
Targeting Cancer with CRISPR/Cas9-Based Therapy


Abstract
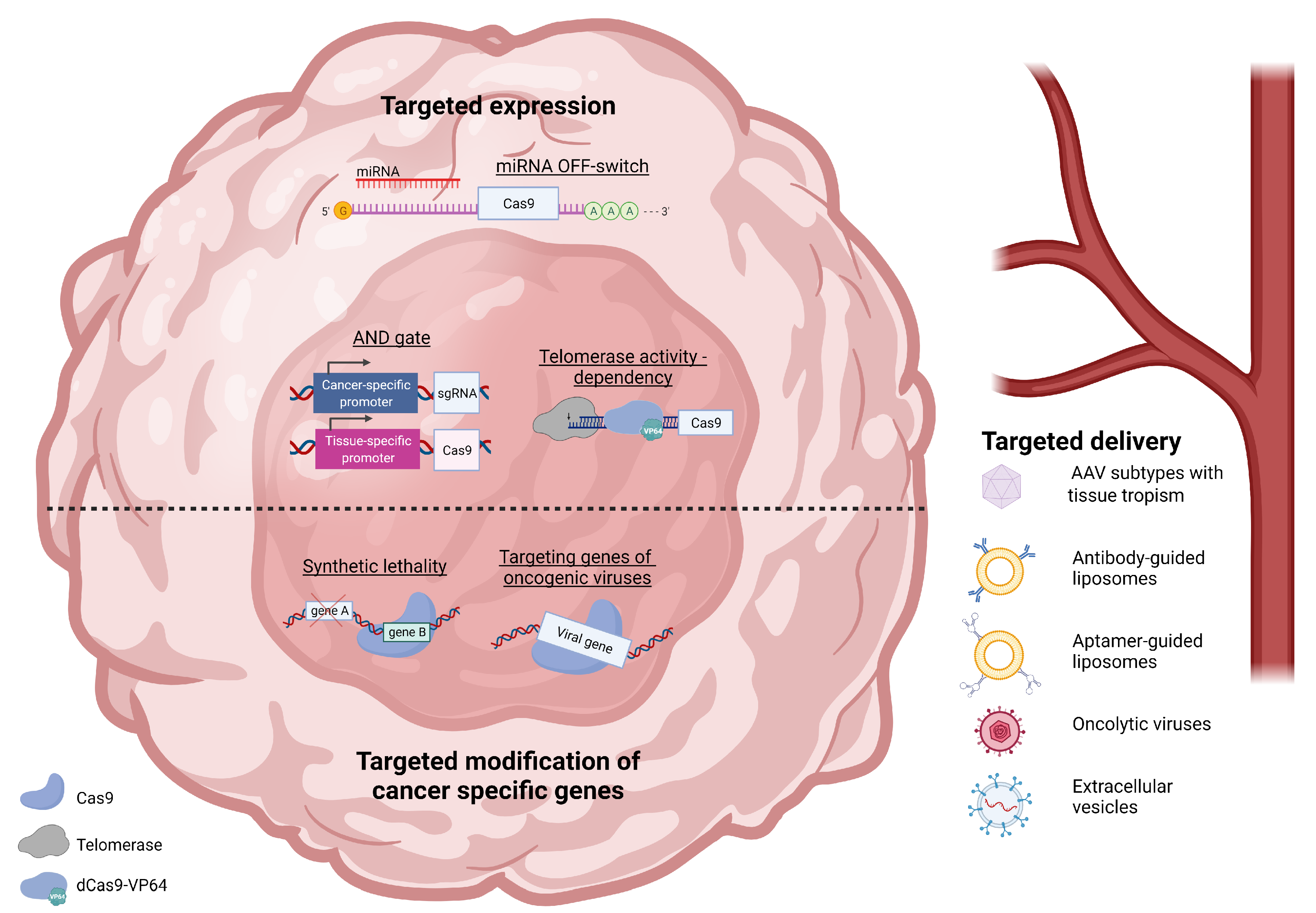
:1. Introduction

{kind=link}
| Cas9 Modification | Application |
|---|---|
| Cas9 nickase | Creates a single-stranded DNA break. Pairing two nickases to target both DNA strands reduces off-target activity and indels introduction |
| deadCas9 (dCas9) | Harbours no nuclease activity, serves as a tool for delivering other active compounds and proteins to selected DNA sequence |
| dCas9 with KRAB domain | Inhibits expression of downstream gene |
| dCas9 with VP64 | Activates expression of downstream gene |
| dCas9 with methylase/demethylase or histone deacethylase | Provides control over epigenetic regulation |
| Cas9 nickase with adenine or cytosine deaminase (Base editors; BEs) | Enables transition substitutions (A>G, T>C, G>A or C>T) with high efficiency and no double-strand DNA breaks |
| Cas9 nickase with a reverse transcription domain (Prime editors; PEs) | Facilitates introduction of any substitution, insertion or deletion mutation with high efficiency and no double-strand DNA breaks |
2. Targeted Delivery
2.1. Adeno-Associated Virus Serotypes
2.2. Oncolytic Viruses
2.3. Antibody- or Aptamer-Guided Lipid-Based Vectors
2.4. Extracellular Vesicles
3. Targeted Expression
3.1. Promoters Specific to Certain Type of Cancer Cells (AND Gate)
3.2. Expression Dependent on Telomerase Activity
3.3. CRISPR/Cas9 Switch Responsive to Micro-RNA
3.4. Expression Activated by Light
4. Targeted Modification of Cancer-Specific Sequences
4.1. Synthetic Lethality
4.2. Oncogenic Viruses
4.3. TERT Promoter Mutation
5. Control over CRISPR/Cas9 Activity with Proteases
6. CRISPR/Cas9 Modification of Non-Cancerous Cells Targeted at Defeating Cancer
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Main Causes of Mortality|Health at a Glance: Europe 2020: State of Health in the EU Cycle|OECD iLibrary. Available online: https://www.oecd-ilibrary.org/sites/82129230-en/1/3/2/1/4/index.html?itemId=/content/publication/82129230-en&_csp_=e7f5d56a7f4dd03271a59acda6e2be1b&itemIGO=oecd&itemContentType=book (accessed on 3 October 2021).
