
Epigenetic Mechanisms in Parenchymal Lung Diseases: Bystanders or Therapeutic Targets?

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
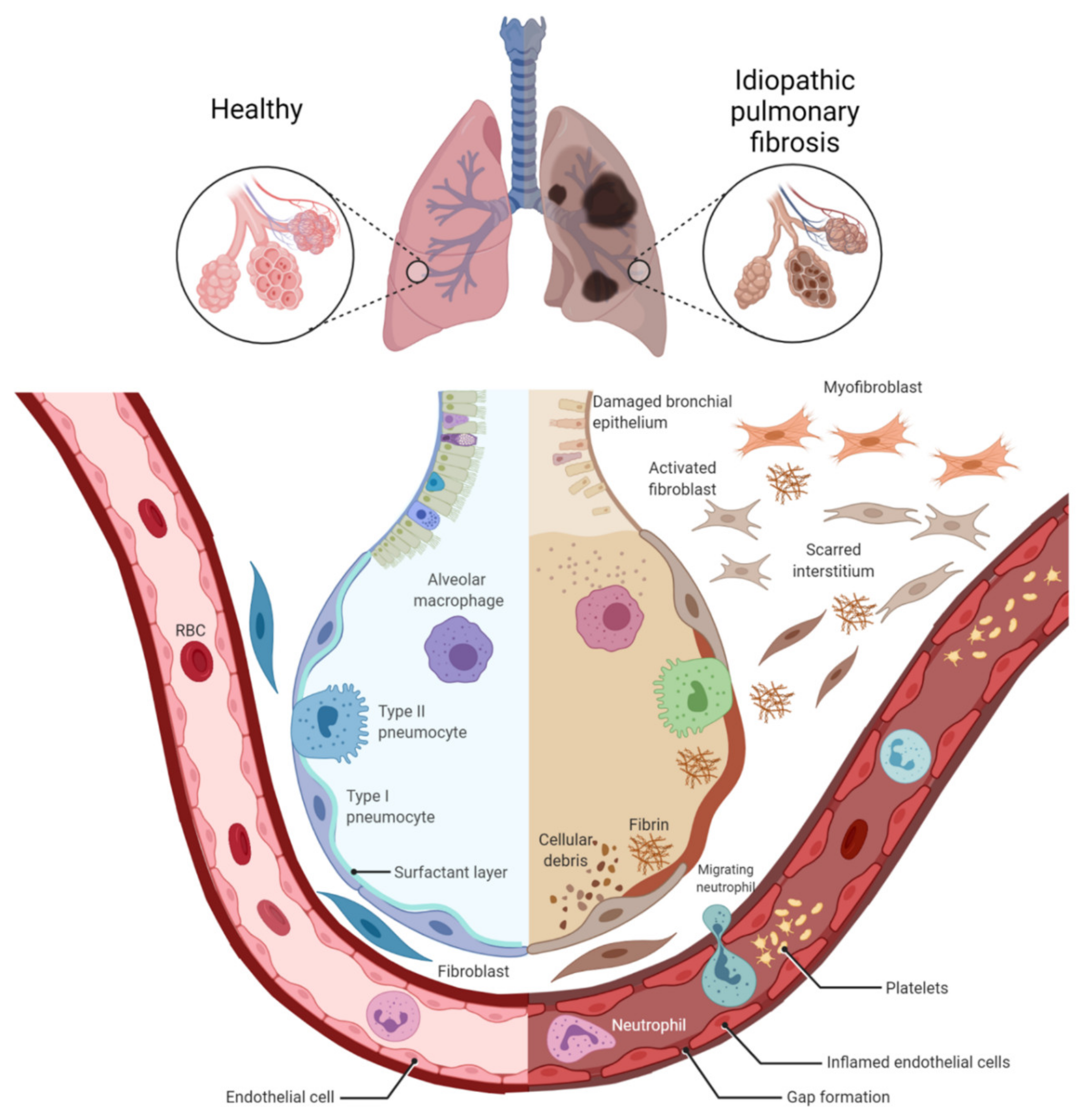
:1. Pathogenesis of IPF and COPD
2. Epigenetic Dysregulation in IPF and COPD
2.1. DNA Methylation in Lung Fibrosis
2.2. DNA Methylation in COPD
2.3. Histone Modifications in Lung Fibrosis
2.4. Histone Modifications in COPD
2.5. MicroRNAs in IPF
2.6. MicroRNAs in COPD
3. Conventional Medications and New Strategies for IPF and COPD
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Marcon, A.; Schievano, E.; Fedeli, U. Mortality Associated with Idiopathic Pulmonary Fibrosis in Northeastern Italy, 2008–2020: A Multiple Cause of Death Analysis. Int. J. Environ. Res. Public Health 2021, 18, 7249. [Google Scholar] [CrossRef]
- Raghu, G.; Chen, S.-Y.; Yeh, W.-S.; Maroni, B.; Li, Q.; Lee, Y.-C.; Collard, H.R. Idiopathic pulmonary fibrosis in US Medicare beneficiaries aged 65 years and older: Incidence, prevalence, and survival, 2001–2011. Lancet Respir. Med. 2014, 2, 566–572. [Google Scholar] [CrossRef]
- Richeldi, L.; Collard, H.R.; Jones, M.G. Idiopathic pulmonary fibrosis. Lancet 2017, 389, 1941–1952. [Google Scholar] [CrossRef]
- Sgalla, G.; Iovene, B.; Calvello, M.; Ori, M.; Varone, F.; Richeldi, L. Idiopathic pulmonary fibrosis: Pathogenesis and management. Respir. Res. 2018, 19, 1–18. [Google Scholar] [CrossRef]
- Birch, J.; Barnes, P.J.; Passos, J.F. Mitochondria, telomeres and cell senescence: Implications for lung ageing and disease. Pharmacol. Ther. 2017, 183, 34–49. [Google Scholar] [CrossRef] [Green Version]
- Bilgili, H.; Białas, A.J.; Górski, P.; Piotrowski, W.J. Telomere Abnormalities in the Pathobiology of Idiopathic Pulmonary Fibrosis. J. Clin. Med. 2019, 8, 1232. [Google Scholar] [CrossRef] [Green Version]
- Vancheri, C. Idiopathic pulmonary fibrosis and cancer: Do they really look similar? BMC Med. 2015, 13, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Vancheri, C.; Failla, M.; Crimi, N.; Raghu, G. Idiopathic pulmonary fibrosis: A disease with similarities and links to cancer biology. Eur. Respir. J. 2010, 35, 496–504. [Google Scholar] [CrossRef] [Green Version]
- Pardo, A.; Selman, M. The Interplay of the Genetic Architecture, Aging, and Environmental Factors in the Pathogenesis of Idiopathic Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2021, 64, 163–172. [Google Scholar] [CrossRef]
- Kabesch, M. Epigenetics in asthma and allergy. Curr. Opin. Allergy Clin. Immunol. 2014, 14, 62–68. [Google Scholar] [CrossRef]
- Schamberger, A.C.; Mise, N.; Meiners, S.; Eickelberg, O. Epigenetic mechanisms in COPD: Implications for pathogenesis and drug discovery. Expert Opin. Drug Discov. 2014, 9, 609–628. [Google Scholar] [CrossRef]
- Vestbo, J.; Hurd, S.S.; Rodríguez-Roisin, R. The 2011 revision of the global strategy for the diagnosis, management and prevention of COPD (GOLD)-why and what? Clin. Respir. J. 2012, 6, 208–214. [Google Scholar] [CrossRef]
- Celli, B.; MacNee, W.; Agusti, A.; Anzueto, A.; Berg, B.; Buist, A.; Calverley, P.; Chavannes, N.; Dillard, T.; Fahy, B.; et al. Standards for the diagnosis and treatment of patients with COPD: A summary of the ATS/ERS position paper. Eur. Respir. J. 2004, 23, 932–946. [Google Scholar] [CrossRef] [Green Version]
- Usmani, O.S. Unravelling the small airways: Structure-function-treatment relationships in Asthma and COPD. Respir. 2012, 84, 1–3. [Google Scholar]
- Shapiro, S.D.; Ingenito, E.P. The pathogenesis of chronic obstructive pulmonary disease: Advances in the past 100 years. Am. J. Respir. Cell Mol. Biol. 2005, 32, 367–372. [Google Scholar] [CrossRef]
- Marwick, J.A.; Kirkham, P.A.; Stevenson, C.S.; Danahay, H.; Giddings, J.; Butler, K.; Donaldson, K.; MacNee, W.; Rahman, I. Cigarette Smoke Alters Chromatin Remodeling and Induces Proinflammatory Genes in Rat Lungs. Am. J. Respir. Cell Mol. Biol. 2004, 31, 633–642. [Google Scholar] [CrossRef] [Green Version]
- Pandey, R. Oxidative/Nitrosative Stress and the Pathobiology of Chronic Obstructive Pulmonary Disease. J. Clin. Diagn. Res. 2013, 7, 580–588. [Google Scholar] [CrossRef]
- Rahman, I. Oxidative Stress in Pathogenesis of Chronic Obstructive Pulmonary Disease: Cellular and Molecular Mechanisms. Cell Biophys. 2005, 43, 167–188. [Google Scholar] [CrossRef]
- Rahman, I.; Adcock, I.M. Oxidative stress and redox regulation of lung inflammation in COPD. Eur. Respir. J. 2006, 28, 219–242. [Google Scholar] [CrossRef]
- Rahman, I. Pharmacological antioxidant strategies as therapeutic interventions for COPD. Biochim. Biophys. Acta Mol. Basis Dis. 2012, 1822, 714–728. [Google Scholar] [CrossRef] [Green Version]
- Chelombitko, M.A. Role of Reactive Oxygen Species in Inflammation: A Minireview. Mosc. Univ. Biol. Sci. Bull. 2018, 73, 199–202. [Google Scholar] [CrossRef] [Green Version]
- Caramori, G.; Adcock, I.; Casolari, P.; Ito, K.; Jazrawi, E.; Tsaprouni, L.; Villetti, G.; Civelli, M.; Carnini, C.; Chung, K.F.; et al. Unbalanced oxidant-induced DNA damage and repair in COPD: A link towards lung cancer. Thorax 2011, 66, 521–527. [Google Scholar] [CrossRef] [Green Version]
- Willemse, B.W.M.; Hacken, N.H.T.T.; Rutgers, B.; Lesman-Leegte, I.G.A.T.; Postma, D.S.; Timens, W. Effect of 1-year smoking cessation on airway inflammation in COPD and asymptomatic smokers. Eur. Respir. J. 2005, 26, 835–845. [Google Scholar] [CrossRef]
