
Small Molecule Arranged Thermal Proximity Coaggregation (smarTPCA)—A Novel Approach to Characterize Protein–Protein Interactions in Living Cells by Similar Isothermal Dose–Responses

Abstract
:1. Introduction
2. Results
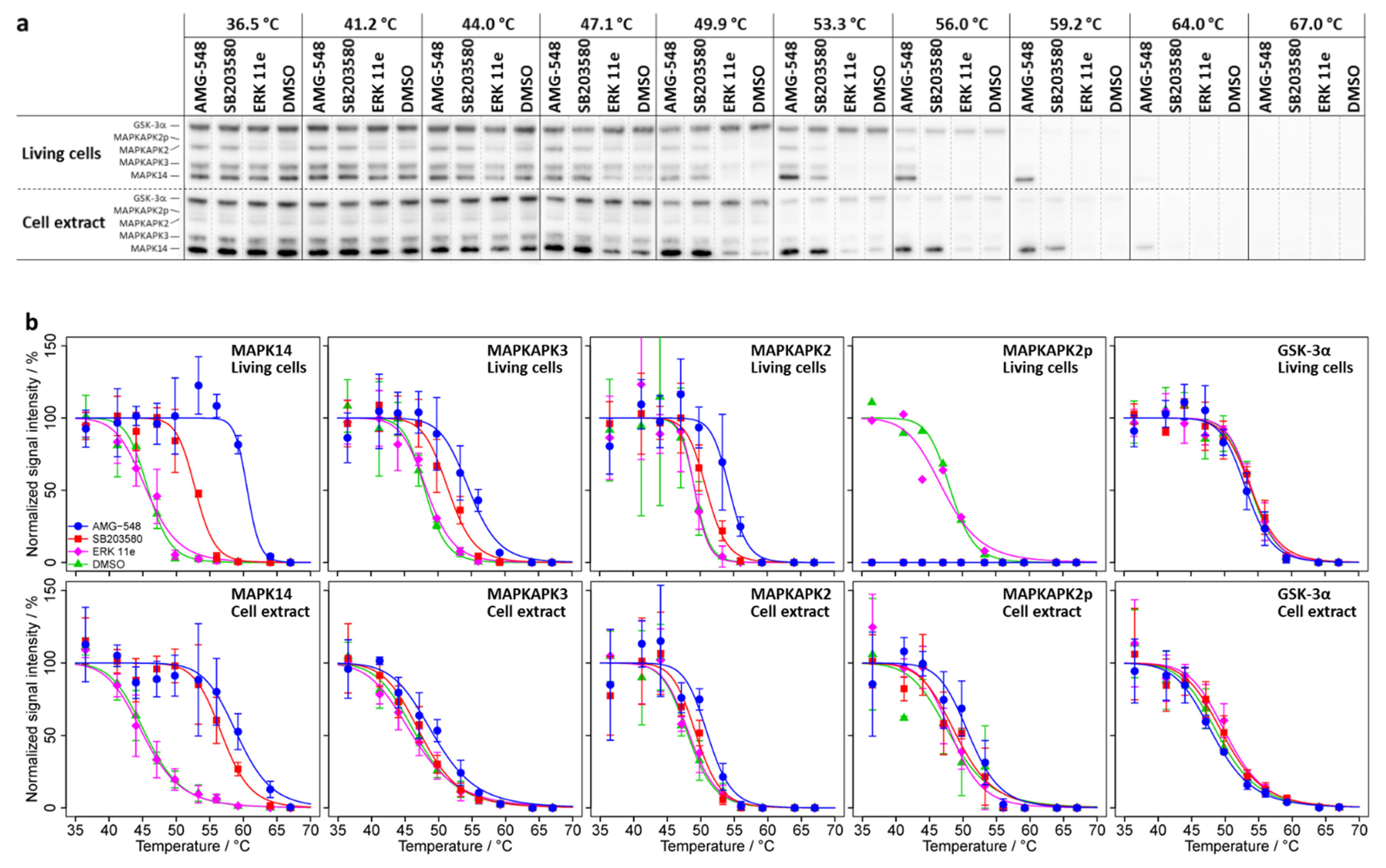
2.1. CETSA—Thermal Stabilization of MAPK14 and Its Interaction Partners MAPKAPK2/3 upon Treatment with MAPK14 Inhibitors AMG-548 and SB203580
2.1.1. CETSA Experiment in Living HL-60 Cells
2.1.2. CETSA Experiment in HL-60 Cell Extract
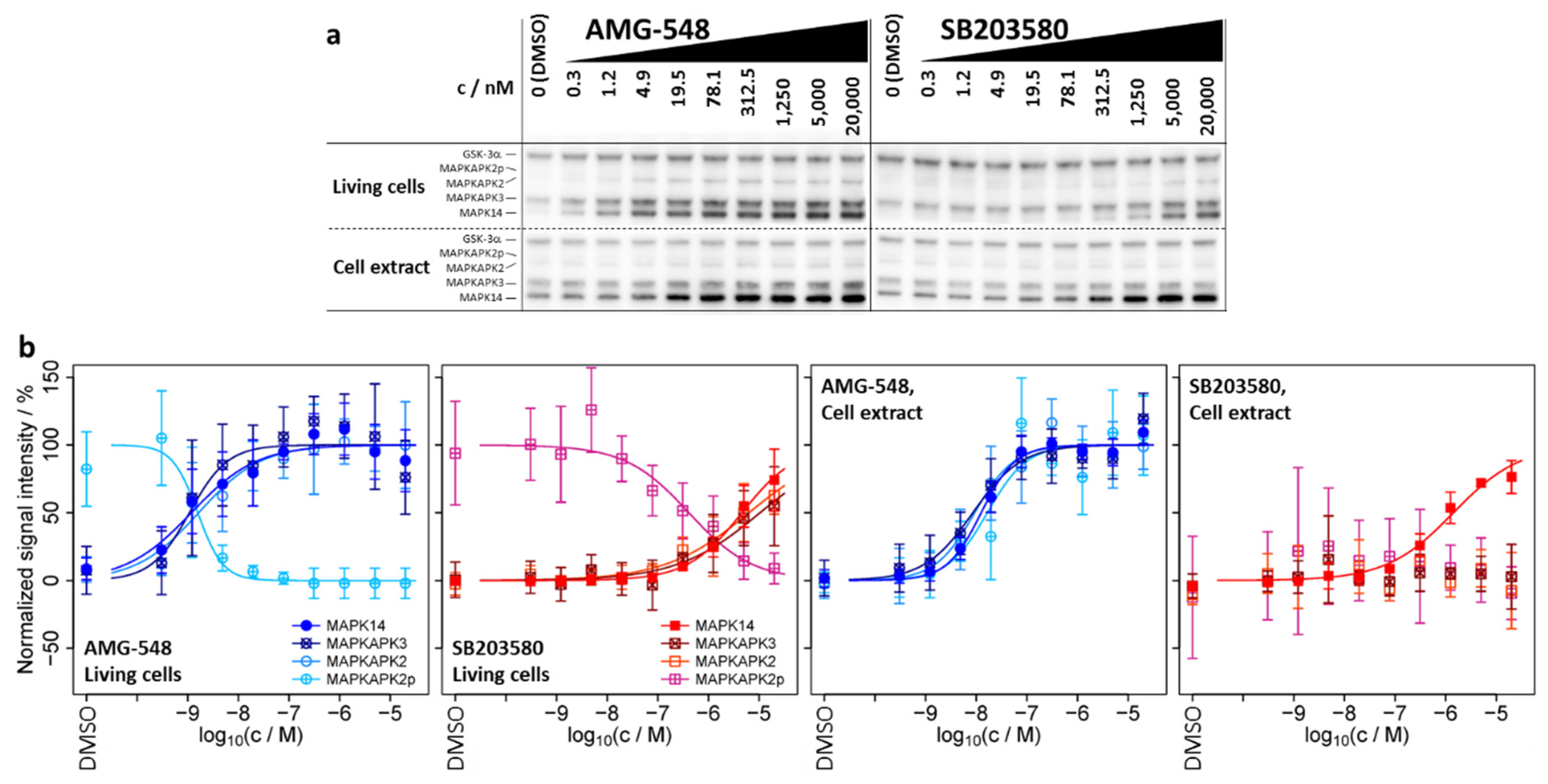
2.2. ITDR-CETSA-MAPK14 and Its Substrates/Interaction Partners MAPKAPK2/3 Exhibit Nearly Identical Dose–Response Characteristics upon Kinase Inhibitor Treatment
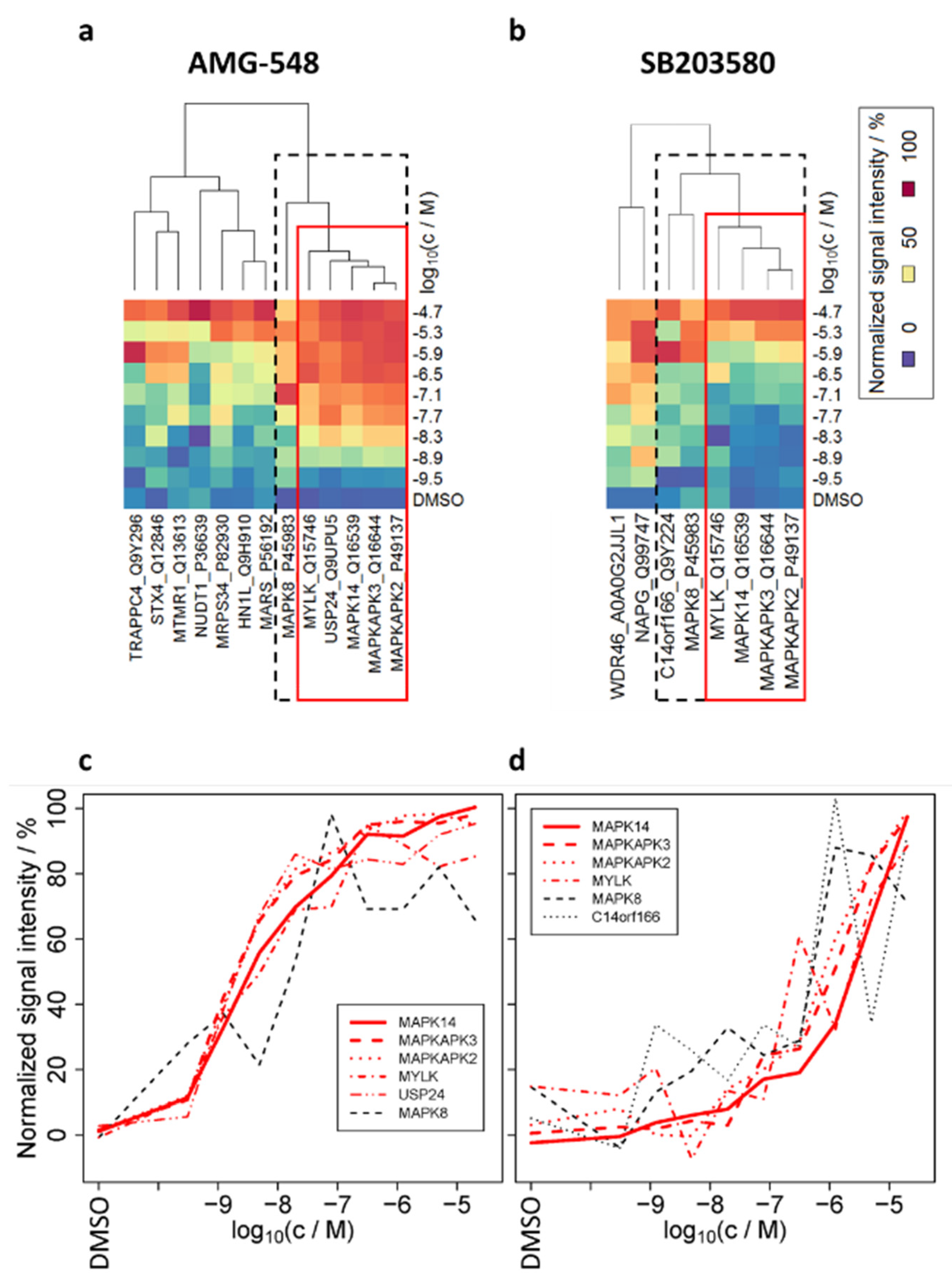
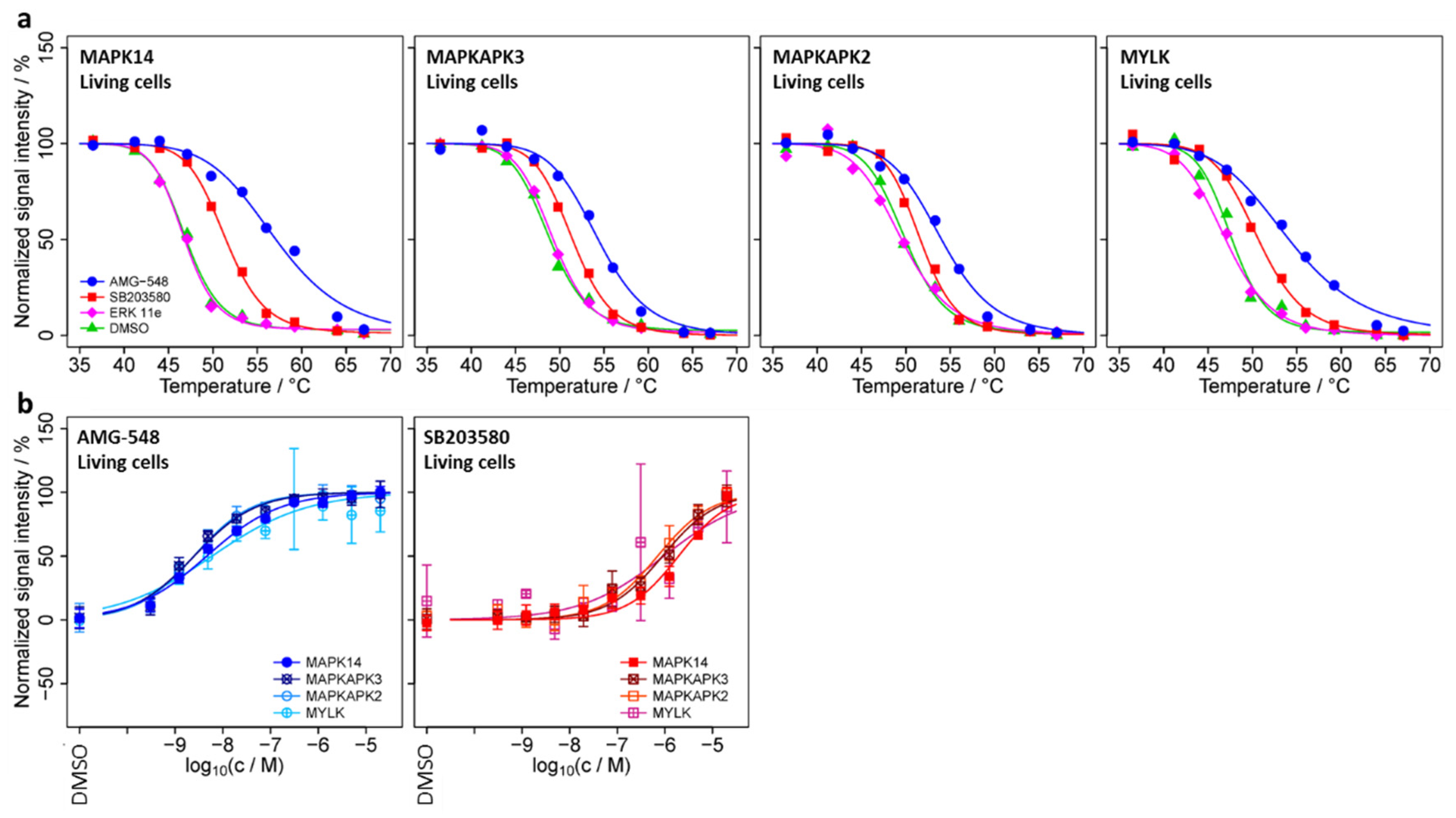
2.3. Determination of Candidate Interaction Partners of MAPK14 in Living HL-60 Cells by Means of smarTCPA
2.4. TPP-TR Confirmation Experiment
3. Discussion
3.1. Phosphorylation Has No Effect on the Thermal Stabilisation of Phospho-MAPKAPK2
3.2. CETSA in Cell Extract Is Not Universally Applicable for the Interrogation of Primary and Secondary Binding Effects
3.3. smarTPCA—A Novel Approach to Interrogate Primarry and Secondary Binding Effects by Means of Dose–Reponse Charateristics
3.4. Application of Small Molecule Arranged Thermal Proximity Coaggregation (smarTPCA) in a Proteome-Wide TPP-CCR Experiment
4. Materials and Methods
4.1. Sample Preparation
4.1.1. Sample Preparation from Living Cells
CETSA and TPP-TR
| PCR strip: | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