- WHO/Europe|Cancer. Available online: https://www.euro.who.int/en/health-topics/noncommunicable-diseases/cancer/cancer (accessed on 3 October 2021).
- Moses, C.; Garcia-Bloj, B.; Harvey, A.R.; Blancafort, P. Hallmarks of cancer: The CRISPR generation. Eur. J. Cancer 2018, 93, 10–18. [Google Scholar] [CrossRef] [Green Version]
- White, M.K.; Khalili, K. CRISPR/Cas9 and cancer targets: Future possibilities and present challenges. Oncotarget 2016, 7, 12305–12317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pon, J.R.; Marra, M.A. Driver and Passenger Mutations in Cancer. Annu. Rev. Pathol. Mech. Dis. 2015, 10, 25–50. [Google Scholar] [CrossRef]
- Fisher, R.; Pusztai, L.; Swanton, C. Cancer heterogeneity: Implications for targeted therapeutics. Br. J. Cancer 2013, 108, 479–485. [Google Scholar] [CrossRef] [Green Version]
- Treatment for Cancer|Cancer in General|Cancer Research UK. Available online: https://www.cancerresearchuk.org/about-cancer/cancer-in-general/treatment (accessed on 26 November 2021).
- Cross, D.; Burmester, J.K. Gene Therapy for Cancer Treatment: Past, Present and Future. Clin. Med. Res. 2006, 4, 218–227. [Google Scholar] [CrossRef] [Green Version]
- Hazafa, A.; Mumtaz, M.; Farooq, M.F.; Bilal, S.; Chaudhry, S.N.; Firdous, M.; Naeem, H.; Ullah, M.O.; Yameen, M.; Mukhtiar, M.S.; et al. CRISPR/Cas9: A powerful genome editing technique for the treatment of cancer cells with present challenges and future directions. Life Sci. 2020, 263, 118525. [Google Scholar] [CrossRef]
- Zhang, M.; Eshraghian, E.A.; Jammal, O.A.; Zhang, Z.; Zhu, X. CRISPR technology: The engine that drives cancer therapy. Biomed. Pharmacother. 2021, 133, 111007. [Google Scholar] [CrossRef] [PubMed]
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR Provides Acquired Resistance Against Viruses in Prokaryotes. Science 2007, 315, 1709–1712. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.D.; Lander, E.S.; Zhang, F. Development and Applications of CRISPR-Cas9 for Genome Engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Mao, A.; Xu, M.; Weng, Q.; Mao, J.; Ji, J. CRISPR-Cas9 for cancer therapy: Opportunities and challenges. Cancer Lett. 2019, 447, 48–55. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Boland, J.; Nedelcu, E. CRISPR/Cas9 for the Clinician: Current uses of gene editing and applications for new therapeutics in oncology. Perm. J. 2020, 24, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.H.; Miller, S.M.; Geurts, M.H.; Tang, W.; Chen, L.; Sun, N.; Zeina, C.M.; Gao, X.; Rees, H.A.; Lin, Z.; et al. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature 2018, 556, 57–63. [Google Scholar] [CrossRef]
- Saber, A.; Liu, B.; Ebrahimi, P.; Haisma, H.J. CRISPR/Cas9 for overcoming drug resistance in solid tumors. DARU J. Pharm. Sci. 2020, 28, 295–304. [Google Scholar] [CrossRef]
- Liu, B.; Saber, A.; Haisma, H.J. CRISPR/Cas9: A powerful tool for identification of new targets for cancer treatment. Drug Discov. Today 2019, 24, 955–970. [Google Scholar] [CrossRef]
- Biagioni, A.; Chillà, A.; Andreucci, E.; Laurenzana, A.; Margheri, F.; Peppicelli, S.; Del Rosso, M.; Fibbi, G. Type II CRISPR/Cas9 approach in the oncological therapy. J. Exp. Clin. Cancer Res. 2017, 36, 80. [Google Scholar] [CrossRef]
- Cheng, X.; Fan, S.; Wen, C.; Du, X. CRISPR/Cas9 for cancer treatment: Technology, clinical applications and challenges. Briefings Funct. Genom. 2020, 19, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Sheriff, A.; Brooks, I.; Jacków, J. Gene Editing Therapies for Genodermatoses. In New Technologies in Dermatological Science and Practice; CRC Press: Boca Raton, FL, USA, 2021; pp. 121–135. [Google Scholar] [CrossRef]
- Skarnes, W.C.; Pellegrino, E.; McDonough, J.A. Improving homology-directed repair efficiency in human stem cells. Methods 2019, 164–165, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Lin, C.Y.; Gootenberg, J.S.; Konermann, S.; Trevino, A.E.; Scott, D.A.; Inoue, A.; Matoba, S.; Zhang, Y.; et al. Double Nicking by RNA-Guided CRISPR Cas9 for Enhanced Genome Editing Specificity. Cell 2013, 154, 1380–1389. [Google Scholar] [CrossRef] [Green Version]
