamara, A.T.; Nencioni, L. Redox-Modulating Agents in the Treatment of Viral Infections. Int. J. Mol. Sci. 2020, 21, 4084. [Google Scholar] [CrossRef]
- Tonk, M.; Růžek, D.; Vilcinskas, A. Compelling Evidence for the Activity of Antiviral Peptides against SARS-CoV-2. Viruses 2021, 13, 912. [Google Scholar] [CrossRef]
- Shanmugaraj, B.; Bulaon, C.J.I.; Malla, A.; Phoolcharoen, W. Biotechnological Insights on the Expression and Production of Antimicrobial Peptides in Plants. Molecules 2021, 26, 4032. [Google Scholar] [CrossRef]
- Rezende, S.B.; Oshiro, K.G.N.; Júnior, N.G.O.; Franco, O.L.; Cardoso, M.H. Advances on Chemically Modified Antimicrobial Peptides for Generating Peptide Antibiotics. Chem. Commun. 2021, 57, 3793. [Google Scholar] [CrossRef]
- Batoni, G.; Maisetta, G.; Esin, S. Antimicrobial Peptides and Their Interaction with Biofilms of Medically Relevant Bacteria. Biochim. Biophys. Acta 2016, 1858, 1044–1060. [Google Scholar] [CrossRef]
- Grassi, L.; Pompilio, A.; Kaya, E.; Rinaldi, A.C.; Sanjust, E.; Maisetta, G.; Crabbé, A.; Di Bonaventura, G.; Batoni, G.; Esin, S. The Anti-Microbial Peptide (Lin-SB056-1)2-K Reduces Pro-Inflammatory Cytokine Release through Interaction with Pseudomonas Aeruginosa Lipopolysaccharide. Antibiotics 2020, 9, 585. [Google Scholar] [CrossRef]
- Grassi, L.; Batoni, G.; Ostyn, L.; Rigole, P.; Van den Bossche, S.; Rinaldi, A.C.; Maisetta, G.; Esin, S.; Coenye, T.; Crabbé, A. The Antimicrobial Peptide Lin-SB056-1 and Its Dendrimeric Derivative Prevent Pseudomonas Aeruginosa Biofilm Formation in Physiologically Relevant Models of Chronic Infections. Front. Microbiol. 2019, 10, 198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, H.; Xie, Z.; Tan, X.; Guo, R.; Song, Y.; Xie, X.; Wang, R.; Li, L.; Wang, M.; Zhang, Y. Temporin-Like Peptides Show Antimicrobial and Anti-Biofilm Activities against Streptococcus Mutans with Reduced Hemolysis. Molecules 2020, 25, 5724. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, A.C.; Conlon, J.M. The Temporins. In Hanbook of Biologically Active Peptides; Kastin, A.J., Ed.; Elsevier: San Diego, CA, USA, 2013; pp. 400–406. [Google Scholar]
- Welch, N.G.; Li, W.; Hossain, M.A.; Separovic, F.; O’Brien-Simpson, N.M.; Wade, J.D. (Re)Defining the Proline-Rich Antimicrobial Peptide Family and the Identification of Putative New Members. Front. Chem. 2020, 8, 607769. [Google Scholar] [CrossRef] [PubMed]
- Holfeld, L.; Herth, N.; Singer, D.; Hoffmann, R.; Knappe, D. Immunogenicity and Pharmacokinetics of Short, Proline-Rich Antimicrobial Peptides. Future Med. Chem. 2015, 7, 1581–1596. [Google Scholar] [CrossRef]
- Holfeld, L.; Knappe, D.; Hoffmann, R. Proline-Rich Antimicrobial Peptides Show a Long-Lasting Post-Antibiotic Effect on Enterobacteriaceae and Pseudomonas Aeruginosa. J. Antimicrob. Chemother. 2018, 73, 933–941. [Google Scholar] [CrossRef]