- Jiménez-Ruiz, C.A.; Andreas, S.; Lewis, K.; Tønnesen, P.; Van Schayck, C.; Hajek, P.; Tonstad, S.; Dautzenberg, B.; Fletcher, M.; Masefield, S.C.; et al. Statement on smoking cessation in COPD and other pulmonary diseases and in smokers with comorbidities who find it difficult to quit. Eur. Respir. J. 2015, 46, 61–79. [Google Scholar] [CrossRef]
- Dupont, C.; Armant, D.R.; Brenner, C.A. Epigenetics: Definition, mechanisms and clinical perspective. Semin. Reprod. Med. 2009, 27, 351–357. [Google Scholar] [CrossRef] [Green Version]
- Sakao, S.; Tatsumi, K. The importance of epigenetics in the development of chronic obstructive pulmonary disease. Respirology 2011, 16, 1056–1063. [Google Scholar] [CrossRef]
- Sanders, Y.Y.; Ambalavanan, N.; Halloran, B.; Zhang, X.; Liu, H.; Crossman, D.K.; Bray, M.; Zhang, K.; Thannickal, V.J.; Hagood, J.S. Altered DNA Methylation Profile in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2012, 186, 525–535. [Google Scholar] [CrossRef]
- Lee, J.-U.; Son, J.-H.; Shim, E.-Y.; Cheong, H.S.; Shin, S.-W.; Shin, H.D.; Baek, A.R.; Ryu, S.; Park, C.-S.; Chang, H.S.; et al. Global DNA Methylation Pattern of Fibroblasts in Idiopathic Pulmonary Fibrosis. DNA Cell Biol. 2019, 38, 905–914. [Google Scholar] [CrossRef]
- Helling, B.A.; Gerber, N.A.; Kadiyala, V.; Sasse, S.K.; Pedersen, B.S.; Sparks, L.; Nakano, Y.; Okamoto, T.; Evans, C.M.; Yang, I.V.; et al. Regulation of MUC5B expression in idiopathic pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2017, 57, 91–99. [Google Scholar] [CrossRef]
- Saco, T.V.; Breitzig, M.T.; Lockey, R.F.; Kolliputi, N. Epigenetics of Mucus Hypersecretion in Chronic Respiratory Diseases. Am. J. Respir. Cell Mol. Biol. 2018, 58, 299–309. [Google Scholar] [CrossRef]
- Rabinovich, E.I.; Kapetanaki, M.G.; Steinfeld, I.; Gibson, K.F.; Pandit, K.; Yu, G.; Yakhini, Z.; Kaminski, N. Global Methylation Patterns in Idiopathic Pulmonary Fibrosis. PLoS ONE 2012, 7, e33770. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.K.; Scruggs, A.M.; McEachin, R.C.; White, E.S.; Peters-Golden, M. Lung Fibroblasts from Patients with Idiopathic Pulmonary Fibrosis Exhibit Genome-Wide Differences in DNA Methylation Compared to Fibroblasts from Nonfibrotic Lung. PLoS ONE 2014, 9, e107055. [Google Scholar] [CrossRef] [Green Version]
- Koh, H.B.; Scruggs, A.M.; Huang, S.K. Transforming growth factor-β1 increases DNA methyltransferase 1 and 3a ex-pression through distinct post-transcriptional mechanisms in lung fibroblasts. J. Biol. Chem. 2016, 291, 19287–19298. [Google Scholar]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef]
- Vucic, E.A.; Chari, R.; Thu, K.L.; Wilson, I.M.; Cotton, A.M.; Kennett, J.Y.; Zhang, M.; Lonergan, K.M.; Steiling, K.; Brown, C.; et al. DNA Methylation Is Globally Disrupted and Associated with Expression Changes in Chronic Obstructive Pulmonary Disease Small Airways. Am. J. Respir. Cell Mol. Biol. 2014, 50, 912–922. [Google Scholar] [CrossRef] [Green Version]
- He, L.-X.; Tang, Z.-H.; Huang, Q.-S.; Li, W.-H. DNA Methylation: A Potential Biomarker of Chronic Obstructive Pulmonary Disease. Front. Cell Dev. Biol. 2020, 8, 585. [Google Scholar] [CrossRef]
- Lee, M.K.; The Bios Consortium; Xu, C.-J.; Carnes, M.U.; Nichols, C.E.; Ward, J.M.; Kwon, S.O.; Kim, S.-Y.; Kim, W.J.; London, S.J. Genome-wide DNA methylation and long-term ambient air pollution exposure in Korean adults. Clin. Epigenet. 2019, 11, 37. [Google Scholar] [CrossRef] [Green Version]
- Tzortzaki, E.G.; Papi, A.; Neofytou, E.; Soulitzis, N.; Siafakas, N.M. Immune and genetic mechanisms in COPD: Possible targets for therapeutic interventions. Curr. Drug Targets 2013, 14, 141–148. [Google Scholar] [CrossRef]
- Adcock, I.M.; Caramori, G.; Barnes, P.J. Chronic Obstructive Pulmonary Disease and Lung Cancer: New Molecular Insights. Respiration 2011, 81, 265–284. [Google Scholar] [CrossRef]
- Zeilinger, S.; Kühnel, B.; Klopp, N.; Baurecht, H.; Kleinschmidt, A.; Gieger, C.; Weidinger, S.; Lattka, E.; Adamski, J.; Peters, A.; et al. Tobacco Smoking Leads to Extensive Genome-Wide Changes in DNA Methylation. PLoS ONE 2013, 8, e63812. [Google Scholar] [CrossRef]
- Gao, X.; Jia, M.; Zhang, Y.; Breitling, L.P.; Brenner, H. DNA methylation changes of whole blood cells in response to active smoking exposure in adults: A systematic review of DNA methylation studies. Clin. Epigenet. 2015, 7, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Meek, P.M.; Sood, A.; Petersen, H.; Belinsky, S.A.; Tesfaigzi, Y. Epigenetic Change (GATA-4 Gene Methylation) Is Associated with Health Status in Chronic Obstructive Pulmonary Disease. Biol. Res. Nurs. 2014, 17, 191–198. [Google Scholar] [CrossRef]
- Bojesen, S.E.; Timpson, N.; Relton, C.; Smith, G.D.; Nordestgaard, B.G. AHRR (cg05575921) hypomethylation marks smoking behaviour, morbidity and mortality. Thorax 2017, 72, 646–653. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.K.; Hong, Y.; Kim, S.Y.; London, S.J.; Kim, W.J. DNA methylation and smoking in Korean adults: Epige-nome-wide association study. Clin. Epigenet. 2016, 8, 103. [Google Scholar] [CrossRef] [Green Version]
- Machin, M.; Amaral, A.F.; Wielscher, M.; Rezwan, F.I.; Imboden, M.; Jarvelin, M.-R.; Adcock, I.M.; Probst-Hensch, N.; Holloway, J.W.; Jarvis, D.L. Systematic review of lung function and COPD with peripheral blood DNA methylation in population based studies. BMC Pulm. Med. 2017, 17, 54. [Google Scholar] [CrossRef] [Green Version]