- Jacków, J.; Guo, Z.; Hansen, C.; Abaci, H.E.; Doucet, Y.S.; Shin, J.U.; Hayashi, R.; DeLorenzo, D.; Kabata, Y.; Shinkuma, S.; et al. CRISPR/Cas9-based targeted genome editing for correction of recessive dystrophic epidermolysis bullosa using iPS cells. Proc. Natl. Acad. Sci. USA 2019, 116, 26846–26852. [Google Scholar] [CrossRef]
- Kocher, T.; Wagner, R.N.; Klausegger, A.; Guttmann-Gruber, C.; Hainzl, S.; Bauer, J.W.; Reichelt, J.; Koller, U. Improved Double-Nicking Strategies for COL7A1-Editing by Homologous Recombination. Mol. Ther.-Nucleic Acids 2019, 18, 496–507. [Google Scholar] [CrossRef] [Green Version]
- Weng, A.P.; Ferrando, A.A.; Lee, W.; Morris, J.P.; Silverman, L.B.; Sanchez-Irizarry, C.; Blacklow, S.C.; Look, A.T.; Aster, J.C. Activating Mutations of NOTCH1 in Human T Cell Acute Lymphoblastic Leukemia. Science 2004, 306, 269–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misiorek, J.O.; Przybyszewska-Podstawka, A.; Kałafut, J.; Paziewska, B.; Rolle, K.; Rivero-Müller, A.; Nees, M. Context Matters: NOTCH Signatures and Pathway in Cancer Progression and Metastasis. Cells 2021, 10, 94. [Google Scholar] [CrossRef] [PubMed]
- Cheung, A.H.K.; Chow, C.; Zhang, J.; Zhou, Y.; Huang, T.; Ng, K.C.K.; Or, T.C.T.; Yao, Y.Y.; Dong, Y.; Fung, J.M.W.; et al. Specific targeting of point mutations in EGFR L858R-positive lung cancer by CRISPR/Cas9. Lab. Investig. 2018, 98, 968–976. [Google Scholar] [CrossRef] [Green Version]
- Perincheri, S.; Hui, P. KRAS mutation testing in clinical practice. Expert Rev. Mol. Diagn. 2015, 15, 375–384. [Google Scholar] [CrossRef]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anzalone, A.V.; Koblan, L.W.; Liu, D.R. Genome editing with CRISPR–Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. 2020, 38, 824–844. [Google Scholar] [CrossRef] [PubMed]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef]
- Matsoukas, I.G. Prime Editing: Genome Editing for Rare Genetic Diseases Without Double-Strand Breaks or Donor DNA. Front. Genet. 2020, 11, 528. [Google Scholar] [CrossRef]
- Chen, Z.H.; Yu, Y.P.; Zuo, Z.H.; Nelson, J.B.; Michalopoulos, G.K.; Monga, S.; Liu, S.; Tseng, G.; Luo, J.H. Targeting genomic rearrangements in tumor cells through Cas9-mediated insertion of a suicide gene. Nat. Biotechnol. 2017, 35, 543–550. [Google Scholar] [CrossRef] [Green Version]
- Kwon, D.Y.; Zhao, Y.T.; Lamonica, J.M.; Zhou, Z. Locus-specific histone deacetylation using a synthetic CRISPR-Cas9-based HDAC. Nat. Commun. 2017, 8, 15315. [Google Scholar] [CrossRef] [PubMed]
- Yao, S.; He, Z.; Chen, C. CRISPR/Cas9-Mediated Genome Editing of Epigenetic Factors for Cancer Therapy. Hum. Gene Ther. 2015, 26, 463–471. [Google Scholar] [CrossRef]
- Wang, J.; Balan, V.; Marincola, F. CRISPR technology for immuno-oncology applications. In Methods in Enzymology, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2020; Volume 635, pp. 251–266. [Google Scholar] [CrossRef]
- Azangou-Khyavy, M.; Ghasemi, M.; Khanali, J.; Boroomand-Saboor, M.; Jamalkhah, M.; Soleimani, M.; Kiani, J. CRISPR/Cas: From Tumor Gene Editing to T Cell-Based Immunotherapy of Cancer. Front. Immunol. 2020, 11, 2062. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zheng, W.; Liu, S.; Li, B.; Jiang, X. Delivery of CRISPR/Cas9 by Novel Strategies for Gene Therapy. ChemBioChem 2018, 20, cbic.201800629. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhang, L.; Liu, H.; Cheng, K. Delivery strategies of the CRISPR-Cas9 gene-editing system for therapeutic applications. J. Control. Release 2017, 266, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Alphonse, M.; Liu, Q. Strategies for nonviral nanoparticle-based delivery of CRISPR/Cas9 therapeutics. WIREs Nanomed. Nanobiotechnol. 2020, 12, e1609. [Google Scholar] [CrossRef]
- Xu, X.; Wan, T.; Xin, H.; Li, D.; Pan, H.; Wu, J.; Ping, Y. Delivery of CRISPR/Cas9 for therapeutic genome editing. J. Gene Med. 2019, 21, e3107. [Google Scholar] [CrossRef] [Green Version]
- Yip, B. Recent Advances in CRISPR/Cas9 Delivery Strategies. Biomolecules 2020, 10, 839. [Google Scholar] [CrossRef]
- Naso, M.F.; Tomkowicz, B.; Perry, W.L.; Strohl, W.R. Adeno-Associated Virus (AAV) as a Vector for Gene Therapy. BioDrugs 2017, 31, 317–334. [Google Scholar] [CrossRef] [Green Version]