| Treatment temp./°C: | 36.5 | 41.2 | 44.0 | 47.1 | 49.8 | 53.3 | 56.0 | 59.2 | 64.0 | 67.0 |
ITDR-CETSA and TPP-CCR
4.1.2. Sample Preparation from Cell Extract
CETSA
ITDR-CETSA
4.2. Protein Detection and Quantification
4.2.1. Detection of Selected Proteins by Immunoblotting after SDS-PAGE Separation
4.2.2. Protein Analysis by LC-MS/MS
TPP-TR
| TMT label: | 131 | 130C | 130N | 129C | 129N | 128C | 128N | 127C | 127N | 126 |
| Treatment temp./°C: | 36.5 | 41.2 | 44.0 | 47.1 | 49.8 | 53.3 | 56.0 | 59.2 | 64.0 | 67.0 |
TPP-CCR
| TMT label: | 131 | 130C | 130N | 129C | 129N | 128C | 128N | 127C | 127N | 126 |
| Inhibitor conc./µM: | 20 | 5 | 1.25 | 0.313 | 0.0781 | 0.0195 | 0.00488 | 0.00122 | 0.000305 | 0 * |
| (* vehicle control) | ||||||||||
4.3. Data Analysis
4.3.1. Immunoblotting
CETSA
ITDR-CETSA
4.3.2. MS Data
TPP-TR
TPP-CCR
5. Conclusions and Outlook
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Koshland, D.E. Application of a Theory of Enzyme Specificity to Protein Synthesis. Proc. Natl. Acad. Sci. USA 1958, 44, 98–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linderstrom-Lang, K.; Schellman, J.A. Protein structure and enzyme activity. In The Enzymes; Boyer, P.D., Lardy, H., Myrback, K., Eds.; Academic Press: New York, NY, USA, 1959; pp. 443–510. [Google Scholar]
- Pantoliano, M.W.; Petrella, E.C.; Kwasnoski, J.D.; Lobanov, V.S.; Myslik, J.; Graf, E.; Carver, T.; Asel, E.; Springer, B.A.; Lane, P.; et al. High-density miniaturized thermal shift assays as a general strategy for drug discovery. J. Biomol. Screen. 2001, 6, 429–440. [Google Scholar] [CrossRef]