- Qiu, W.; Baccarelli, A.; Carey, V.J.; Boutaoui, N.; Bacherman, H.; Klanderman, B.; Rennard, S.; Agusti, A.; Anderson, W.; Lomas, D.A.; et al. Variable DNA Methylation Is Associated with Chronic Obstructive Pulmonary Disease and Lung Function. Am. J. Respir. Crit. Care Med. 2012, 185, 373–381. [Google Scholar] [CrossRef] [Green Version]
- Sundar, I.K.; Yin, Q.; Baier, B.S.; Yan, L.; Mazur, W.; Li, D.; Susiarjo, M.; Rahman, I. DNA methylation profiling in peripheral lung tissues of smokers and patients with COPD. Clin. Epigenet. 2017, 9, 38. [Google Scholar] [CrossRef]
- Bruse, S.; Petersen, H.; Weissfeld, J.; Picchi, M.; Willink, R.; Do, K.; Siegfried, J.; Belinsky, S.A.; Tesfaigzi, Y. Increased methylation of lung cancer-associated genes in sputum DNA of former smokers with chronic mucous hypersecretion. Respir. Res. 2014, 15, 2. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, D.A.; Chen, Y.; Dessaint, J.A.; Aridgides, D.S.; Channon, J.Y.; Mellinger, D.L.; Christensen, B.C.; Ashare, A. DNA Methylation Changes in Regional Lung Macrophages Are Associated with Metabolic Differences. ImmunoHorizons 2019, 3, 274–281. [Google Scholar] [CrossRef]
- Peng, H.; Guo, T.; Chen, Z.; Zhang, H.; Cai, S.; Yang, M.; Chen, P.; Guan, C.; Fang, X. Hypermethylation of mitochondrial transcription factor A induced by cigarette smoke is associated with chronic obstructive pulmonary disease. Exp. Lung Res. 2019, 45, 101–111. [Google Scholar] [CrossRef]
- Liu, F.; Killian, J.K.; Yang, M.; Walker, R.L.; Hong, J.A.; Zhang, M.; Davis, S.; Zhang, Y.; Hussain, M.U.; Xi, S.; et al. Epigenomic alterations and gene expression profiles in respiratory epithelia exposed to cigarette smoke condensate. Oncogene 2010, 29, 3650–3664. [Google Scholar] [CrossRef]
- Song, J.; Heijink, I.H.; Kistemaker, L.E.M.; Reinders-Luinge, M.; Kooistra, W.; Noordhoek, J.A.; Gosens, R.; Brandsma, C.A.; Timens, W.; Hiemstra, P.S.; et al. Aberrant DNA methylation and expression of SPDEF and FOXA2 in airway epithelium of patients with COPD. Clin. Epigenet. 2017, 9, 42. [Google Scholar] [CrossRef] [Green Version]
- Clifford, R.L.; Fishbane, N.; Patel, J.; MacIsaac, J.L.; McEwen, L.M.; Fisher, A.J.; Brandsma, C.-A.; Nair, P.; Kobor, M.S.; Hackett, T.-L.; et al. Altered DNA methylation is associated with aberrant gene expression in parenchymal but not airway fibroblasts isolated from individuals with COPD. Clin. Epigenet. 2018, 10, 32. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, M.; Wada, H.M.; Tian, L.; Shigematsu, H.; Suzuki, H.; Alaa, M.; Tamura, H.; Fujiwara, T.; Nagato, K.; Motohashi, S.; et al. Molecular characterization of chronic obstructive pulmonary disease-related nonsmall cell lung cancer through aberrant methylation and alterations of EGFR signaling. Ann. Surg. Oncol. 2010, 17, 878–888. [Google Scholar] [CrossRef]
- Adam, M.; Schikowski, T.; Carsin, A.E.; Cai, Y.; Jacquemin, B.; Sanchez, M.; Vierkötter, A.; Marcon, A.; Keidel, D.; Sugiri, D.; et al. Adult lung function and long-term air pollution exposure. ESCAPE: A multicentre cohort study and meta-analysis. Eur. Respir. J. 2015, 45, 38–50. [Google Scholar] [CrossRef] [Green Version]
- Lam, H.C.; Cloonan, S.M.; Bhashyam, A.R.; Hasper, J.A.; Singh, A.; Sathirapongsasuti, J.F.; Cervo, M.; Yao, H.; Chung, A.L.; Mizumura, K.; et al. Histone deacetylase 6-mediated selective autophagy regulates COPD associated cilia dysfunction. J. Clin. Investig. 2013, 123, 5212–5230. [Google Scholar]
- Santos-Rosa, H.; Schneider, R.; Bannister, A.; Sherriff, J.; Bernstein, B.E.; Emre, T.; Schreiber, S.L.; Mellor, J.; Kouzarides, T. Active genes are tri-methylated at K4 of histone H3. Nature 2002, 419, 407–411. [Google Scholar] [CrossRef]
- Wagner, E.J.; Carpenter, P.B. Understanding the language of Lys36 methylation at histone H3. Nat. Rev. Mol. Cell Biol. 2012, 13, 115–126. [Google Scholar] [CrossRef] [Green Version]
- Lehnertz, B.; Ueda, Y.; Derijck, A.A.; Braunschweig, U.; Perez-Burgos, L.; Kubicek, S.; Chen, T.; Li, E.; Jenuwein, T.; Peters, A.H. Suv39h-Mediated Histone H3 Lysine 9 Methylation Directs DNA Methylation to Major Satellite Repeats at Pericentric Heterochromatin. Curr. Biol. 2003, 13, 1192–1200. [Google Scholar] [CrossRef] [Green Version]
- Jørgensen, S.; Schotta, G.; Sørensen, C.S. Histone H4 Lysine 20 methylation: Key player in epigenetic regulation of genomic integrity. Nucleic Acids Res. 2013, 41, 2797–2806. [Google Scholar] [CrossRef]
- Cao, R.; Wang, L.; Wang, H.; Xia, L.; Erdjument-Bromage, H.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of Histone H3 Lysine 27 Methylation in Polycomb-Group Silencing. Science 2002, 298, 1039–1043. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, A.T.; Zhang, Y. The diverse functions of Dot1 and H3K79 methylation. Genes Dev. 2011, 25, 1345–1358. [Google Scholar] [CrossRef] [Green Version]
- Greer, E.; Shi, Y. Histone methylation: A dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012, 13, 343–357. [Google Scholar] [CrossRef] [Green Version]
- Rubio, K.; Singh, I.; Dobersch, S.; Sarvari, P.; Günther, S.; Cordero, J.; Mehta, A.; Wujak, L.; Cabrera-Fuentes, H.; Chao, C.-M.; et al. Inactivation of nuclear histone deacetylases by EP300 disrupts the MiCEE complex in idiopathic pulmonary fibrosis. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Huang, Z.; Bao, Y.; Qi, D.; Huang, D. Increased p300/CBP expression in acute respiratory distress syndrome is associated with interleukin-17 and prognosis. Clin. Respir. J. 2020, 14, 791–799. [Google Scholar] [CrossRef]