- Zincarelli, C.; Soltys, S.; Rengo, G.; Rabinowitz, J.E. Analysis of AAV Serotypes 1–9 Mediated Gene Expression and Tropism in Mice After Systemic Injection. Mol. Ther. 2008, 16, 1073–1080. [Google Scholar] [CrossRef]
- Chamberlain, K.; Riyad, J.M.; Weber, T. Expressing Transgenes That Exceed the Packaging Capacity of Adeno-Associated Virus Capsids. Hum. Gene Ther. Methods 2016, 27, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Ran, F.A.; Cong, L.; Yan, W.X.; Scott, D.A.; Gootenberg, J.S.; Kriz, A.J.; Zetsche, B.; Shalem, O.; Wu, X.; Makarova, K.S.; et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature 2015, 520, 186–191. [Google Scholar] [CrossRef]
- Yang, Y.; Xu, J.; Ge, S.; Lai, L. CRISPR/Cas: Advances, Limitations, and Applications for Precision Cancer Research. Front. Med. 2021, 8, 649896. [Google Scholar] [CrossRef]
- Kim, E.; Koo, T.; Park, S.W.; Kim, D.; Kim, K.; Cho, H.Y.; Song, D.W.; Lee, K.J.; Jung, M.H.; Kim, S.; et al. In vivo genome editing with a small Cas9 orthologue derived from Campylobacter jejuni. Nat. Commun. 2017, 8, 14500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mondal, M.; Guo, J.; He, P.; Zhou, D. Recent advances of oncolytic virus in cancer therapy. Hum. Vaccines Immunother. 2020, 16, 2389–2402. [Google Scholar] [CrossRef] [PubMed]
- Raja, J.; Ludwig, J.M.; Gettinger, S.N.; Schalper, K.A.; Kim, H.S. Oncolytic virus immunotherapy: Future prospects for oncology. J. Immunother. Cancer 2018, 6, 140. [Google Scholar] [CrossRef]
- Fukuhara, H.; Ino, Y.; Todo, T. Oncolytic virus therapy: A new era of cancer treatment at dawn. Cancer Sci. 2016, 107, 1373–1379. [Google Scholar] [CrossRef] [PubMed]
- Phelps, M.P.; Yang, H.; Patel, S.; Rahman, M.M.; McFadden, G.; Chen, E. Oncolytic Virus-Mediated RAS Targeting in Rhabdomyosarcoma. Mol. Ther.-Oncolytics 2018, 11, 52–61. [Google Scholar] [CrossRef] [Green Version]
- Zylberberg, C.; Gaskill, K.; Pasley, S.; Matosevic, S. Engineering liposomal nanoparticles for targeted gene therapy. Gene Ther. 2017, 24, 441–452. [Google Scholar] [CrossRef] [PubMed]
- Finn, J.D.; Smith, A.R.; Patel, M.C.; Shaw, L.; Youniss, M.R.; van Heteren, J.; Dirstine, T.; Ciullo, C.; Lescarbeau, R.; Seitzer, J.; et al. A Single Administration of CRISPR/Cas9 Lipid Nanoparticles Achieves Robust and Persistent In Vivo Genome Editing. Cell Rep. 2018, 22, 2227–2235. [Google Scholar] [CrossRef] [Green Version]
- Rosenblum, D.; Gutkin, A.; Kedmi, R.; Ramishetti, S.; Veiga, N.; Jacobi, A.M.; Schubert, M.S.; Friedmann-Morvinski, D.; Cohen, Z.R.; Behlke, M.A.; et al. CRISPR-Cas9 genome editing using targeted lipid nanoparticles for cancer therapy. Sci. Adv. 2020, 6, 9450–9468. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Rossi, J.J. Cell-Specific Aptamer-Mediated Targeted Drug Delivery. Oligonucleotides 2011, 21, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Hong, H.; Cai, W. Tumor-Targeted Drug Delivery with Aptamers. Curr. Med. Chem. 2011, 18, 4185–4194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, C.; Li, F.; Wang, L.; Zhang, Z.K.; Wang, C.; He, B.; Li, J.; Chen, Z.; Shaikh, A.B.; Liu, J.; et al. Tumor cell-targeted delivery of CRISPR/Cas9 by aptamer-functionalized lipopolymer for therapeutic genome editing of VEGFA in osteosarcoma. Biomaterials 2017, 147, 68–85. [Google Scholar] [CrossRef] [Green Version]
- Zhen, S.; Takahashi, Y.; Narita, S.; Yang, Y.C.; Li, X. Targeted delivery of CRISPR/Cas9 to prostate cancer by modified gRNA using a flexible aptamer-cationic liposome. Oncotarget 2017, 8, 9375–9387. [Google Scholar] [CrossRef] [Green Version]
- Aksoy, Y.A.; Yang, B.; Chen, W.; Hung, T.; Kuchel, R.P.; Zammit, N.W.; Grey, S.T.; Goldys, E.M.; Deng, W. Spatial and Temporal Control of CRISPR-Cas9-Mediated Gene Editing Delivered via a Light-Triggered Liposome System. ACS Appl. Mater. Interfaces 2020, 12, 52433–52444. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.; Li, X.; Liu, S.; Chen, J.; Li, M.; Chew, S.Y.; Leong, K.W.; Cheng, D. Codelivery of CRISPR-Cas9 and chlorin e6 for spatially controlled tumor-specific gene editing with synergistic drug effects. Sci. Adv. 2020, 6, 4005. [Google Scholar] [CrossRef]