- Ericsson, U.B.; Hallberg, B.M.; Detitta, G.T.; Dekker, N.; Nordlund, P. Thermofluor-based high-throughput stability optimization of proteins for structural studies. Anal. Biochem. 2006, 357, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Vedadi, M.; Niesen, F.H.; Allali-Hassani, A.; Fedorov, O.Y.; Finerty, P.J., Jr.; Wasney, G.A.; Yeung, R.; Arrowsmith, C.; Ball, L.J.; Berglund, H.; et al. Chemical screening methods to identify ligands that promote protein stability, protein crystallization, and structure determination. Proc. Natl. Acad. Sci. USA 2006, 103, 15835–15840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez Molina, D.; Nordlund, P. The Cellular Thermal Shift Assay: A Novel Biophysical Assay for In Situ Drug Target Engagement and Mechanistic Biomarker Studies. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 141–161. [Google Scholar] [CrossRef] [PubMed]
- Martinez Molina, D.; Jafari, R.; Ignatushchenko, M.; Seki, T.; Larsson, E.A.; Dan, C.; Sreekumar, L.; Cao, Y.; Nordlund, P. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science 2013, 341, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Jafari, R.; Almqvist, H.; Axelsson, H.; Ignatushchenko, M.; Lundback, T.; Nordlund, P.; Martinez Molina, D. The cellular thermal shift assay for evaluating drug target interactions in cells. Nat. Protoc. 2014, 9, 2100–2122. [Google Scholar] [CrossRef]
- Ruiz-Garcia, A.; Bermejo, M.; Moss, A.; Casabo, V.G. Pharmacokinetics in drug discovery. J. Pharm. Sci. 2008, 97, 654–690. [Google Scholar] [CrossRef]