- Xiao, X.-S.; Cai, M.-Y.; Chen, J.-W.; Guan, X.-Y.; Kung, H.-F.; Zeng, Y.-X.; Xie, D. High expression of p300 in human breast cancer correlates with tumor recurrence and predicts adverse prognosis. Chin. J. Cancer Res. 2011, 23, 201–207. [Google Scholar] [CrossRef] [Green Version]
- Liao, Z.-W.; Zhou, T.-C.; Tan, X.-J.; Song, X.-L.; Liu, Y.; Shi, X.-Y.; Huang, W.-J.; Du, L.-L.; Tu, B.-J.; Lin, X.-D. High expression of p300 is linked to aggressive features and poor prognosis of Nasopharyngeal Carcinoma. J. Transl. Med. 2012, 10, 110. [Google Scholar] [CrossRef] [Green Version]
- Yokomizo, C.; Yamaguchi, K.; Itoh, Y.; Nishimura, T.; Umemura, A.; Minami, M.; Yasui, K.; Mitsuyoshi, H.; Fujii, H.; Tochiki, N.; et al. High expression of p300 in HCC predicts shortened overall survival in association with enhanced epithelial mesenchymal transition of HCC cells. Cancer Lett. 2011, 310, 140–147. [Google Scholar] [CrossRef]
- Tao, J.; Zhang, M.; Wen, Z.; Wang, B.; Zhang, L.; Ou, Y.; Tang, X.; Yu, X.; Jiang, Q. Inhibition of EP300 and DDR1 synergistically alleviates pulmonary fibrosis in vitro and in vivo. Biomed. Pharmacother. 2018, 106, 1727–1733. [Google Scholar] [CrossRef]
- Yang, X.-J.; Seto, E. The Rpd3/Hda1 family of lysine deacetylases: From bacteria and yeast to mice and men. Nat. Rev. Mol. Cell Biol. 2008, 9, 206–218. [Google Scholar] [CrossRef] [Green Version]
- Majdzadeh, N.; Morrison, B.E.; D’Mello, S.R. Class IIA HDACs in the regulation of neurodegeneration. Front. Biosci. 2008, 13, 1072. [Google Scholar] [CrossRef] [Green Version]
- Park, S.-Y.; Kim, J.-S. A short guide to histone deacetylases including recent progress on class II enzymes. Exp. Mol. Med. 2020, 52, 204–212. [Google Scholar] [CrossRef]
- Haberland, M.; Montgomery, R.L.; Olson, E.N. The many roles of histone deacetylases in development and physiology: Implications for disease and therapy. Nat. Rev. Genet. 2009, 10, 32–42. [Google Scholar] [CrossRef]
- Minucci, S.; Pelicci, P.G. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat. Cancer 2006, 6, 38–51. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.; Bisson, W.H.; Löhr, C.V.; Williams, D.E.; Ho, E.; Dashwood, R.H.; Rajendran, P. Histone and Non-Histone Targets of Dietary Deacetylase Inhibitors. Curr. Top. Med. Chem. 2015, 16, 714–731. [Google Scholar] [CrossRef]
- Ropero, S.; Esteller, M. The role of histone deacetylases (HDACs) in human cancer. Mol. Oncol. 2007, 1, 19–25. [Google Scholar] [CrossRef]
- Singh, B.N.; Zhang, G.; Hwa, Y.L.; Li, J.; Dowdy, S.C.; Jiang, S.-W. Nonhistone protein acetylation as cancer therapy targets. Expert Rev. Anticancer. Ther. 2010, 10, 935–954. [Google Scholar] [CrossRef] [Green Version]
- Milazzo, G.; Mercatelli, D.; Di Muzio, G.; Triboli, L.; De Rosa, P.; Perini, G.; Giorgi, F.M. Histone Deacetylases (HDACs): Evolution, Specificity, Role in Transcriptional Complexes, and Pharmacological Actionability. Genes 2020, 11, 556. [Google Scholar] [CrossRef]
- Choi, J.-H.; Kwon, H.J.; Yoon, B.-I.; Kim, J.-H.; Han, S.U.; Joo, H.J.; Kim, D.-Y. Expression Profile of Histone Deacetylase 1 in Gastric Cancer Tissues. Jpn. J. Cancer Res. 2001, 92, 1300–1304. [Google Scholar] [CrossRef]
- Halkidou, K.; Gaughan, L.; Cook, S.; Leung, H.Y.; Neal, D.E.; Robson, C.N. Upregulation and Nuclear Recruitment of HDAC1 in Hormone Refractory Prostate Cancer. Prostate 2004, 59, 177–189. [Google Scholar] [CrossRef]
- Wilson, A.J.; Byun, D.-S.; Popova, N.; Murray, L.B.; L’Italien, K.; Sowa, Y.; Arango, D.; Velcich, A.; Augenlicht, L.H.; Mariadason, J. Histone Deacetylase 3 (HDAC3) and Other Class I HDACs Regulate Colon Cell Maturation and p21 Expression and Are Deregulated in Human Colon Cancer. J. Biol. Chem. 2006, 281, 13548–13558. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Yamashita, H.; Toyama, T.; Sugiura, H.; Ando, Y.; Mita, K.; Hamaguchi, M.; Hara, Y.; Kobayashi, S.; Iwase, H. Quantitation of HDAC1 mRNA Expression in Invasive Carcinoma of the Breast*. Breast Cancer Res. Treat. 2005, 94, 11–16. [Google Scholar] [CrossRef]
- Huang, B.; Laban, M.; Leung, C.H.-W.; Lee, L.; Salto-Tellez, M.; Raju, G.C.; Hooi, S.C. Inhibition of histone deacetylase 2 increases apoptosis and p21 Cip1/WAF1 expression, independent of histone deacetylase. Cell Death Differ. 2005, 12, 395–404. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; Noh, J.H.; Lee, J.H.; Eun, J.W.; Ahn, Y.M.; Kim, S.Y.; Lee, S.H.; Park, W.S.; Yoo, N.J.; Lee, J.Y.; et al. Increased expression of histone deacetylase 2 is found in human gastric cancer. APMIS 2005, 113, 264–268. [Google Scholar] [CrossRef]
- Zhu, P.; Martin, E.; Mengwasser, J.; Schlag, P.; Janssen, K.-P.; Göttlicher, M. Induction of HDAC2 expression upon loss of APC in colorectal tumorigenesis. Cancer Cell 2004, 5, 455–463. [Google Scholar] [CrossRef] [Green Version]
- Wilson, A.J.; Byun, D.-S.; Nasser, S.; Murray, L.B.; Ayyanar, K.; Arango, D.; Figueroa, M.; Melnick, A.; Kao, G.D.; Augenlicht, L.H.; et al. HDAC4 Promotes Growth of Colon Cancer Cells via Repression of p21. Mol. Biol. Cell 2008, 19, 4062–4075. [Google Scholar] [CrossRef] [Green Version]
- Pang, M.; Kothapally, J.; Mao, H.; Ponnusamy, M.; Chin, Y.E.; Zhuang, S. Inhibition of histone deacetylase activity attenuates renal fibroblast activation and interstitial fibrosis in ob-structive nephropathy. Am. J. Physiol. Renal Physiol. 2009, 297, F996–F1005. [Google Scholar] [CrossRef] [Green Version]