- Wang, J.; Zhu, M.; Nie, G. Biomembrane-based nanostructures for cancer targeting and therapy: From synthetic liposomes to natural biomembranes and membrane-vesicles. Adv. Drug Deliv. Rev. 2021, 178, 113974. [Google Scholar] [CrossRef]
- Yao, X.; Lyu, P.; Yoo, K.; Yadav, M.K.; Singh, R.; Atala, A.; Lu, B. Engineered extracellular vesicles as versatile ribonucleoprotein delivery vehicles for efficient and safe CRISPR genome editing. J. Extracell. Vesicles 2021, 10, e12076. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Zhang, Z.; Zhao, L.; Qin, Y.; Cai, H.; Geng, Z.; Zhu, X.; Zhang, W.; Zhang, Y.; Tan, J.; et al. Tropism-facilitated delivery of CRISPR/Cas9 system with chimeric antigen receptor-extracellular vesicles against B-cell malignancies. J. Control. Release 2020, 326, 455–467. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.; Xu, L.; Xu, X.; Qin, Z.; Zhou, X.; Xiao, Y.; Liang, Y.; Xia, J. Exosome-mediated delivery of gene vectors for gene therapy. Nanoscale 2021, 13, 1387–1397. [Google Scholar] [CrossRef] [PubMed]
- Bowman, K.R.; Kim, J.H.; Lim, C.S. Narrowing the field: Cancer-specific promoters for mitochondrially-targeted p53-BH3 fusion gene therapy in ovarian cancer. J. Ovarian Res. 2019, 12, 38. [Google Scholar] [CrossRef]
- Chen, X.; Scapa, J.E.; Liu, D.X.; Godbey, W.T. Cancer-specific promoters for expression-targeted gene therapy: Ran, brms1 and mcm5. J. Gene Med. 2016, 18, 89–101. [Google Scholar] [CrossRef]
- Rama, A.R.; Aguilera, A.; Melguizo, C.; Caba, O.; Prados, J. Tissue Specific Promoters in Colorectal Cancer. Dis. Markers 2015, 2015, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Zeng, Y.; Liu, L.; Zhuang, C.; Fu, X.; Huang, W.; Cai, Z. Synthesizing AND gate genetic circuits based on CRISPR-Cas9 for identification of bladder cancer cells. Nat. Commun. 2014, 5, 5393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zvereva, M.I.; Shcherbakova, D.M.; Dontsova, O.A. Telomerase: Structure, functions, and activity regulation. Biochem. (Mosc.) 2010, 75, 1563–1583. [Google Scholar] [CrossRef]
- Mizukoshi, E.; Kaneko, S. Telomerase-Targeted Cancer Immunotherapy. Int. J. Mol. Sci. 2019, 20, 1823. [Google Scholar] [CrossRef] [Green Version]
- Akincilar, S.C.; Unal, B.; Tergaonkar, V. Reactivation of telomerase in cancer. Cell. Mol. Life Sci. 2016, 73, 1659–1670. [Google Scholar] [CrossRef] [Green Version]
- Roake, C.M.; Artandi, S.E. Regulation of human telomerase in homeostasis and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 384–397. [Google Scholar] [CrossRef]
- Dai, W.; Xu, X.; Wang, D.; Wu, J.; Wang, J. Cancer therapy with a CRISPR-assisted telomerase-activating gene expression system. Oncogene 2019, 38, 4110–4124. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Meng, X.; Pan, J.; Jiang, N.; Zhou, C.; Wu, Z.; Gong, Z. CRISPR/Cas9-mediated noncoding RNA editing in human cancers. RNA Biol. 2018, 15, 35–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, S.; Yadav, T.; Rani, V. Exploring miRNA based approaches in cancer diagnostics and therapeutics. Crit. Rev. Oncol./Hematol. 2016, 98, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Ali Syeda, Z.; Langden, S.S.S.; Munkhzul, C.; Lee, M.; Song, S.J. Regulatory Mechanism of MicroRNA Expression in Cancer. Int. J. Mol. Sci. 2020, 21, 1723. [Google Scholar] [CrossRef] [Green Version]
- Hirosawa, M.; Fujita, Y.; Parr, C.J.C.; Hayashi, K.; Kashida, S.; Hotta, A.; Woltjen, K.; Saito, H. Cell-type-specific genome editing with a microRNA-responsive CRISPR—Cas9 switch. Nucleic Acids Res. 2017, 45, e118. [Google Scholar] [CrossRef] [Green Version]
- Hynes, A.P.; Rousseau, G.M.; Agudelo, D.; Goulet, A.; Amigues, B.; Loehr, J.; Romero, D.A.; Fremaux, C.; Horvath, P.; Doyon, Y.; et al. Widespread anti-CRISPR proteins in virulent bacteriophages inhibit a range of Cas9 proteins. Nat. Commun. 2018, 9, 2919. [Google Scholar] [CrossRef] [Green Version]
- Trasanidou, D.; Gerós, A.S.; Mohanraju, P.; Nieuwenweg, A.C.; Nobrega, F.L.; Staals, R.H.J. Keeping crispr in check: Diverse mechanisms of phage-encoded anti-crisprs. FEMS Microbiol. Lett. 2019, 366, 98. [Google Scholar] [CrossRef] [Green Version]
- Harrington, L.B.; Doxzen, K.W.; Ma, E.; Liu, J.J.; Knott, G.J.; Edraki, A.; Garcia, B.; Amrani, N.; Chen, J.S.; Cofsky, J.C.; et al. A Broad-Spectrum Inhibitor of CRISPR-Cas9. Cell 2017, 170, 1224–1233.e15. [Google Scholar] [CrossRef] [Green Version]
