
Adipose Tissue Plasticity in Response to Pathophysiological Cues: A Connecting Link between Obesity and Its Associated Comorbidities

 , and
, and 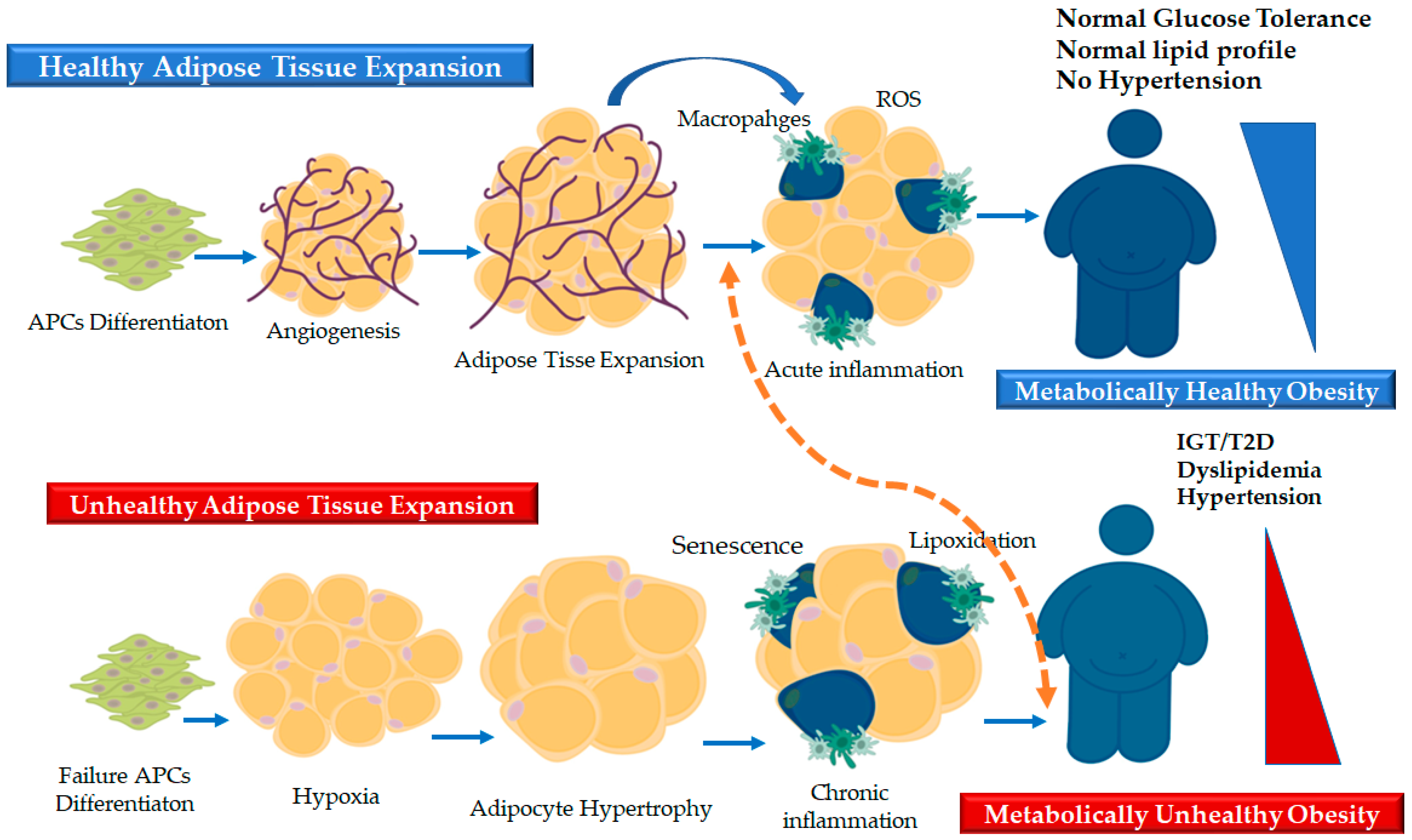{kind=link}
{kind=link}
Abstract
:1. Introduction: Obesity Not Only a Matter of Fat Mass Accrual
2. The Adipose Tissue: A Remarkably Active and Plastic Organ
3. The Dynamics of AT Remodeling: Healthy and “Pathological” Expansion
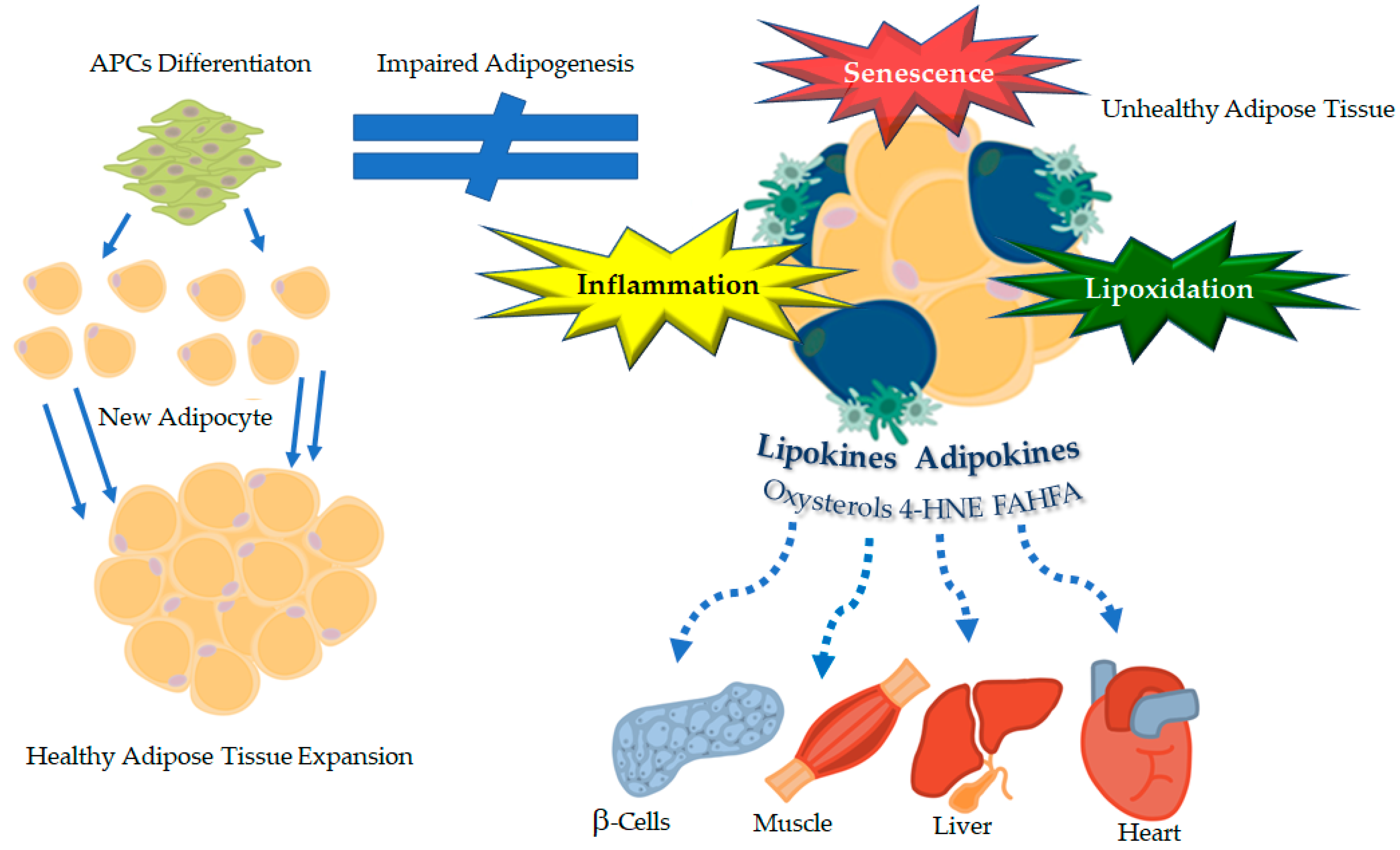
4. Impaired Adipogenesis and Metabolic Dysfunctions: Role of Novel Lipids as Signaling Hormones (“Lipokines”)
4.1. Hypertrophic Obesity and Impaired Adipose Precursor Cells Differentiation: The “Revised” Expandability Hypothesis
4.2. Restricted Adipogenesis in the SAT Triggers Metabolic Dysfunctions
5. Oxidative Stress and Cellular Senescence: Novel Actors in the Scene of Adipose Plasticity
5.1. Oxidative Stress and Lipid Peroxidation in Dysfunctional Fat: Oxysterols and 4-Hydroxynonenal as Novel “Lipokines”
5.2. Cellular Senescence: An Emerging Hallmark of the Dysfunctional Fat
6. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- GBD 2015 Obesity Collaborators; Afshin, A.; Forouzanfar, M.H.; Reitsma, M.B.; Sur, P.; Estep, K.; Lee, A.; Marczak, L.; Mokdad, A.H.; Moradi-Lakeh, M.; et al. Health Effects of Overweight and Obesity in 195 Countries over 25 Years. N. Engl. J. Med. 2017, 377, 13–27. [Google Scholar] [PubMed]
- Despres, J.P.; Lemieux, I. Abdominal obesity and metabolic syndrome. Nature 2006, 444, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Goossens, G.H. The role of adipose tissue dysfunction in the pathogenesis of obesity-related insulin resistance. Physiol. Behav. 2008, 94, 206–218. [Google Scholar] [CrossRef] [PubMed]
- Andersson, C.X.; Gustafson, B.; Hammarstedt, A.; Hedjazifar, S.; Smith, U. Inflamed adipose tissue, insulin resistance and vascular injury. Diabetes Metab. Res. Rev. 2008, 24, 595–603. [Google Scholar] [CrossRef]
- Ruderman, N.; Chisholm, D.; Pi-Sunyer, X.; Schneider, S. The metabolically obese, normal weight individual revisited. Diabetes 1998, 47, 699–713. [Google Scholar] [CrossRef]
- Bluher, M. The distinction of metabolically ‘healthy’ from ‘unhealthy’ obese individuals. Curr. Opin. Lipidol. 2010, 21, 38–43. [Google Scholar] [CrossRef]
- Klöting, N.; Fasshauer, M.; Dietrich, A.; Kovacs, P.; Schön, M.; Kern, M.; Stumvoll, M.; Blüher, M. Insulin-sensitive obesity. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E506–E515. [Google Scholar] [CrossRef]
- Craveiro, V.; Ramos, E.; Araujo, J. Metabolically healthy overweight in young adulthood: Is it a matter of duration and degree of overweight? Nutr. Metab. Cardiovasc. Dis. 2021, 31, 455–463. [Google Scholar] [CrossRef]
- Caleyachetty, R.; Thomas, G.N.; Toulis, K.A.; Mohammed, N.; Gokhale, K.M.; Balachandran, K.; Nirantharakumar, K. Metabolically Healthy Obese and Incident Cardiovascular Disease Events Among 3.5 Million Men and Women. J. Am. Coll. Cardiol. 2017, 70, 1429–1437. [Google Scholar] [CrossRef]
- Scherer, P.E. The many secret lives of adipocytes: Implications for diabetes. Diabetologia 2019, 62, 223–232. [Google Scholar] [CrossRef] [Green Version]
- Cypess, A.M. Reassessing Human Adipose Tissue. N. Engl. J. Med. 2022, 386, 768–779. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Christine, M.; Kusminski, M.; Scherer, P. Adipose tissue remodelling in obesity. J. Clin. Investig. 2011, 121, 2094–2101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cinti, S. The adipose organ at a glance. Dis. Model. Mech. 2012, 5, 588–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giordano, A.; Smorlesi, A.; Frontini, A.; Barbatelli, G.; Cinti, S. White, brown and pink adipocytes: The extraordinary plasticity of the adipose organ. Eur. J. Endocrinol. 2014, 170, R159–R171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghaben, A.L.; Scherer, P.E. Adipogenesis and metabolic health. Nat. Rev. Mol. Cell Biol. 2019, 20, 242–258. [Google Scholar] [CrossRef] [PubMed]