- Ellmers, L.J.; Scott, N.J.A.; Piuhola, J.; Maeda, N.; Smithies, O.; Frampton, C.M.; Richards, A.M.; Cameron, V.A. Npr1-regulated gene pathways contributing to cardiac hypertrophy and fibrosis. J. Mol. Endocrinol. 2007, 38, 245–257. [Google Scholar] [CrossRef] [Green Version]
- Qin, L.; Han, Y.-P. Epigenetic Repression of Matrix Metalloproteinases in Myofibroblastic Hepatic Stellate Cells through Histone Deacetylases 4: Implication in Tissue Fibrosis. Am. J. Pathol. 2010, 177, 1915–1928. [Google Scholar] [CrossRef]
- Sanders, Y.Y.; Tollefsbol, T.O.; Varisco, B.M.; Hagood, J.S. Epigenetic Regulation of Thy-1 by Histone Deacetylase Inhibitor in Rat Lung Fibroblasts. Am. J. Respir. Cell Mol. Biol. 2011, 45, 16–23. [Google Scholar] [CrossRef] [Green Version]
- Guo, W.; Shan, B.; Klingsberg, R.C.; Qin, X.; Lasky, J.A. Abrogation of TGF-β1-induced fibroblast-myofibroblast differentiation by histone deacetylase inhibition. Am. J. Physiol. Cell. Mol. Physiol. 2009, 297, L864–L870. [Google Scholar] [CrossRef] [Green Version]
- Glenisson, W.; Castronovo, V.; Waltregny, D. Histone deacetylase 4 is required for TGFβ1-induced myofibroblastic dif-ferentiation. Biochim. Biophys. Acta Mol. Cell Res. 2007, 1773, 1572–1582. [Google Scholar] [CrossRef] [Green Version]
- Korfei, M.; Skwarna, S.; Henneke, I.; MacKenzie, B.; Klymenko, O.; Saito, S.; Ruppert, C.; von der Beck, D.; Mahavadi, P.; Klepetko, W.; et al. Aberrant expression and activity of histone deacetylases in sporadic idiopathic pulmonary fibrosis. Thorax 2015, 70, 1022–1032. [Google Scholar] [CrossRef] [Green Version]
- Ito, K.; Ito, M.; Elliott, W.M.; Cosio, B.G.; Caramori, G.; Kon, O.M.; Barczyk, A.; Hayashi, S.; Adcock, I.; Hogg, J.C.; et al. Decreased Histone Deacetylase Activity in Chronic Obstructive Pulmonary Disease. N. Engl. J. Med. 2005, 352, 1967–1976. [Google Scholar] [CrossRef] [Green Version]
- Stypula-Cyrus, Y.; Damania, D.; Kunte, D.P.; Cruz, M.D.; Subramanian, H.; Roy, H.K.; Backman, V. HDAC Up-Regulation in Early Colon Field Carcinogenesis Is Involved in Cell Tumorigenicity through Regulation of Chromatin Structure. PLoS ONE 2013, 8, e64600. [Google Scholar] [CrossRef] [Green Version]
- Dekker, F.J.; Haisma, H.J. Histone acetyl transferases as emerging drug targets. Drug Discov. Today 2009, 14, 942–948. [Google Scholar] [CrossRef]
- Adenuga, D.; Yao, H.; March, T.H.; Seagrave, J.; Rahman, I. Histone deacetylase 2 is phosphorylated, ubiquitinated, and degraded by cigarette smoke. Am. J. Respir. Cell Mol. Biol. 2009, 40, 464–473. [Google Scholar]
- Chung, K.F.; Adcock, I. Multifaceted mechanisms in COPD: Inflammation, immunity, and tissue repair and destruction. Eur. Respir. J. 2008, 31, 1334–1356. [Google Scholar] [CrossRef]
- Lai, T.; Tian, B.; Cao, C.; Hu, Y.; Zhou, J.; Wang, Y.; Wu, Y.; Li, Z.; Xu, X.; Zhang, M.; et al. HDAC2 Suppresses IL17A-Mediated Airway Remodeling in Human and Experimental Modeling of COPD. Chest 2017, 153, 863–875. [Google Scholar] [CrossRef]
- Gan, L.; Li, C.; Wang, J.; Guo, X. Curcumin modulates the effect of histone modification on the expression of chemokines by type II alveolar epithelial cells in a rat COPD model. Int. J. Chronic Obstr. Pulm. Dis. 2016, 11, 2765–2773. [Google Scholar] [CrossRef] [Green Version]
- Ding, J.; Li, F.; Cong, Y.; Miao, J.; Wu, D.; Liu, B.; Wang, L. Trichostatin A inhibits skeletal muscle atrophy induced by cigarette smoke exposure in mice. Life Sci. 2019, 235, 116800. [Google Scholar] [CrossRef]
- Leus, N.G.; van den Bosch, T.; van der Wouden, P.E.; Krist, K.; Ourailidou, M.E.; Eleftheriadis, N.; Kistemaker, L.E.M.; Bos, S.; Gjaltema, R.A.F.; Mekonnen, S.A.; et al. HDAC1-3 inhibitor MS-275 enhances IL10 expression in RAW264. 7 macrophages and reduces cigarette smoke-induced airway in-flammation in mice. Sci. Rep. 2017, 7, 45047. [Google Scholar] [CrossRef]
- Kim, S.-Y.; Zhang, Q.; Brunmeir, R.; Han, W.; Xu, F. SIRT1 Interacts with and Deacetylates ATP6V1B2 in Mature Adipocytes. PLoS ONE 2015, 10, e0133448. [Google Scholar] [CrossRef] [Green Version]
- Ma, N.; Deng, T.-T.; Wang, Q.; Luo, Z.-L.; Zhu, C.-F.; Qiu, J.-F.; Tang, X.-J.; Huang, M.; Bai, J.; He, Z.-Y.; et al. Erythromycin Regulates Cigarette Smoke-Induced Proinflammatory Mediator Release Through Sirtuin 1-Nuclear Factor κB Axis in Macrophages and Mice Lungs. Pathobiology 2019, 86, 237–247. [Google Scholar] [CrossRef]
- Di Vincenzo, S.; Heijink, I.H.; Noordhoek, J.A.; Cipollina, C.; Siena, L.; Bruno, A.; Ferraro, M.; Postma, D.S.; Gjomarkaj, M.; Pace, E. SIRT 1/FoxO3 axis alteration leads to aberrant immune responses in bronchial epithelial cells. J. Cell. Mol. Med. 2018, 22, 2272–2282. [Google Scholar] [CrossRef] [Green Version]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [Green Version]
- Bernstein, B.E.; Mikkelsen, T.S.; Xie, X.; Kamal, M.; Huebert, D.J.; Cuff, J.; Fry, B.; Meissner, A.; Wernig, M.; Plath, K.; et al. A Bivalent Chromatin Structure Marks Key Developmental Genes in Embryonic Stem Cells. Cell 2006, 125, 315–326. [Google Scholar] [CrossRef] [Green Version]
- Kaneda, R.; Takada, S.; Yamashita, Y.; Choi, Y.L.; Nonaka-Sarukawa, M.; Soda, M.; Misawa, Y.; Isomura, T.; Shimada, K.; Mano, H. Genome-wide histone methylation profile for heart failure. Genes Cells 2008, 14, 69–77. [Google Scholar] [CrossRef]