- Shivram, H.; Cress, B.F.; Knott, G.J.; Doudna, J.A. Controlling and enhancing CRISPR systems. Nat. Chem. Biol. 2021, 17, 10–19. [Google Scholar] [CrossRef]
- Hoffmann, M.D.; Aschenbrenner, S.; Grosse, S.; Rapti, K.; Domenger, C.; Fakhiri, J.; Mastel, M.; Börner, K.; Eils, R.; Grimm, D.; et al. Cell-specific CRISPR–Cas9 activation by microRNA-dependent expression of anti-CRISPR proteins. Nucleic Acids Res. 2019, 47, e75. [Google Scholar] [CrossRef] [Green Version]
- Qi, F.; Tan, B.; Ma, F.; Zhu, B.; Zhang, L.; Liu, X.; Li, H.; Yang, J.; Cheng, B. A Synthetic Light-switchable System based on CRISPR Cas13a Regulates the Expression of LncRNA MALAT1 and Affects the Malignant Phenotype of Bladder Cancer Cells. Int. J. Biol. Sci. 2019, 15, 1630–1636. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Wu, X.; Guan, N.; Shao, J.; Li, H.; Chen, Y.; Ping, Y.; Li, D.; Ye, H. Engineering a far-red light–activated split-Cas9 system for remote-controlled genome editing of internal organs and tumors. Sci. Adv. 2020, 6, 1777. [Google Scholar] [CrossRef]
- Zhan, T.; Rindtorff, N.; Betge, J.; Ebert, M.P.; Boutros, M. CRISPR/Cas9 for cancer research and therapy. Semin. Cancer Biol. 2019, 55, 106–119. [Google Scholar] [CrossRef] [PubMed]
- Topatana, W.; Juengpanich, S.; Li, S.; Cao, J.; Hu, J.; Lee, J.; Suliyanto, K.; Ma, D.; Zhang, B.; Chen, M.; et al. Advances in synthetic lethality for cancer therapy: Cellular mechanism and clinical translation. J. Hematol. Oncol. 2020, 13, 118. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.; Garraway, L.A.; Ashworth, A.; Weber, B. Synthetic lethality as an engine for cancer drug target discovery. Nat. Rev. Drug Discov. 2020, 19, 23–38. [Google Scholar] [CrossRef]
- Li, J.; Huang, C.; Xiong, T.; Zhuang, C.; Zhuang, C.; Li, Y.; Ye, J.; Gui, Y. A CRISPR Interference of CBP and p300 Selectively Induced Synthetic Lethality in Bladder Cancer Cells In Vitro. Int. J. Biol. Sci. 2019, 15, 1276–1286. [Google Scholar] [CrossRef] [Green Version]
- Zhen, S.; Li, X. Oncogenic Human Papillomavirus: Application of CRISPR/Cas9 Therapeutic Strategies for Cervical Cancer. Cell. Physiol. Biochem. 2017, 44, 2455–2466. [Google Scholar] [CrossRef] [PubMed]
- Zhen, S.; Hua, L.; Takahashi, Y.; Narita, S.; Liu, Y.H.; Li, Y. In vitro and in vivo growth suppression of human papillomavirus 16-positive cervical cancer cells by CRISPR/Cas9. Biochem. Biophys. Res. Commun. 2014, 450, 1422–1426. [Google Scholar] [CrossRef]
- Hsu, D.S.; Kornepati, A.V.; Glover, W.; Kennedy, E.M.; Cullen, B.R. Targeting HPV16 DNA using CRISPR/Cas inhibits anal cancer growth in vivo. Future Virol. 2018, 13, 475–482. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, E.M.; Kornepati, A.V.R.; Goldstein, M.; Bogerd, H.P.; Poling, B.C.; Whisnant, A.W.; Kastan, M.B.; Cullen, B.R. Inactivation of the Human Papillomavirus E6 or E7 Gene in Cervical Carcinoma Cells by Using a Bacterial CRISPR/Cas RNA-Guided Endonuclease. J. Virol. 2014, 88, 11965–11972. [Google Scholar] [CrossRef] [Green Version]
- Yuen, K.S.; Chan, C.P.; Wong, N.H.M.; Ho, C.H.; Ho, T.H.; Lei, T.; Deng, W.; Tsao, S.W.; Chen, H.; Kok, K.H.; et al. CRISPR/Cas9-mediated genome editing of Epstein–Barr virus in human cells. J. Gen. Virol. 2015, 96, 626–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huo, H.; Hu, G. CRISPR/Cas9-mediated LMP1 knockout inhibits Epstein-Barr virus infection and nasopharyngeal carcinoma cell growth. Infect. Agents Cancer 2019, 14, 30. [Google Scholar] [CrossRef]
- Dong, C.; Qu, L.; Wang, H.; Wei, L.; Dong, Y.; Xiong, S. Targeting hepatitis B virus cccDNA by CRISPR/Cas9 nuclease efficiently inhibits viral replication. Antivir. Res. 2015, 118, 110–117. [Google Scholar] [CrossRef]
- Liu, X.; Hao, R.; Chen, S.; Guo, D.; Chen, Y. Inhibition of hepatitis B virus by the CRISPR/Cas9 system via targeting the conserved regions of the viral genome. J. Gen. Virol. 2015, 96, 2252–2261. [Google Scholar] [CrossRef]
- Price, A.A.; Sampson, T.R.; Ratner, H.K.; Grakoui, A.; Weiss, D.S. Cas9-mediated targeting of viral RNA in eukaryotic cells. Proc. Natl. Acad. Sci. USA 2015, 112, 6164–6169. [Google Scholar] [CrossRef] [Green Version]