- Dai, L.; Prabhu, N.; Yu, L.Y.; Bacanu, S.; Ramos, A.D.; Nordlund, P. Horizontal Cell Biology: Monitoring Global Changes of Protein Interaction States with the Proteome-Wide Cellular Thermal Shift Assay (CETSA). Annu. Rev. Biochem. 2019, 88, 383–408. [Google Scholar] [CrossRef]
- Schurmann, M.; Janning, P.; Ziegler, S.; Waldmann, H. Small-Molecule Target Engagement in Cells. Cell Chem. Biol. 2016, 23, 435–441. [Google Scholar] [CrossRef] [Green Version]
- Seashore-Ludlow, B.; Axelsson, H.; Lundback, T. Perspective on CETSA Literature: Toward More Quantitative Data Interpretation. SLAS Discov. 2020, 25, 118–126. [Google Scholar] [CrossRef]
- Durham, T.B.; Blanco, M.J. Target engagement in lead generation. Bioorg. Med. Chem. Lett. 2015, 25, 998–1008. [Google Scholar] [CrossRef] [Green Version]
- Werner, T.; Becher, I.; Sweetman, G.; Doce, C.; Savitski, M.M.; Bantscheff, M. High-resolution enabled TMT 8-plexing. Anal. Chem. 2012, 84, 7188–7194. [Google Scholar] [CrossRef]
- Werner, T.; Sweetman, G.; Savitski, M.F.; Mathieson, T.; Bantscheff, M.; Savitski, M.M. Ion coalescence of neutron encoded TMT 10-plex reporter ions. Anal. Chem. 2014, 86, 3594–3601. [Google Scholar] [CrossRef]
- Savitski, M.M.; Reinhard, F.B.; Franken, H.; Werner, T.; Savitski, M.F.; Eberhard, D.; Martinez Molina, D.; Jafari, R.; Dovega, R.B.; Klaeger, S.; et al. Tracking cancer drugs in living cells by thermal profiling of the proteome. Science 2014, 346, 1255784. [Google Scholar] [CrossRef] [Green Version]