- Sethi, J.K. Activatin’ human adipose progenitors in obesity. Diabetes 2010, 59, 2354–2357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Virtue, S.; Vidal-Puig, A. Adipose tissue expandability, lipotoxicity and the Metabolic Syndrome. An allostatic perspective. Biochim. Biophys. Acta 2010, 1801, 338–349. [Google Scholar] [CrossRef]
- Gomez-Ambrosi, J.; Silva, C.; Galofre, J.C.; Escalada, J.; Santos, S.; Millan, D.; Vila, N.; Ibanez, P.; Gil, M.J.; Valenti, V.; et al. Body mass index classification misses subjects with increased cardiometabolic risk factors related to elevated adiposity. Int. J. Obes. 2012, 36, 286–294. [Google Scholar] [CrossRef] [Green Version]
- Padwal, R.S.; Pajewski, N.M.; Allison, D.B.; Sharma, A.M. Using the Edmonton obesity staging system to predict mortality in a population-representative cohort of people with overweight and obesity. CMAJ 2011, 183, E1059–E1066. [Google Scholar] [CrossRef] [Green Version]
- Danforth, E., Jr. Failure of adipocyte differentiation causes type II diabetes mellitus? Nat. Genet. 2000, 26, 13. [Google Scholar] [CrossRef]
- Christodoulides, C.; Lagathu, C.; Sethi, J.K.; Vidal-Puig, A. Adipogenesis and WNT signalling. Trends Endocrinol. Metab. 2009, 20, 16–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammarstedt, A.; Gogg, S.; Hedjazifar, S.; Nerstedt, A.; Smith, U. Impaired Adipogenesis and Dysfunctional Adipose Tissue in Human Hypertrophic Obesity. Physiol. Rev. 2018, 98, 1911–1941. [Google Scholar] [CrossRef] [PubMed]
- Arner, E.; Westermark, P.O.; Spalding, K.L.; Britton, T.; Ryden, M.; Frisen, J.; Bernard, S.; Arner, P. Adipocyte turnover: Relevance to human adipose tissue morphology. Diabetes 2010, 59, 105–109. [Google Scholar] [CrossRef] [Green Version]
- Spalding, K.L.; Arner, E.; Westermark, P.O.; Bernard, S.; Buchholz, B.A.; Bergmann, O.; Blomqvist, L.; Hoffstedt, J.; Naslund, E.; Britton, T.; et al. Dynamics of fat cell turnover in humans. Nature 2008, 453, 783–787. [Google Scholar] [CrossRef] [PubMed]
- Arner, P.; Arner, E.; Hammarstedt, A.; Smith, U. Genetic predisposition for Type 2 diabetes, but not for overweight/obesity, is associated with a restricted adipogenesis. PLoS ONE 2011, 6, e18284. [Google Scholar] [CrossRef]
- Smith, U. Impaired (‘diabetic’) insulin signaling and action occur in fat cells long before glucose intolerance--is insulin resistance initiated in the adipose tissue? Int. J. Obes. Relat. Metab. Disord. 2002, 26, 897–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, U.; Axelsen, M.; Carvalho, E.; Eliasson, B.; Jansson, P.A.; Wesslau, C. Insulin signaling and action in fat cells: Associations with insulin resistance and type 2 diabetes. Ann. N. Y. Acad. Sci. 1999, 892, 119–126. [Google Scholar] [CrossRef]
- Smith, U.; Kahn, B.B. Adipose tissue regulates insulin sensitivity: Role of adipogenesis, de novo lipogenesis and novel lipids. J. Intern. Med. 2016, 280, 465–475. [Google Scholar] [CrossRef] [Green Version]
- Ryden, M.; Andersson, D.P.; Bergstrom, I.B.; Arner, P. Adipose tissue and metabolic alterations: Regional differences in fat cell size and number matter, but differently: A cross-sectional study. J. Clin. Endocrinol. Metab. 2014, 99, E1870–E1876. [Google Scholar] [CrossRef] [Green Version]
- Stefan, N.; Kantartzis, S.; Machann, J.; Schick, F.; Thamer, C.; Ritting, K.; Balletshofer, B.; Machicao, F.; Fritsche, A.; Haring, H. Identification and characterization of metabolically benign obesity in humans. Arch. Intern. Med. 2008, 168, 1609–1616. [Google Scholar] [CrossRef]
- Lundgren, M.; Svensson, M.; Lindmark, S.; Rendtrom, M.; Ruge, T.; Eriksson, J.W. Fat cell enlargement is an independent marker of insulin resistance and “hyperleptinemia”. Diabetologia 2007, 50, 625–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weyer, C.; Foley, J.; Bogardus, C.; Howard, B.; Ravussin, E. Enlarged subcutaneous abdominal adipocyte size, but not obesity itself, predicts type II diabetes independent of insulin resistance. Diabetologia 2000, 43, 1498–1506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammarstedt, A.; Jansson, P.A.; Wesslau, C.; Yang, X.; Smith, U. Reduced expression of PGC-1 and insulin-signaling molecules in adipose tissue is associated with insulin resistance. Biochem. Biophys. Res. Commun. 2003, 301, 578–582. [Google Scholar] [CrossRef]