- Liang, Z.; Qin, Z.; Riker, A.I.; Xi, Y. CRISPR/Cas9 ablating viral microRNA promotes lytic reactivation of Kaposi’s sarcoma-associated herpesvirus. Biochem. Biophys. Res. Commun. 2020, 533, 1400–1405. [Google Scholar] [CrossRef]
- Tso, F.Y.; West, J.T.; Wood, C. Reduction of Kaposi’s Sarcoma-Associated Herpesvirus Latency Using CRISPR-Cas9 To Edit the Latency-Associated Nuclear Antigen Gene. J. Virol. 2019, 93, e02183-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panfil, A.R.; Green, P.L.; Yoder, K.E. CRISPR Genome Editing Applied to the Pathogenic Retrovirus HTLV-1. Front. Cell. Infect. Microbiol. 2020, 10, 580371. [Google Scholar] [CrossRef] [PubMed]
- Gavvovidis, I.; Leisegang, M.; Willimsky, G.; Miller, N.; Nghiem, P.; Blankenstein, T. Targeting Merkel Cell Carcinoma by Engineered T Cells Specific to T-Antigens of Merkel Cell Polyomavirus. Clin. Cancer Res. 2018, 24, 3644–3655. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Qian, X.; Wang, B.; Xia, Y.; Zheng, Y.; Du, L.; Xu, D.; Xing, D.; DePinho, R.A.; Lu, Z. Programmable base editing of mutated TERT promoter inhibits brain tumour growth. Nat. Cell Biol. 2020, 22, 282–288. [Google Scholar] [CrossRef]
- Oakes, B.L.; Fellmann, C.; Rishi, H.; Taylor, K.L.; Ren, S.M.; Nadler, D.C.; Yokoo, R.; Arkin, A.P.; Doudna, J.A.; Savage, D.F. CRISPR-Cas9 Circular Permutants as Programmable Scaffolds for Genome Modification. Cell 2019, 176, 254–267.e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef]
- Huang, C.H.; Lee, K.C.; Doudna, J.A. Applications of CRISPR-Cas Enzymes in Cancer Therapeutics and Detection. Trends Cancer 2018, 4, 499–512. [Google Scholar] [CrossRef]
- Yang, L.; Achreja, A.; Yeung, T.L.; Mangala, L.S.; Jiang, D.; Han, C.; Baddour, J.; Marini, J.C.; Ni, J.; Nakahara, R.; et al. Targeting Stromal Glutamine Synthetase in Tumors Disrupts Tumor Microenvironment-Regulated Cancer Cell Growth. Cell Metab. 2016, 24, 685–700. [Google Scholar] [CrossRef] [Green Version]
- Legut, M.; Dolton, G.; Mian, A.A.; Ottmann, O.G.; Sewell, A.K. CRISPR-mediated TCR replacement generates superior anticancer transgenic T cells. Blood 2018, 131, 311–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Xue, J.; Deng, T.; Zhou, X.; Yu, K.; Deng, L.; Huang, M.; Yi, X.; Liang, M.; Wang, Y.; et al. Safety and feasibility of CRISPR-edited T cells in patients with refractory non-small-cell lung cancer. Nat. Med. 2020, 26, 732–740. [Google Scholar] [CrossRef]
- He, S. The first human trial of CRISPR-based cell therapy clears safety concerns as new treatment for late-stage lung cancer. Signal Transduct. Target. Ther. 2020, 5, 168. [Google Scholar] [CrossRef] [PubMed]
- Lacey, S.F.; Fraietta, J.A. First Trial of CRISPR-Edited T cells in Lung Cancer. Trends Mol. Med. 2020, 26, 713–715. [Google Scholar] [CrossRef]
- Delgado, E.; Kiani, S. A CRISPR Odyssey into Cancer Immunotherapy. CRISPR J. 2020, 3, 73–75. [Google Scholar] [CrossRef]
- Tang, N.; Cheng, C.; Zhang, X.; Qiao, M.; Li, N.; Mu, W.; Wei, X.F.; Han, W.; Wang, H. TGF-β inhibition via CRISPR promotes the long-term efficacy of CAR T cells against solid tumors. JCI Insight 2020, 5, e133977. [Google Scholar] [CrossRef]
- Wu, H.Y.; Cao, C.Y. The application of CRISPR-Cas9 genome editing tool in cancer immunotherapy. Briefings Funct. Genom. 2019, 18, 129–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mollanoori, H.; Shahraki, H.; Rahmati, Y.; Teimourian, S. CRISPR/Cas9 and CAR-T cell, collaboration of two revolutionary technologies in cancer immunotherapy, an instruction for successful cancer treatment. Hum. Immunol. 2018, 79, 876–882. [Google Scholar] [CrossRef] [PubMed]
- Reinshagen, C.; Bhere, D.; Choi, S.H.; Hutten, S.; Nesterenko, I.; Wakimoto, H.; Le Roux, E.; Rizvi, A.; Du, W.; Minicucci, C.; et al. CRISPR-enhanced engineering of therapy-sensitive cancer cells for self-targeting of primary and metastatic tumors. Sci. Transl. Med. 2018, 10, 3240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ClinicalTrials.gov. A Safety and Efficacy Study of TALEN and CRISPR/Cas9 in the Treatment of HPV-Related Cervical Intraepithelial NeoplasiaI. In Identifier: NCT03057912.; 2017. Available online: https://clinicaltrials.gov/ct2/show/NCT03057912 (accessed on 9 December 2021).