- Huber, K.V.; Olek, K.M.; Muller, A.C.; Tan, C.S.; Bennett, K.L.; Colinge, J.; Superti-Furga, G. Proteome-wide drug and metabolite interaction mapping by thermal-stability profiling. Nat. Methods 2015, 12, 1055–1057. [Google Scholar] [CrossRef] [Green Version]
- Franken, H.; Mathieson, T.; Childs, D.; Sweetman, G.M.; Werner, T.; Togel, I.; Doce, C.; Gade, S.; Bantscheff, M.; Drewes, G.; et al. Thermal proteome profiling for unbiased identification of direct and indirect drug targets using multiplexed quantitative mass spectrometry. Nat. Protoc. 2015, 10, 1567–1593. [Google Scholar]
- Savitski, M.M.; Zinn, N.; Faelth-Savitski, M.; Poeckel, D.; Gade, S.; Becher, I.; Muelbaier, M.; Wagner, A.J.; Strohmer, K.; Werner, T.; et al. Multiplexed Proteome Dynamics Profiling Reveals Mechanisms Controlling Protein Homeostasis. Cell 2018, 173, 260–274.e25. [Google Scholar] [CrossRef] [Green Version]
- Reinhard, F.B.; Eberhard, D.; Werner, T.; Franken, H.; Childs, D.; Doce, C.; Savitski, M.F.; Huber, W.; Bantscheff, M.; Savitski, M.M.; et al. Thermal proteome profiling monitors ligand interactions with cellular membrane proteins. Nat. Methods 2015, 12, 1129–1131. [Google Scholar] [CrossRef]
- Becher, I.; Werner, T.; Doce, C.; Zaal, E.A.; Togel, I.; Khan, C.A.; Rueger, A.; Muelbaier, M.; Salzer, E.; Berkers, C.R.; et al. Thermal profiling reveals phenylalanine hydroxylase as an off-target of panobinostat. Nat. Chem. Biol. 2016, 12, 908–910. [Google Scholar] [CrossRef]