- Dubois, S.G.; Heilbronn, L.K.; Smith, S.R.; Albu, J.B.; Kelley, D.E.; Ravussin, E. Decreased expression of adipogenic genes in obese subjects with type 2 diabetes. Obesity 2006, 14, 1543–1552. [Google Scholar] [CrossRef]
- van Tienen, F.H.; van der Kallen, C.J.; Lindsey, P.J.; Wanders, R.J.; van Greevenbroek, M.M.; Smeets, H.J. Preadipocytes of type 2 diabetes subjects display an intrinsic gene expression profile of decreased differentiation capacity. Int. J. Obes. 2011, 35, 1154–1164. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.Y.; Grayburn, P.; Chen, S.; Ravazzola, M.; Orci, L.; Unger, R.H. Adipogenic capacity and the susceptibility to type 2 diabetes and metabolic syndrome. Proc. Natl. Acad. Sci. USA 2008, 105, 6139–6144. [Google Scholar] [CrossRef] [Green Version]
- Gustafson, B.; Smith, U. The Wnt inhibitor Dicckopf 1 and Bone Mprphogenetic Protein 4 rescue adipogenesis in hypertrpphic obesity in human. Diabetes 2012, 61, 1217–1224. [Google Scholar] [CrossRef] [Green Version]
- Park, H.T.; Lee, E.S.; Cheon, Y.P.; Lee, D.R.; Yang, K.S.; Kim, Y.T.; Hur, J.Y.; Kim, S.H.; Lee, K.W.; Kim, T. The relationship between fat depot-specific preadipocyte differentiation and metabolic syndrome in obese women. Clin. Endocrinol. 2012, 76, 59–66. [Google Scholar] [CrossRef]
- Gustafson, B.; Hammarstedt, A.; Andersson, C.X.; Smith, U. Inflamed adipose tissue: A culprit underlying the metabolic syndrome and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2276–2283. [Google Scholar] [CrossRef]
- Murdolo, G.; Smith, U. The dysregulated adipose tissue: A connecting link between insulin resistance, type 2 diabetes mellitus and atherosclerosis. Nutr. Metab. Cardiovasc. Dis. 2006, 16 (Suppl. 1), S35–S38. [Google Scholar] [CrossRef]
- Tan, C.Y.; Vidal-Puig, A. Adipose tissue expandability: The metabolic problems of obesity may arise from the inability to become more obese. Biochem. Soc. Trans. 2008, 36 Pt 5, 935–940. [Google Scholar] [CrossRef] [PubMed]
- Virtue, S.; Vidal-Puig, A. It’s not how fat you are, it’s what you do with it that counts. PLoS Biol. 2008, 6, e237. [Google Scholar] [CrossRef] [PubMed]
- Milan, G.; Conci, S.; Sanna, M.; Favaretto, F.; Bettini, S.; Vettor, R. ASCs and their role in obesity and metabolic diseases. Trends Endocrinol. Metab. 2021, 32, 994–1006. [Google Scholar] [CrossRef]
- Jiang, Y.; Berry, D.C.; Jo, A.; Tang, W.; Arpke, R.W.; Kyba, M.; Graff, J.M. A PPARgamma transcriptional cascade directs adipose progenitor cell-niche interaction and niche expansion. Nat. Commun. 2017, 8, 15926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosen, E.; MacDougald, O. Adipocyte differentiation from the inside out. Nature 2006, 7, 885–896. [Google Scholar] [CrossRef]
- Feve, B. Adipogenesis: Cellular and molecular aspects. Best Pract. Res. Clin. Endocrinol. Metab. 2005, 19, 483–499. [Google Scholar] [CrossRef]
- Gustafson, B.; Hammarstedt, A.; Hedjazifar, S.; Smith, U. Restricted adipogenesis in hypertrophic obesity: The role of WISP2, WNT, and BMP4. Diabetes 2013, 62, 2997–3004. [Google Scholar] [CrossRef] [Green Version]
- Gustafson, B.; Nerstedt, A.; Smith, U. Reduced subcutaneous adipogenesis in human hypertrophic obesity is linked to senescent precursor cells. Nat. Commun. 2019, 10, 2757. [Google Scholar] [CrossRef]
- Gustafson, B.; Gogg, S.; Hedjazifar, S.; Jenndahl, L.; Hammarstedt, A.; Smith, U. Inflammation and impaired adipogenesis in hypertrophic obesity in man. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E999–E1003. [Google Scholar] [CrossRef] [Green Version]
- Smith, U.; Hammarstedt, A. Antagonistic effects of thiazolidinediones and cytokines in lipotoxicity. Biochim. Biophys. Acta. 2010, 1801, 377–380. [Google Scholar] [CrossRef]