- Ke, X.-S.; Qu, Y.; Rostad, K.; Li, W.-C.; Lin, B.; Halvorsen, O.J.; Haukaas, S.A.; Jonassen, I.; Petersen, K.; Goldfinger, N.; et al. Genome-Wide Profiling of Histone H3 Lysine 4 and Lysine 27 Trimethylation Reveals an Epigenetic Signature in Prostate Carcinogenesis. PLoS ONE 2009, 4, e4687. [Google Scholar] [CrossRef] [Green Version]
- Sandgren, J.; Andersson, R.; Rada-Iglesias, A.; Enroth, S.; Åkerström, G.; Dumanski, J.P.; Komorowski, J.; Westin, G.; Wadelius, C. Integrative epigenomic and genomic analysis of malignant pheochromocytoma. Exp. Mol. Med. 2010, 42, 484–502. [Google Scholar] [CrossRef] [Green Version]
- Yildirim, A.O.; Bulau, P.; Zakrzewicz, D.; Kitowska, K.E.; Weissmann, N.; Grimminger, F. Increased protein arginine methylation in chronic hypoxia: Role of protein arginine methyltransferases. Am. J. Respir. Cell Mol. 2006, 35, 436–443. [Google Scholar] [CrossRef]
- Andresen, E.; Günther, G.; Bullwinkel, J.; Lange, C.; Heine, H. Increased Expression of Beta-Defensin 1 (DEFB1) in Chronic Obstructive Pulmonary Disease. PLoS ONE 2011, 6, e21898. [Google Scholar] [CrossRef]
- He, X.; Li, T.; Luo, L.; Zeng, H.; Chen, Y.; Cai, S. PRMT6 mediates inflammation via activation of the NF-κB/p65 pathway on a cigarette smoke extract-induced murine emphysema model. Tob. Induc. Dis. 2020, 18. [Google Scholar] [CrossRef]
- Sessa, R.; Hata, A. Role of microRNAs in Lung Development and Pulmonary Diseases. Pulm. Circ. 2013, 3, 315–328. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Zhao, X.; Shan, H.; Liang, H. MicroRNAs in idiopathic pulmonary fibrosis: Involvement in pathogenesis and potential use in diagnosis and therapeutics. Acta Pharm. Sin. B 2016, 6, 531–539. [Google Scholar] [CrossRef] [Green Version]
- Miao, C.; Xiong, Y.; Zhang, G.; Chang, J. MicroRNAs in idiopathic pulmonary fibrosis, new research progress and their pathophysiological implication. Exp. Lung Res. 2018, 44, 178–190. [Google Scholar] [CrossRef]
- Li, Y.; Huang, J.; Hu, C.; Zhou, J.; Xu, D.; Hou, Y.; Wu, C.; Zhao, J.; Li, M.; Zeng, X.; et al. MicroRNA-320a: An important regulator in the fibrotic process in interstitial lung disease of systemic sclerosis. Arthritis Res. 2021, 23, 1–11. [Google Scholar] [CrossRef]
- Bartczak, K.; Białas, A.J.; Kotecki, M.J.; Górski, P.; Piotrowski, W.J. More than a Genetic Code: Epigenetics of Lung Fibrosis. Mol. Diagn. Ther. 2020, 24, 665–681. [Google Scholar] [CrossRef]
- Yang, G.; Yang, L.; Wang, W.; Wang, J.; Wang, J.; Xu, Z. Discovery and validation of extracellular/circulating microRNAs during idiopathic pulmonary fibrosis disease progression. Gene 2015, 562, 138–144. [Google Scholar] [CrossRef]
- Ezzie, M.E.; Crawford, M.; Cho, J.-H.; Orellana, R.; Zhang, S.; Gelinas, R.; Batte, K.; Yu, L.; Nuovo, G.; Galas, D.; et al. Gene expression networks in COPD: microRNA and mRNA regulation. Thorax 2011, 67, 122–131. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.; Xin, Q.; Chai, R.; Liu, L.; Ma, Z. Ectopic expressed miR-203 contributes to chronic obstructive pulmonary disease via targeting TAK1 and PIK3CA. Int. J. Clin. Exp. Pathol. 2015, 8, 10662–10670. [Google Scholar]
- Shen, W.; Liu, J.; Zhao, G.; Fan, M.; Song, G.; Zhang, Y.; Weng, Z.; Zhang, Y. Repression of Toll-like receptor-4 by microRNA-149-3p is associated with smoking-related COPD. Int. J. Chronic Obstr. Pulm. Dis. 2017, 12, 705–715. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Feng, J.; Zhang, L.; Zhao, F.; Li, M.; Du, Y.; Li, Y.; Wu, Q.; Li, G. One functional variant in the 3′-untranslated region of TLR4 is associated with the elevated risk of ventilator-associated pneumonia in the patients with chronic obstructive pulmonary disease. J. Cell. Physiol. 2019, 234, 18879–18886. [Google Scholar] [CrossRef]
- Pottelberge, G.R.V.; Mestdagh, P.; Bracke, K.R.; Thas, O.; Durme, Y.M.V.; Joos, G.F.; Vandesompele, J.; Brusselle, G.G. MicroRNA expression in induced sputum of smokers and patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2011, 183, 898–906. [Google Scholar] [CrossRef]
- Sun, Y.; An, N.; Li, J.; Xia, J.; Tian, Y.; Zhao, P.; Liu, X.; Huang, H.; Gao, J.; Zhang, X. miRNA-206 regulates human pulmonary microvascular endothelial cell apoptosis via targeting in chronic obstructive pulmonary disease. J. Cell. Biochem. 2018, 120, 6223–6236. [Google Scholar] [CrossRef]
- Long, Y.-J.; Liu, X.-P.; Chen, S.-S.; Zong, D.-D.; Chen, Y.; Chen, P. miR-34a is involved in CSE-induced apoptosis of human pulmonary microvascular endothelial cells by targeting Notch-1 receptor protein. Respir. Res. 2018, 19, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Sekine, Y.; Katsura, H.; Koh, E.; Hiroshima, K.; Fujisawa, T. Early detection of COPD is important for lung cancer surveillance. Eur. Respir. J. 2011, 39, 1230–1240. [Google Scholar] [CrossRef] [Green Version]
- Tellez, C.S.; Juri, D.E.; Do, K.; Bernauer, A.M.; Thomas, C.L.; Damiani, L.A.; Tessema, M.; Leng, S.; Belinsky, S.A. EMT and Stem Cell–Like Properties Associated with miR-205 and miR-200 Epigenetic Silencing Are Early Manifestations during Carcinogen-Induced Transformation of Human Lung Epithelial Cells. Cancer Res. 2011, 71, 3087–3097. [Google Scholar] [CrossRef] [Green Version]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol. 2008, 10, 593–601. [Google Scholar] [CrossRef]
- Christenson, S.A.; Brandsma, C.A.; Campbell, J.D.; Knit, D.A.; Pechkovsky, D.V.; Hogg, J.C.; Times, W.; Postma, D.S.; Lenburg, M.; Spira, A. miR-638 regulates gene expression networks associated with emphysematous lung destruction. Genome Med. 2013, 5, 114. [Google Scholar] [CrossRef] [Green Version]