- Mateus, A.; Maatta, T.A.; Savitski, M.M. Thermal proteome profiling: Unbiased assessment of protein state through heat-induced stability changes. Proteome Sci. 2016, 15, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mateus, A.; Bobonis, J.; Kurzawa, N.; Stein, F.; Helm, D.; Hevler, J.; Typas, A.; Savitski, M.M. Thermal proteome profiling in bacteria: Probing protein state in vivo. Mol. Syst. Biol. 2018, 14, e8242. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, M.; Liao, P.J.; Lee, K.H.; Wong, J.; Shang, S.C.; Minami, N.; Sampetrean, O.; Saya, H.; Lingyun, D.; Prabhu, N.; et al. Dual blockade of the lipid kinase PIP4Ks and mitotic pathways leads to cancer-selective lethality. Nat. Commun. 2017, 8, 2200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azimi, A.; Caramuta, S.; Seashore-Ludlow, B.; Bostrom, J.; Robinson, J.L.; Edfors, F.; Tuominen, R.; Kemper, K.; Krijgsman, O.; Peeper, D.S.; et al. Targeting CDK2 overcomes melanoma resistance against BRAF and Hsp90 inhibitors. Mol. Syst. Biol. 2018, 14, e7858. [Google Scholar] [CrossRef]
- Hu, D.; Yang, C.; Lok, C.N.; Xing, F.; Lee, P.Y.; Fung, Y.M.E.; Jiang, H.; Che, C.M. An Antitumor Bis(N-Heterocyclic Carbene)Platinum(II) Complex That Engages Asparagine Synthetase as an Anticancer Target. Angew. Chem. Int. Ed. Engl. 2019, 58, 10914–10918. [Google Scholar] [CrossRef]
- Sridharan, S.; Günthner, I.; Becher, I.; Savitski, M.; Bantscheff, M. Target Discovery Using Thermal Proteome Profiling. In Mass Spectrometry-Based Chemical Proteomics; Tao, W.A., Zhang, Y., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2019; pp. 267–291. [Google Scholar]
- Mateus, A.; Kurzawa, N.; Becher, I.; Sridharan, S.; Helm, D.; Stein, F.; Typas, A.; Savitski, M.M. Thermal proteome profiling for interrogating protein interactions. Mol. Syst. Biol. 2020, 16, e9232. [Google Scholar] [CrossRef]