- Cao, H.; Gerhold, K.; Mayers, J.R.; Wiest, M.M.; Watkins, S.M.; Hotamisligil, G.S. Identification of a lipokine, a lipid hormone linking adipose tissue to systemic metabolism. Cell 2008, 134, 933–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mozaffarian, D.; Cao, H.; King, I.B.; Lemaitre, R.N.; Song, X.; Siscovick, D.S.; Hotamisligil, G.S. Circulating palmitoleic acid and risk of metabolic abnormalities and new-onset diabetes. Am. J. Clin. Nutr. 2010, 92, 1350–1358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefan, N.; Kantartzis, K.; Celebi, N.; Staiger, H.; Machann, J.; Schick, F.; Cegan, A.; Elcnerova, M.; Schleicher, E.; Fritsche, A.; et al. Circulating palmitoleate strongly and independently predicts insulin sensitivity in humans. Diabetes Care 2010, 33, 405–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yore, M.M.; Syed, I.; Moraes-Vieira, P.M.; Zhang, T.; Herman, M.A.; Homan, E.A.; Patel, R.T.; Lee, J.; Chen, S.; Peroni, O.D.; et al. Discovery of a class of endogenous mammalian lipids with anti-diabetic and anti-inflammatory effects. Cell 2014, 159, 318–332. [Google Scholar] [CrossRef] [Green Version]
- Hodson, L.; Skeaff, C.M.; Fielding, B.A. Fatty acid composition of adipose tissue and blood in humans and its use as a biomarker of dietary intake. Prog. Lipid Res. 2008, 47, 348–380. [Google Scholar] [CrossRef] [PubMed]
- Herman, M.A.; Kahn, B.B. Glucose transport and sensing in the maintenance of glucose homeostasis and metabolic harmony. J Clin. Investig. 2006, 116, 1767–1775. [Google Scholar] [CrossRef] [Green Version]
- Gustafson, B.; Smith, U. Cytokines promote Wnt signaling and inflammation and impair the normal differentiation and lipid accumulation in 3T3-L1 preadipocytes. J. Biol. Chem. 2006, 281, 9507–9516. [Google Scholar] [CrossRef] [Green Version]
- Hammarstedt, A.; Isakson, P.; Gustafson, B.; Smith, U. Wnt-signaling is maintained and adipogenesis inhibited by TNFalpha but not MCP-1 and resistin. Biochem. Biophys. Res. Commun. 2007, 357, 700–706. [Google Scholar] [CrossRef]
- Isakson, P.; Hammarstedt, A.; Gustafson, B.; Smith, U. Impaired preadipocyte differentiation in human abdominal obesity: Role of Wnt, tumor necrosis factor-alpha, and inflammation. Diabetes 2009, 58, 1550–1557. [Google Scholar] [CrossRef] [Green Version]
- Puri, V.; Ranjit, S.; Konda, S.; Nicoloro, S.M.; Straubhaar, J.; Chawla, A.; Chouinard, M.; Lin, C.; Burkart, A.; Corvera, S.; et al. Cidea is associated with lipid droplets and insulin sensitivity in humans. Proc. Natl. Acad. Sci. USA 2008, 105, 7833–7838. [Google Scholar] [CrossRef] [Green Version]
- Wernstedt Asterholm, I.; Tao, C.; Morley, T.S.; Wang, Q.A.; Delgado-Lopez, F.; Wang, Z.V.; Scherer, P.E. Adipocyte inflammation is essential for healthy adipose tissue expansion and remodeling. Cell Metab. 2014, 20, 103–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iuliano, L. Pathways of cholesterol oxidation via non-enzymatic mechanisms. Chem. Phys. Lipids 2011, 164, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Galli, F.; Piroddi, M.; Annetti, C.; Aisa, C.; Floridi, E.; Floridi, A. Oxidative stress and reactive oxygen species. Contrib. Nephrol. 2005, 149, 240–260. [Google Scholar]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, S.; Fujita, T.; Shimabukuro, M.; Iwaki, M.; Yamada, Y.; Nakajima, Y.; Nakayama, O.; Makishima, M.; Matsuda, M.; Shimomura, I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Investig. 2004, 114, 1752–1761. [Google Scholar] [CrossRef] [PubMed]
- Houstis, N.; Rosen, E.D.; Lander, E.S. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature 2006, 440, 944–948. [Google Scholar] [CrossRef]
- Lee, H.; Lee, Y.J.; Choi, H.; Ko, E.H.; Kim, J.W. Reactive oxygen species facilitate adipocyte differentiation by accelerating mitotic clonal expansion. J. Biol. Chem. 2009, 284, 10601–10609. [Google Scholar] [CrossRef] [Green Version]
- Findeisen, H.M.; Gizard, F.; Zhao, Y.; Qing, H.; Jones, K.L.; Cohn, D.; Heywood, E.B.; Bruemmer, D. Glutathione depletion prevents diet-induced obesity and enhances insulin sensitivity. Obesity 2011, 19, 2429–2432. [Google Scholar] [CrossRef]
- Murdolo, G.; Piroddi, M.; Luchetti, F.; Tortoioli, C.; Canonico, B.; Zerbinati, C.; Galli, F.; Iuliano, L. Oxidative stress and lipid peroxidation by-products at the crossroad between adipose organ dysregulation and obesity-linked insulin resistance. Biochimie 2013, 95, 585–594. [Google Scholar] [CrossRef]