- Francis, S.M.S.; Davidson, M.R.; Tan, M.E.; Wright, C.M.; Clarke, B.E.; Duhig, E.E.; Bowman, R.V.; Hayward, N.; Fong, K.M.; Yang, I.A. MicroRNA-34c is associated with emphysema severity and modulates SERPINE1 expression. BMC Genom. 2014, 15, 88. [Google Scholar] [CrossRef] [Green Version]
- Avci, E.; Balci-Peynircioglu, B. An overview of exosomes: From biology to emerging roles in immune response. Acta Medica. 2016, 5, 2–10. [Google Scholar]
- Beckler, M.D.; Higginbotham, J.N.; Franklin, J.L.; Ham, A.-J.; Halvey, P.J.; Imasuen, I.E.; Whitwell, C.; Li, M.; Liebler, D.; Coffey, R.J. Proteomic Analysis of Exosomes from Mutant KRAS Colon Cancer Cells Identifies Intercellular Transfer of Mutant KRAS. Mol. Cell. Proteom. 2013, 12, 343–355. [Google Scholar] [CrossRef] [Green Version]
- Skogberg, G.; Gudmundsdottir, J.; van der Post, S.; Sandström, K.; Bruhn, S.; Benson, M.; Mincheva-Nilsson, L.; Baranov, V.; Telemo, S.; Ekwall, O. Characterization of human thymic exosomes. PLoS ONE 2013, 8, e675548. [Google Scholar] [CrossRef]
- Moon, H.-G.; Kim, S.-H.; Gao, J.; Quan, T.; Qin, Z.; Osorio, J.C.; Rosas, I.O.; Wu, M.; Tesfaigzi, Y.; Jin, Y. CCN1 secretion and cleavage regulate the lung epithelial cell functions after cigarette smoke. Am. J. Physiol. Cell. Mol. Physiol. 2014, 307, L326–L337. [Google Scholar] [CrossRef] [Green Version]
- Cordazzo, C.; Petrini, S.; Neri, T.; Lombardi, S.; Carmazzi, Y.; Pedrinelli, R.; Paggiaro, P.; Celi, A. Rapid shedding of proin-flammatory microparticles by human mononuclear cells exposed to cigarette smoke is dependent on Ca2+ mobilization. In-flamm. Res. 2014, 63, 539–547. [Google Scholar] [CrossRef]
- Fujita, Y.; Araya, J.; Ito, S.; Kobayashi, K.; Kosaka, N.; Yoshioka, Y.; Kadota, T.; Hara, H.; Kuwano, K.; Ochiya, T. Suppression of autophagy by extracellular vesicles promotes myofibroblast differentiation in COPD pathogenesis. J. Extracell. Vesicles 2015, 4, 28388. [Google Scholar] [CrossRef]
- Serban, K.A.; Rezania, S.; Petrusca, D.N.; Poirier, C.; Cao, D.; Justice, M.J.; Patel, M.; Tsvetkova, I.; Kamocki, K.; Mikosz, A.; et al. Structural and functional characterization of endothelial microparticles released by cigarette smoke. Sci. Rep. 2016, 6, 31596. [Google Scholar] [CrossRef]
- Collard, H.R.; Ward, A.J.; Lanes, S.; Hayflinger, D.C.; Rosenberg, D.M.; Hunsche, E. Burden of illness in idiopathic pulmonary fibrosis. J. Med. Econ. 2012, 15, 829–835. [Google Scholar] [CrossRef]
- Archontogeorgis, K.; Steiropoulos, P.; Tzouvelekis, A.; Nena, E.; Bouros, D. Lung Cancer and Interstitial Lung Diseases: A Systematic Review. Pulm. Med. 2012, 2012, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Kim, D.S.; Shim, T.S.; Lim, C.M.; Koh, Y.; Lee, S.D.; Kim, W.S.; Kim, W.D.; Lee, J.S.; Song, K. Lung cancer in patients with idiopathic pulmonary fibrosis. Eur. Respir. J. 2001, 17, 1216–1219. [Google Scholar] [CrossRef] [Green Version]
- Nathan, S.D.; Noble, P.W.; Tuder, R.M. Idiopathic pulmonary fibrosis and pulmonary hypertension: Connecting the dots. Am. J. Respir. Crit. Care Med. 2007, 175, 875–880. [Google Scholar] [CrossRef]
- Cottin, V.; Nunes, H.; Brillet, P.-Y.; Devouassoux, G.; Tillie-Leblond, I.; Israel-Biet, D.; Court-Fortune, I.; Valeyre, D.; Cordier, J.-F. Combined pulmonary fibrosis and emphysema: A distinct underrecognised entity. Eur. Respir. J. 2005, 26, 586–593. [Google Scholar] [CrossRef]
- Nathan, S.D.; Basavaraj, A.; Reichner, C.; Shlobin, O.A.; Ahmad, S.; Kiernan, J.; Burton, N.; Barnett, S.D. Prevalence and impact of coronary artery disease in idiopathic pulmonary fibrosis. Respir. Med. 2010, 104, 1035–1041. [Google Scholar] [CrossRef] [Green Version]
- Zisman, D.A.; Kawut, S.M. Idiopathic pulmonary fibrosis: A shot through the heart? Am. J. Respir. Crit. Care Med. 2008, 178, 1192–1193. [Google Scholar] [CrossRef]
- Shorr, A.F.; Wainright, J.L.; Cors, C.S.; Lettieri, C.J.; Nathan, S.D. Pulmonary hypertension in patients with pulmonary fibrosis awaiting lung transplant. Eur. Respir. J. 2007, 30, 715–721. [Google Scholar] [CrossRef] [Green Version]
- Somogyi, V.; Chaudhuri, N.; Torrisi, N.; Kahn, S.E.; Müller, V.; Kreuter, M. The therapy of idiopathic pulmonary fibrosis: What is next? Eur. Respir. Rev. 2019, 28, 190021. [Google Scholar] [CrossRef] [Green Version]
- Falkenberg, K.J.; Johnstone, R. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat. Rev. Drug Discov. 2014, 13, 673–691. [Google Scholar] [CrossRef]
- Liao, D. Profiling technologies for the identification and characterization of small-molecule histone deacetylase inhibitors. Drug Discov. Today: Technol. 2015, 18, 24–28. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Hao, D.; Wang, L.; Wang, H.; Wang, Y.; Zhao, Z.; Li, P.; Deng, C.; Di, L.-J. Epigenetic targeting drugs potentiate chemotherapeutic effects in solid tumor therapy. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [Green Version]
- De Groote, M.L.; Verschure, P.J.; Rots, M.G. Epigenetic Editing: Targeted rewriting of epigenetic marks to modulate expression of selected target genes. Nucleic Acids Res. 2012, 40, 10596–10613. [Google Scholar] [CrossRef]
- Wu, D.-D.; Song, J.; Bartel, S.; Krauss-Etschmann, S.; Rots, M.G.; Hylkema, M.N. The potential for targeted rewriting of epigenetic marks in COPD as a new therapeutic approach. Pharmacol. Ther. 2018, 182, 1–14. [Google Scholar] [CrossRef]