- Tan, C.S.H.; Go, K.D.; Bisteau, X.; Dai, L.; Yong, C.H.; Prabhu, N.; Ozturk, M.B.; Lim, Y.T.; Sreekumar, L.; Lengqvist, J.; et al. Thermal proximity coaggregation for system-wide profiling of protein complex dynamics in cells. Science 2018, 359, 1170–1177. [Google Scholar] [CrossRef] [Green Version]
- Becher, I.; Andres-Pons, A.; Romanov, N.; Stein, F.; Schramm, M.; Baudin, F.; Helm, D.; Kurzawa, N.; Mateus, A.; Mackmull, M.T.; et al. Pervasive Protein Thermal Stability Variation during the Cell Cycle. Cell 2018, 173, 1495–1507.e18. [Google Scholar] [CrossRef] [Green Version]
- Dai, L.; Zhao, T.; Bisteau, X.; Sun, W.; Prabhu, N.; Lim, Y.T.; Sobota, R.M.; Kaldis, P.; Nordlund, P. Modulation of Protein-Interaction States through the Cell Cycle. Cell 2018, 173, 1481–1494.e13. [Google Scholar] [CrossRef] [Green Version]
- Miettinen, T.P.; Peltier, J.; Hartlova, A.; Gierlinski, M.; Jansen, V.M.; Trost, M.; Bjorklund, M. Thermal proteome profiling of breast cancer cells reveals proteasomal activation by CDK4/6 inhibitor palbociclib. EMBO J. 2018, 37, e98359. [Google Scholar] [CrossRef]
- Dominguez, C.; Powers, D.A.; Tamayo, N. p38 MAP kinase inhibitors: Many are made, but few are chosen. Curr. Opin. Drug Discov. Devel. 2005, 8, 421–430. [Google Scholar] [PubMed]
- Lee, M.R.; Dominguez, C. MAP kinase p38 inhibitors: Clinical results and an intimate look at their interactions with p38alpha protein. Curr. Med. Chem. 2005, 12, 2979–2994. [Google Scholar] [CrossRef] [PubMed]