- Iuliano, L.; Praticò, D.; Ghiselli, A.; Bonavita, M.S.; Violi, F. Reaction of dipyridamole with the hydroxyl radical. Lipids 1992, 27, 349–353. [Google Scholar] [CrossRef]
- Mattson, M.P. Roles of the lipid peroxidation product 4-hydroxynonenal in obesity, the metabolic syndrome, and associated vascular and neurodegenerative disorders. Exp. Gerontol. 2009, 44, 625–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poli, G.; Schaur, R.J.; Siems, W.G.; Leonarduzzi, G. 4-hydroxynonenal: A membrane lipid oxidation product of medicinal interest. Med. Res. Rev. 2008, 28, 569–631. [Google Scholar] [CrossRef] [PubMed]
- Riahi, Y.; Cohen, G.; Shamni, O.; Sasson, S. Signaling and cytotoxic functions of 4-hydroxyalkenals. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E879–E886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pillon, N.J.; Soulere, L.; Vella, R.E.; Croze, M.; Care, B.R.; Soula, H.A.; Doutheau, A.; Lagarde, M.; Soulage, C.O. Quantitative structure-activity relationship for 4-hydroxy-2-alkenal induced cytotoxicity in L6 muscle cells. Chem. Biol. Interact. 2010, 188, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Giusepponi, D.; Galarini, R.; Barola, C.; Torquato, P.; Bartolini, D.; Moretti, S.; Saluti, G.; Gioiello, A.; Libetta, C.; Galli, F. LC-MS/MS assay for the simultaneous determination of tocopherols, polyunsaturated fatty acids and their metabolites in human plasma and serum. Free Radic. Biol. Med. 2019, 144, 134–143. [Google Scholar] [CrossRef]
- Torquato, P.; Giusepponi, D.; Bartolini, D.; Barola, C.; Marinelli, R.; Sebastiani, B.; Galarini, R.; Galli, F. Pre-analytical monitoring and protection of oxidizable lipids in human plasma (vitamin E and ω-3 and ω-6 fatty acids): An update for redox-lipidomics methods. Free Radic. Biol. Med. 2021, 176, 142–148. [Google Scholar] [CrossRef]
- Svegliati-Baroni, G.; Pierantonelli, I.; Torquato, P.; Marinelli, R.; Ferreri, C.; Chatgilialoglu, C.; Bartolini, D.; Galli, F. Lipidomic biomarkers and mechanisms of lipotoxicity in non-alcoholic fatty liver disease. Free Radic. Biol. Med. 2019, 144, 293–309. [Google Scholar] [CrossRef]
- Torquato, P.; Bartolini, D.; Giusepponi, D.; Piroddi, M.; Sebastiani, B.; Saluti, G.; Galarini, R.; Galli, F. Increased plasma levels of the lipoperoxyl radical-derived vitamin E metabolite α-tocopheryl quinone are an early indicator of lipotoxicity in fatty liver subjects. Free Radic. Biol. Med. 2019, 131, 115–125. [Google Scholar] [CrossRef]
- Torquato, P.; Giusepponi, D.; Galarini, R.; Bartolini, D.; Piroddi, M.; Galli, F. Analysis of Vitamin E Metabolites. In Vitamin E: Chemistry and Nutritional Benefits; Niki, E., Ed.; The Royal Society of Chemistry: London, UK, 2019. [Google Scholar]
- Otaegui-Arrazola, A.; Menendez-Carreno, M.; Ansorena, D.; Astiasaran, I. Oxysterols: A world to explore. Food Chem. Toxicol. 2010, 48, 3289–3303. [Google Scholar] [CrossRef]
- Demozay, D.; Mas, J.C.; Rocchi, S.; Van Obberghen, E. FALDH reverses the deleterious action of oxidative stress induced by lipid peroxidation product 4-hydroxynonenal on insulin signaling in 3T3-L1 adipocytes. Diabetes 2008, 57, 1216–1226. [Google Scholar] [CrossRef] [Green Version]
- Pillon, N.J.; Croze, M.L.; Vella, R.E.; Soulere, L.; Lagarde, M.; Soulage, C.O. The Lipid Peroxidation By-Product 4-Hydroxy-2-Nonenal (4-HNE) Induces Insulin Resistance in Skeletal Muscle through Both Carbonyl and Oxidative Stress. Endocrinology 2012, 153, 2099–2111. [Google Scholar] [CrossRef] [PubMed]
- Pillon, N.J.; Vella, R.E.; Souleere, L.; Becchi, M.; Lagarde, M.; Soulage, C.O. Structural and functional changes in human insulin induced by the lipid peroxidation byproducts 4-hydroxy-2-nonenal and 4-hydroxy-2-hexenal. Chem. Res. Toxicol. 2011, 24, 752–762. [Google Scholar] [CrossRef] [PubMed]
- Endo, K.; Oyama, T.; Saiki, A.; Ban, N.; Ohira, M.; Koide, N.; Murano, T.; Watanabe, H.; Nishii, M.; Miura, M.; et al. Determination of serum 7-ketocholesterol concentrations and their relationships with coronary multiple risks in diabetes mellitus. Diabetes Res. Clin. Pract. 2008, 80, 63–68. [Google Scholar] [CrossRef]