- Wang, Y.; Su, H.H.; Yang, Y.; Hu, Y.; Zhang, L.; Blancafort, P.; Huang, L. Systemic delivery of modified mRNA encoding herpes simplex virus 1 thymidine kinase for targeted cancer gene therapy. Mol. Ther. 2021, 21, 358–367. [Google Scholar] [CrossRef]
- Chu, H.W.; Rios, C.; Huang, C.; Wesolowska-Andersen, A.; Burchard, E.G.; O’Connor, B.P.; Fingerlin, T.E.; Nichols, D.; Reynolds, S.D.; Seibold, M.A. CRISPR–Cas9-mediated gene knockout in primary human airway epithelial cells reveals a proinflammatory role for MUC18. Gene Ther. 2015, 22, 822–829. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.-H.; Wu, L.-Z.; Liang, H.-L.; Yang, Y.; Qiu, J.; Kan, Q.; Zhu, W.; Ma, C.-L.; Zhou, X.-Y. Pulmonary surfactant synthesis in miRNA-26a-1/miRNA-26a-2 double knockout mice generated using the CRISPR/Cas9 system. Am. J. Transl. Resv. 2017, 9, 355–365. [Google Scholar]
- Wu, Q.; Jiang, D.; Matsuda, J.; Ternyak, K.; Zhang, B.; Chu, H.W. Cigarette Smoke Induces Human Airway Epithelial Senescence via Growth Differentiation Factor 15 Production. Am. J. Respir. Cell Mol. Biol. 2016, 55, 429–438. [Google Scholar] [CrossRef]
- Song, J.; Cano-Rodriquez, D.; Winkle, M.; Gjaltema, R.A.F.; Goubert, D.; Jurkowski, T.P.; Heijink, I.H.; Rots, M.G.; Hylkema, M.N. Targeted epigenetic editing of SPDEF reduces mucus production in lung epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 312, L334–L347. [Google Scholar] [CrossRef] [Green Version]
- Sondhi, D.; Stiles, K.M.; De, B.P.; Crystal, R.G. Genetic Modification of the Lung Directed Toward Treatment of Human Disease. Hum. Gene Ther. 2017, 28, 3–84. [Google Scholar] [CrossRef]
- Nemerow, G.R.; Stewart, P.L. Insights into Adenovirus Uncoating from Interactions with Integrins and Mediators of Host Immunity. Viruses 2016, 8, 337. [Google Scholar] [CrossRef] [Green Version]
- Alton, E.W.F.W.; Beekman, J.M.; Boyd, A.C.; Brand, J.; Carlon, M.S.; Connolly, M.M.; Chan, M.; Conlon, S.; Davidson, H.E.; Davies, J.C.; et al. Preparation for a first-in-man lentivirus trial in patients with cystic fibrosis. Thorax 2016, 72, 137–147. [Google Scholar] [CrossRef] [Green Version]
- Mastorakos, P.; da Silva, A.L.; Chisholm, J.; Song, E.; Choi, W.K.; Boyle, M.; Morales, M.M.; Hanes, J.; Suk, J.S. Highly compacted biodegradable DNA nanoparticles capable of overcoming the mucus barrier for inhaled lung gene therapy. Proc. Natl. Acad. Sci. USA 2015, 112, 8720–8725. [Google Scholar] [CrossRef] [Green Version]
- Suk, J.S.; Kim, A.J.; Trehan, K.; Schneider, C.S.; Cebotaru, L.; Woodward, O.M.; Boylan, N.J.; Boyle, M.; Lai, S.K.; Guggino, W.B.; et al. Lung gene therapy with highly compacted DNA nanoparticles that overcome the mucus barrier. J. Control. Release 2014, 178, 8–17. [Google Scholar] [CrossRef] [Green Version]
- Mahiny, A.J.; Dewerth, A.; Mays, L.E.; Alkhaled, M.; Mothes, B.; Malaeksefat, E.; Loretz, B.; Rottenberger, J.; Brosch, D.M.; Reautschnig, P.; et al. In vivo genome editing using nuclease-encoding mRNA corrects SP-B deficiency. Nat. Biotechnol. 2015, 33, 584–586. [Google Scholar] [CrossRef]
- Al-Qadi, S.; Grenha, A.; Carrión-Recio, D.; Seijo, B.; Remuñán-López, C. Microencapsulated chitosan nanoparticles for pulmonary protein delivery: In vivo evaluation of insulin-loaded formulations. J. Control. Release 2011, 157, 383–390. [Google Scholar] [CrossRef]
- Mohamed, A.; Pekoz, A.Y.; Ross, K.; Hutcheon, G.A.; Saleem, I.Y. Pulmonary delivery of Nanocomposite Microparticles (NCMPs) incorporating miR-146a for treatment of COPD. Int. J. Pharm. 2019, 569, 118524. [Google Scholar] [CrossRef]
- Kim, J.; Kim, D.Y.; Heo, H.-R.; Choi, S.S.; Hong, S.-H.; Kim, W.J. Role of miRNA-181a-2-3p in cadmium-induced inflammatory responses of human bronchial epithelial cells. J. Thorac. Dis. 2019, 11, 3055–3069. [Google Scholar] [CrossRef]
- Musri, M.M.; Coll-Bonfill, N.; Maron, B.A.; Peinado, V.I.; Wang, R.-S.; Altirriba, J.; Blanco, I.; Oldham, W.M.; Tura-Ceide, O.; García-Lucio, J.; et al. MicroRNA Dysregulation in Pulmonary Arteries from Chronic Obstructive Pulmonary Disease. Relationships with Vascular Remodeling. Am. J. Respir. Cell Mol. Biol. 2018, 59, 490–499. [Google Scholar] [CrossRef]
- Wang, R.; Xu, J.; Liu, H.; Zhao, Z. Peripheral leukocyte microRNAs as novel biomarkers for COPD. Int. J. Chronic Obstr. Pulm. Dis. 2017, 12, 1101–1112. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Avci, E.; Sarvari, P.; Savai, R.; Seeger, W.; Pullamsetti, S.S. Epigenetic Mechanisms in Parenchymal Lung Diseases: Bystanders or Therapeutic Targets? Int. J. Mol. Sci. 2022, 23, 546. https://doi.org/10.3390/ijms23010546
Avci E, Sarvari P, Savai R, Seeger W, Pullamsetti SS. Epigenetic Mechanisms in Parenchymal Lung Diseases: Bystanders or Therapeutic Targets? International Journal of Molecular Sciences. 2022; 23(1):546. https://doi.org/10.3390/ijms23010546
Chicago/Turabian StyleAvci, Edibe, Pouya Sarvari, Rajkumar Savai, Werner Seeger, and Soni S. Pullamsetti. 2022. "Epigenetic Mechanisms in Parenchymal Lung Diseases: Bystanders or Therapeutic Targets?" International Journal of Molecular Sciences 23, no. 1: 546. https://doi.org/10.3390/ijms23010546
APA StyleAvci, E., Sarvari, P., Savai, R., Seeger, W., & Pullamsetti, S. S. (2022). Epigenetic Mechanisms in Parenchymal Lung Diseases: Bystanders or Therapeutic Targets? International Journal of Molecular Sciences, 23(1), 546. https://doi.org/10.3390/ijms23010546