- ClinicalTrials.gov. A Study of Metastatic Gastrointestinal Cancers Treated with Tumor Infiltrating Lymphocytes in Which the Gene Encoding the Intracellular Immune Checkpoint CISH Is Inhibited Using CRISPR Genetic Engineering. In Identifier: NCT04426669.; 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT04426669 (accessed on 9 December 2021).
- ClinicalTrials.gov. TGFβR-KO CAR-EGFR T Cells in Advanced Biliary Tract Cancer. In Identifier: NCT04976218.; 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT04976218 (accessed on 9 December 2021).
- ClinicalTrials.gov. Study of CRISPR-Cas9 Mediated PD-1 and TCR Gene-Knocked out Mesothelin-Directed CAR-T Cells in Patients with Mesothelin Positive Multiple Solid Tumors. In Identifier: NCT03545815.; 2018. Available online: https://clinicaltrials.gov/ct2/show/NCT03545815 (accessed on 9 December 2021).
- ClinicalTrials.gov. Programmed Allogeneic CRISPR-Edited T Cells Engineered to Express Anti-CD19 Chimeric Antigen Receptor (PACE CART19) in Patients with Relapsed or Refractory CD19+ Leukemia and Lymphoma. Identifier: NCT05037669; 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT05037669 (accessed on 9 December 2021).

| Delivery Method | Pros | Cons | |
|---|---|---|---|
| Capsule | Viral vector | Wide variety of available viruses; high effectiveness | Immunogenicity; pathogenicity; integration to genome |
| AAV | Low immunogenicity; non-integrating; serotypes with tissue tropism | Low packaging capacity | |
| Lipid-based vector | Available surface modifications for targeted delivery | Low effectiveness; toxicity | |
| Cas9 form | Plasmid | Stable; long lasting transfection | Slow generation of effects; requires delivery to nucleus |
| mRNA | Effects observable faster; short-lived in cell (lower off-target activity) | Unstable; quickly degraded | |
| Protein | Editing immediately upon delivery | Difficult to deliver; contaminated with bacterial particles | |
| Identifier | Study Title | Start Date | Phase | Disease/Condition |
|---|---|---|---|---|
| NCT05037669 | Programmed Allogeneic CRISPR-edited T Cells Engineered to Express Anti-CD19 Chimeric Antigen Receptor (PACE CART19) in Patients With Relapsed Or Refractory CD19+ Leukemia and Lymphoma | January 2022 | I | CD19+ Leukemia and Lymphoma |
| NCT03545815 | Study of CRISPR-Cas9 Mediated PD-1 and TCR Gene-knocked Out Mesothelin- directed CAR-T Cells in Patients With Mesothelin Positive Multiple Solid Tumors | 19 March 2018 | I | Mesothelin Positive Solid Tumors |
| NCT04976218 | TGF R-KO CAR-EGFR T Cells in Advanced Biliary Tract Cancer | 1 August 2021 | I | Advanced Biliary Tract Cancer |
| NCT04426669 | A Study of Metastatic Gastrointestinal Cancers Treated With Tumor Infiltrating Lymphocytes in Which the Gene Encoding the Intracellular Immune Checkpoint CISH Is Inhibited Using CRISPR Genetic Engineering | 15 May 2020 | I/II | Gastrointestinal Cancer |
| NCT03057912 | A Safety and Efficacy Study of TALEN and CRISPR/Cas9 in the Treatment of HPV-related Cervical Intraepithelial NeoplasiaI | 15 January 2018 | I | Human Papillomavirus- Related Malignant Neoplasm |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balon, K.; Sheriff, A.; Jacków, J.; Łaczmański, Ł. Targeting Cancer with CRISPR/Cas9-Based Therapy. Int. J. Mol. Sci. 2022, 23, 573. https://doi.org/10.3390/ijms23010573
Balon K, Sheriff A, Jacków J, Łaczmański Ł. Targeting Cancer with CRISPR/Cas9-Based Therapy. International Journal of Molecular Sciences. 2022; 23(1):573. https://doi.org/10.3390/ijms23010573
Chicago/Turabian StyleBalon, Katarzyna, Adam Sheriff, Joanna Jacków, and Łukasz Łaczmański. 2022. "Targeting Cancer with CRISPR/Cas9-Based Therapy" International Journal of Molecular Sciences 23, no. 1: 573. https://doi.org/10.3390/ijms23010573
APA StyleBalon, K., Sheriff, A., Jacków, J., & Łaczmański, Ł. (2022). Targeting Cancer with CRISPR/Cas9-Based Therapy. International Journal of Molecular Sciences, 23(1), 573. https://doi.org/10.3390/ijms23010573