- Wamil, M.; Andrew, R.; Chapman, K.E.; Street, J.; Morton, N.M.; Seckl, J.R. 7-oxysterols modulate glucocorticoid activity in adipocytes through competition for 11beta-hydroxysteroid dehydrogenase type. Endocrinology 2008, 149, 5909–5918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.P.; Niemczyk, M.; Saini, D.; Awasthi, Y.C.; Zimniak, L.; Zimniak, P. Role of the electrophilic lipid peroxidation product 4-hydroxynonenal in the development and maintenance of obesity in mice. Biochemistry 2008, 47, 3900–3911. [Google Scholar] [CrossRef] [PubMed]
- Toyokuni, S.; Yamada, S.; Kashima, M.; Ihara, Y.; Yamada, Y.; Tanaka, T.; Hiai, H.; Seino, Y.; Uchida, K. Serum 4-hydroxy-2-nonenal-modified albumin is elevated in patients with type 2 diabetes mellitus. Antioxid. Redox Signal. 2000, 2, 681–685. [Google Scholar] [CrossRef] [PubMed]
- Janowski, B.A.; Willy, P.J.; Devi, T.R.; Falck, J.R.; Mangelsdorf, D.J. An oxysterol signalling pathway mediated by the nuclear receptor LXR alpha. Nature 1996, 383, 728–731. [Google Scholar] [CrossRef]
- Murphy, C.; Murray, A.M.; Meaney, S.; Gafvels, M. Regulation by SREBP-2 defines a potential link between isoprenoid and adenosylcobalamin metabolism. Biochem. Biophys. Res. Commun. 2007, 355, 359–364. [Google Scholar] [CrossRef]
- Olkkonen, V.M.; Hynynen, R. Interactions of oxysterols with membranes and proteins. Mol. Aspects. Med. 2009, 30, 123–133. [Google Scholar] [CrossRef]
- Mann, R.K.; Beachy, P.A. Novel lipid modifications of secreted protein signals. Annu. Rev. Biochem. 2004, 73, 891–923. [Google Scholar] [CrossRef]
- Waki, H.; Park, K.W.; Mitro, N.; Pei, L.; Damoiseaux, R.; Wilpitz, D.C.; Reue, K.; Saez, E.; Tontonoz, P. The small molecule harmine is an antidiabetic cell-type-specific regulator of PPARgamma expression. Cell Metab. 2007, 5, 357–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chui, P.C.; Guan, H.P.; Lehrke, M.; Lazar, M.A. PPARgamma regulates adipocyte cholesterol metabolism via oxidized LDL receptor 1. J. Clin. Investig. 2005, 115, 2244–2256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krause, B.R.; Hartman, A.D. Adipose tissue and cholesterol metabolism. J. Lipid Res. 1984, 25, 97–110. [Google Scholar] [CrossRef]
- Le Lay, S.; Krief, S.; Farnier, C.; Lefrere, I.; Le Liepvre, X.; Bazin, R.; Ferre, P.; Dugail, I. Cholesterol, a cell size-dependent signal that regulates glucose metabolism and gene expression in adipocytes. J. Biol. Chem. 2001, 276, 16904–16910. [Google Scholar] [CrossRef] [Green Version]
- Brown, A.J.; Leong, S.L.; Dean, R.T.; Jessup, W. 7-Hydroperoxycholesterol and its products in oxidized low density lipoprotein and human atherosclerotic plaque. J. Lipid Res. 1997, 38, 1730–1745. [Google Scholar] [CrossRef]
- Ferderbar, S.; Pereira, E.C.; Apolinario, E.; Bertolami, M.C.; Faludi, A.; Monte, O.; Calliari, L.E.; Sales, J.E.; Gagliardi, A.R.; Xavier, H.T.; et al. Cholesterol oxides as biomarkers of oxidative stress in type 1 and type 2 diabetes mellitus. Diabetes Metab. Res. Rev. 2007, 23, 35–42. [Google Scholar] [CrossRef]
- Murdolo, G.; Piroddi, M.; Tortoioli, C.; Bartolini, D.; Schmelz, M.; Luchetti, F.; Canonico, B.; Papa, S.; Zerbinati, C.; Iuliano, L.; et al. Free Radical-derived Oxysterols: Novel Adipokines Modulating Adipogenic Differentiation of Adipose Precursor Cells. J. Clin. Endocrinol. Metab. 2016, 101, 4974–4983. [Google Scholar] [CrossRef]
- Ingram, K.H.; Hill, H.; Moellering, D.R.; Hill, B.G.; Lara-Castro, C.; Newcomer, B.; Brandon, L.J.; Ingalls, C.P.; Penumetcha, M.; Rupp, J.C.; et al. Skeletal Muscle Lipid Peroxidation and Insulin Resistance in Humans. J. Clin. Endocrinol. Metab. 2012, 97, E1182–E1186. [Google Scholar] [CrossRef] [Green Version]
- Mogensen, M.; Sahlin, K.; Fernstrom, M.; Glintborg, D.; Vind, B.F.; Beck-Nielsen, H.; Hojlund, K. Mitochondrial respiration is decreased in skeletal muscle of patients with type 2 diabetes. Diabetes 2007, 56, 1592–1599. [Google Scholar] [CrossRef] [Green Version]