- Nuri, R.; Shprung, T.; Shai, Y. Defensive Remodeling: How Bacterial Surface Properties and Biofilm Formation Promote Resistance to Antimicrobial Peptides. Biochim. Biophys. Acta 2015, 1848, 3089–3100. [Google Scholar] [CrossRef] [Green Version]
- Band, V.I.; Weiss, D.S. Mechanisms of Antimicrobial Peptide Resistance in Gram-Negative Bacteria. Antibiotics 2015, 4, 18–41. [Google Scholar] [CrossRef] [Green Version]
- Joo, H.-S.; Fu, C.-I.; Otto, M. Bacterial Strategies of Resistance to Antimicrobial Peptides. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2016, 371, 20150292. [Google Scholar] [CrossRef] [Green Version]
- Magana, M.; Pushpanathan, M.; Santos, A.L.; Leanse, L.; Fernandez, M.; Ioannidis, A.; Giulianotti, M.A.; Apidianakis, Y.; Bradfute, S.; Ferguson, A.L.; et al. The Value of Antimicrobial Peptides in the Age of Resistance. Lancet Infect. Dis. 2020, 20, e216–e230. [Google Scholar] [CrossRef]
- Nuding, S.; Gersemann, M.; Hosaka, Y.; Konietzny, S.; Schaefer, C.; Beisner, J.; Schroeder, B.O.; Ostaff, M.J.; Saigenji, K.; Ott, G.; et al. Gastric Antimicrobial Peptides Fail to Eradicate Helicobacter Pylori Infection Due to Selective Induction and Resistance. PLoS ONE 2013, 8, e73867. [Google Scholar] [CrossRef] [Green Version]
- Annunziato, G.; Costantino, G. Antimicrobial Peptides (AMPs): A Patent Review (2015–2020). Expert Opin. Ther. Pat. 2020, 30, 931–947. [Google Scholar] [CrossRef]
- Guan, Q.; Huang, S.; Jin, Y.; Campagne, R.; Alezra, V.; Wan, Y. Recent Advances in the Exploration of Therapeutic Analogues of Gramicidin S, an Old but Still Potent Antimicrobial Peptide. J. Med. Chem. 2019, 62, 7603–7617. [Google Scholar] [CrossRef]
- Rodríguez-Santiago, J.; Cornejo-Juárez, P.; Silva-Sánchez, J.; Garza-Ramos, U. Polymyxin Resistance in Enterobacterales: Overview and Epidemiology in the Americas. Int. J. Antimicrob. Agents 2021, 16, 106426. [Google Scholar] [CrossRef]
- Gogry, F.A.; Siddiqui, M.T.; Sultan, I.; Haq, Q.M.R. Current Update on Intrinsic and Acquired Colistin Resistance Mechanisms in Bacteria. Front. Med. 2021, 8, 677720. [Google Scholar] [CrossRef]
- Huang, H.W. DAPTOMYCIN, Its Membrane-Active Mechanism vs. That of Other Antimicrobial Peptides. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183395. [Google Scholar] [CrossRef]
- Liu, W.-T.; Chen, E.-Z.; Yang, L.; Peng, C.; Wang, Q.; Xu, Z.; Chen, D.-Q. Emerging Resistance Mechanisms for 4 Types of Common Anti-MRSA Antibiotics in Staphylococcus Aureus: A Comprehensive Review. Microb. Pathog. 2021, 156, 104915. [Google Scholar] [CrossRef]
- Fariñas, M.C.; González-Rico, C.; Fernández-Martínez, M.; Fortún, J.; Escudero-Sanchez, R.; Moreno, A.; Bodro, M.; Muñoz, P.; Valerio, M.; Montejo, M.; et al. Oral Decontamination with Colistin plus Neomycin in Solid Organ Transplant Recipients Colonized by Multidrug-Resistant Enterobacterales: A Multicentre, Randomized, Controlled, Open-Label, Parallel-Group Clinical Trial. Clin. Microbiol. Infect. 2021, 27, 856–863. [Google Scholar] [CrossRef]