- Miwa, I.; Ichimura, N.; Sugiura, M.; Hamada, Y.; Taniguchi, S. Inhibition of glucose-induced insulin secretion by 4-hydroxy-2-nonenal and other lipid peroxidation products. Endocrinology 2000, 141, 2767–2772. [Google Scholar] [CrossRef]
- Grimsrud, P.A.; Picklo, M.J.S.; Griffin, T.J.; Bernlohr, D.A. Carbonylation of adipose proteins in obesity and insulin resistance: Identification of adipocyte fatty acid-binding protein as a cellular target of 4-hydroxynonenal. Mol. Cell Proteom. 2007, 6, 624–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demozay, D.; Rocchi, S.; Mas, J.C.; Grillo, S.; Pirola, L.; Chavey, C.; Van Obberghen, E. Fatty aldehyde dehydrogenase: Potential role in oxidative stress protection and regulation of its gene expression by insulin. J. Biol. Chem. 2004, 279, 6261–6270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Descamps, S.; Arzouk, H.; Bacou, F.; Bernardi, H.; Fedon, Y.; Gay, S.; Reyne, Y.; Rossano, B.; Levin, J. Inhibition of myoblast differentiation by Sfrp1 and Sfrp2. Cell Tissue Res. 2008, 332, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Soares, A.F.; Guichardant, M.; Cozzone, D.; Bernoud-Hubac, N.; Bouzaidi-Tiali, N.; Lagarde, M.; Geloen, A. Effects of oxidative stress on adiponectin secretion and lactate production in 3T3-L1 adipocytes. Free Radic. Biol. Med. 2005, 38, 882–889. [Google Scholar] [CrossRef] [PubMed]
- Zarrouki, B.; Soares, A.F.; Guichardant, M.; Lagarde, M.; Geloen, A. The lipid peroxidation end-product 4-HNE induces COX-2 expression through p38MAPK activation in 3T3-L1 adipose cell. FEBS Lett. 2007, 581, 2394–2400. [Google Scholar] [CrossRef] [Green Version]
- Bost, F.; Aouadi, M.; Caron, L.; Binetruy, B. The role of MAPKs in adipocyte differentiation and obesity. Biochimie 2005, 87, 51–56. [Google Scholar] [CrossRef] [Green Version]
- Jaganjac, M.; Almuraikhy, S.; Al-Khelaifi, F.; Al-Jaber, M.; Bashah, M.; Mazloum, N.A.; Zarkovic, K.; Zarkovic, N.; Waeg, G.; Kafienah, W.; et al. Combined metformin and insulin treatment reverses metabolically impaired omental adipogenesis and accumulation of 4-hydroxynonenal in obese diabetic patients. Redox Biol. 2017, 12, 483–490. [Google Scholar] [CrossRef] [Green Version]
- Jankovic, A.; Korac, A.; Srdic-Galic, B.; Buzadzic, B.; Otasevic, V.; Stancic, A.; Vucetic, M.; Markelic, M.; Velickovic, K.; Golic, I.; et al. Differences in the redox status of human visceral and subcutaneous adipose tissues--relationships to obesity and metabolic risk. Metabolism 2014, 63, 661–671. [Google Scholar] [CrossRef]
- Elrayess, M.A.; Almuraikhy, S.; Kafienah, W.; Al-Menhali, A.; Al-Khelaifi, F.; Bashah, M.; Zarkovic, K.; Zarkovic, N.; Waeg, G.; Alsayrafi, M.; et al. 4-hydroxynonenal causes impairment of human subcutaneous adipogenesis and induction of adipocyte insulin resistance. Free Radic. Biol. Med. 2017, 104, 129–137. [Google Scholar] [CrossRef] [Green Version]
- Curzio, M.; Di Mauro, C.; Esterbauer, H.; Dianzani, M.U. Chemotactic activity of aldehydes. Structural requirements. Role in inflammatory process. Biomed. Pharmacother. 1987, 41, 304–314. [Google Scholar]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef] [PubMed]
- Riahi, Y.; Kaiser, N.; Cohen, G.; Abd-Elrahman, I.; Blum, G.; Shapira, O.M.; Koler, T.; Simionescu, M.; Sima, A.V.; Zarkovic, N.; et al. Foam cell-derived 4-hydroxynonenal induces endothelial cell senescence in a TXNIP-dependent manner. J. Cell. Mol. Med. 2015, 19, 1887–1899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burton, D.G.A.; Faragher, R.G.A. Obesity and type-2 diabetes as inducers of premature cellular senescence and ageing. Biogerontology 2018, 19, 447–459. [Google Scholar] [CrossRef] [Green Version]
- Smith, U.; Li, Q.; Ryden, M.; Spalding, K.L. Cellular senescence and its role in white adipose tissue. Int. J. Obes. 2021, 45, 934–943. [Google Scholar] [CrossRef] [PubMed]
- Khosla, S.; Farr, J.N.; Tchkonia, T.; Kirkland, J.L. The role of cellular senescence in ageing and endocrine disease. Nat. Rev. Endocrinol. 2020, 16, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Tabula Muris, C. A single-cell transcriptomic atlas characterizes ageing tissues in the mouse. Nature 2020, 583, 590–595. [Google Scholar] [CrossRef] [PubMed]
- Ademowo, O.S.; Dias, H.K.I.; Burton, D.G.A.; Griffiths, H.R. Lipid (per) oxidation in mitochondria: An emerging target in the ageing process? Biogerontology 2017, 18, 859–879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flor, A.C.; Kron, S.J. Lipid-derived reactive aldehydes link oxidative stress to cell senescence. Cell Death Dis. 2016, 7, e2366. [Google Scholar] [CrossRef] [Green Version]
- Gogg, S.; Nerstedt, A.; Boren, J.; Smith, U. Human adipose tissue microvascular endothelial cells secrete PPARgamma ligands and regulate adipose tissue lipid uptake. JCI Insight 2019, 4, e125914. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Hagberg, C.E.; Silva Cascales, H.; Lang, S.; Hyvonen, M.T.; Salehzadeh, F.; Chen, P.; Alexandersson, I.; Terezaki, E.; Harms, M.J.; et al. Obesity and hyperinsulinemia drive adipocytes to activate a cell cycle program and senesce. Nat. Med. 2021, 27, 1941–1953. [Google Scholar] [CrossRef]
- Chen, Y.W.; Harris, R.A.; Hatahet, Z.; Chou, K.M. Ablation of XP-V gene causes adipose tissue senescence and metabolic abnormalities. Proc. Natl. Acad. Sci. USA 2015, 112, E4556–E4564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rouault, C.; Marcelin, G.; Adriouch, S.; Rose, C.; Genser, L.; Ambrosini, M.; Bichet, J.C.; Zhang, Y.; Marquet, F.; Aron-Wisnewsky, J.; et al. Senescence-associated beta-galactosidase in subcutaneous adipose tissue associates with altered glycaemic status and truncal fat in severe obesity. Diabetologia 2021, 64, 240–254. [Google Scholar] [CrossRef] [PubMed]
- Hickson, L.J.; Langhi Prata, L.G.P.; Bobart, S.A.; Evans, T.K.; Giorgadze, N.; Hashmi, S.K.; Herrmann, S.M.; Jensen, M.D.; Jia, Q.; Jordan, K.L.; et al. Senolytics decrease senescent cells in humans: Preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine 2019, 47, 446–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmer, A.K.; Xu, M.; Zhu, Y.; Pirtskhalava, T.; Weivoda, M.M.; Hachfeld, C.M.; Prata, L.G.; van Dijk, T.H.; Verkade, E.; Casaclang-Verzosa, G.; et al. Targeting senescent cells alleviates obesity-induced metabolic dysfunction. Aging Cell 2019, 18, e12950. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Luo, X.; Zhong, Y.; Deng, L.; Feng, J. New insights into the role of melatonin in diabetic cardiomyopathy. Pharmacol. Res. Perspect. 2022, 10, e00904. [Google Scholar] [CrossRef]
- Knani, L.; Bartolini, D.; Kechiche, S.; Tortoioli, C.; Murdolo, G.; Moretti, M.; Messaoudi, I.; Reiter, R.J.; Galli, F. Melatonin prevents cadmium-induced bone damage: First evidence on an improved osteogenic/adipogenic differentiation balance of mesenchymal stem cells as underlying mechanism. J. Pineal Res. 2019, 67, e12597. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Fano, M.; Bartolini, D.; Tortoioli, C.; Vermigli, C.; Malara, M.; Galli, F.; Murdolo, G. Adipose Tissue Plasticity in Response to Pathophysiological Cues: A Connecting Link between Obesity and Its Associated Comorbidities. Int. J. Mol. Sci. 2022, 23, 5511. https://doi.org/10.3390/ijms23105511
De Fano M, Bartolini D, Tortoioli C, Vermigli C, Malara M, Galli F, Murdolo G. Adipose Tissue Plasticity in Response to Pathophysiological Cues: A Connecting Link between Obesity and Its Associated Comorbidities. International Journal of Molecular Sciences. 2022; 23(10):5511. https://doi.org/10.3390/ijms23105511
Chicago/Turabian StyleDe Fano, Michelatonio, Desirèe Bartolini, Cristina Tortoioli, Cristiana Vermigli, Massimo Malara, Francesco Galli, and Giuseppe Murdolo. 2022. "Adipose Tissue Plasticity in Response to Pathophysiological Cues: A Connecting Link between Obesity and Its Associated Comorbidities" International Journal of Molecular Sciences 23, no. 10: 5511. https://doi.org/10.3390/ijms23105511
APA StyleDe Fano, M., Bartolini, D., Tortoioli, C., Vermigli, C., Malara, M., Galli, F., & Murdolo, G. (2022). Adipose Tissue Plasticity in Response to Pathophysiological Cues: A Connecting Link between Obesity and Its Associated Comorbidities. International Journal of Molecular Sciences, 23(10), 5511. https://doi.org/10.3390/ijms23105511