- Jovcic, B.; Novovic, K.; Dekic, S.; Hrenovic, J. Colistin Resistance in Environmental Isolates of Acinetobacter Baumannii. Microb. Drug Resist. 2021, 27, 328–336. [Google Scholar] [CrossRef]
- Chow, H.Y.; Po, K.H.L.; Jin, K.; Qiao, G.; Sun, Z.; Ma, W.; Ye, X.; Zhou, N.; Chen, S.; Li, X. Establishing the Structure-Activity Relationship of Daptomycin. ACS Med. Chem. Lett. 2020, 11, 1442–1449. [Google Scholar] [CrossRef]
- Blasco, P.; Zhang, C.; Chow, H.Y.; Chen, G.; Wu, Y.; Li, X. An Atomic Perspective on Improving Daptomycin’s Activity. Biochim. Biophys. Acta Gen. Subj. 2021, 1865, 129918. [Google Scholar] [CrossRef]
- Jiang, Y.; Chen, Y.; Song, Z.; Tan, Z.; Cheng, J. Recent Advances in Design of Antimicrobial Peptides and Polypeptides toward Clinical Translation. Adv. Drug. Deliv. Rev. 2021, 170, 261–280. [Google Scholar] [CrossRef]
- Provenzani, A.; Hospodar, A.R.; Meyer, A.L.; Leonardi Vinci, D.; Hwang, E.Y.; Butrus, C.M.; Polidori, P. Multidrug-Resistant Gram-Negative Organisms: A Review of Recently Approved Antibiotics and Novel Pipeline Agents. Int. J. Clin. Pharm. 2020, 42, 1016–1025. [Google Scholar] [CrossRef]
- Jabbour, J.-F.; Sharara, S.L.; Kanj, S.S. Treatment of Multidrug-Resistant Gram-Negative Skin and Soft Tissue Infections. Curr. Opin. Infect. Dis. 2020, 33, 146–154. [Google Scholar] [CrossRef]
- Farrell, D.J.; Robbins, M.; Rhys-Williams, W.; Love, W.G. In Vitro Activity of XF-73, a Novel Antibacterial Agent, against Antibiotic-Sensitive and -Resistant Gram-Positive and Gram-Negative Bacterial Species. Int. J. Antimicrob. Agents 2010, 35, 531–536. [Google Scholar] [CrossRef] [Green Version]
- MacLean, R.C. Assessing the Potential for Staphylococcus Aureus to Evolve Resistance to XF-73. Trends Microbiol. 2020, 28, 432–435. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vanzolini, T.; Bruschi, M.; Rinaldi, A.C.; Magnani, M.; Fraternale, A. Multitalented Synthetic Antimicrobial Peptides and Their Antibacterial, Antifungal and Antiviral Mechanisms. Int. J. Mol. Sci. 2022, 23, 545. https://doi.org/10.3390/ijms23010545
Vanzolini T, Bruschi M, Rinaldi AC, Magnani M, Fraternale A. Multitalented Synthetic Antimicrobial Peptides and Their Antibacterial, Antifungal and Antiviral Mechanisms. International Journal of Molecular Sciences. 2022; 23(1):545. https://doi.org/10.3390/ijms23010545
Chicago/Turabian StyleVanzolini, Tania, Michela Bruschi, Andrea C. Rinaldi, Mauro Magnani, and Alessandra Fraternale. 2022. "Multitalented Synthetic Antimicrobial Peptides and Their Antibacterial, Antifungal and Antiviral Mechanisms" International Journal of Molecular Sciences 23, no. 1: 545. https://doi.org/10.3390/ijms23010545
APA StyleVanzolini, T., Bruschi, M., Rinaldi, A. C., Magnani, M., & Fraternale, A. (2022). Multitalented Synthetic Antimicrobial Peptides and Their Antibacterial, Antifungal and Antiviral Mechanisms. International Journal of Molecular Sciences, 23(1), 545. https://doi.org/10.3390/ijms23010545