- Saklatvala, J.; Rawlinson, L.; Waller, R.J.; Sarsfield, S.; Lee, J.C.; Morton, L.F.; Barnes, M.J.; Farndale, R.W. Role for p38 mitogen-activated protein kinase in platelet aggregation caused by collagen or a thromboxane analogue. J. Biol. Chem. 1996, 271, 6586–6589. [Google Scholar] [CrossRef] [Green Version]
- Rouse, J.; Cohen, P.; Trigon, S.; Morange, M.; Alonso-Llamazares, A.; Zamanillo, D.; Hunt, T.; Nebreda, A.R. A novel kinase cascade triggered by stress and heat shock that stimulates MAPKAP kinase-2 and phosphorylation of the small heat shock proteins. Cell 1994, 78, 1027–1037. [Google Scholar] [CrossRef]
- Ben-Levy, R.; Leighton, I.A.; Doza, Y.N.; Attwood, P.; Morrice, N.; Marshall, C.J.; Cohen, P. Identification of novel phosphorylation sites required for activation of MAPKAP kinase-2. EMBO J. 1995, 14, 5920–5930. [Google Scholar] [CrossRef] [PubMed]
- Ronkina, N.; Kotlyarov, A.; Dittrich-Breiholz, O.; Kracht, M.; Hitti, E.; Milarski, K.; Askew, R.; Marusic, S.; Lin, L.L.; Gaestel, M.; et al. The mitogen-activated protein kinase (MAPK)-activated protein kinases MK2 and MK3 cooperate in stimulation of tumor necrosis factor biosynthesis and stabilization of p38 MAPK. Mol. Cell Biol. 2007, 27, 170–181. [Google Scholar] [CrossRef] [Green Version]
- Hughes, C.S.; Moggridge, S.; Muller, T.; Sorensen, P.H.; Morin, G.B.; Krijgsveld, J. Single-pot, solid-phase-enhanced sample preparation for proteomics experiments. Nat. Protoc. 2019, 14, 68–85. [Google Scholar] [CrossRef]
- Aronov, A.M.; Tang, Q.; Martinez-Botella, G.; Bemis, G.W.; Cao, J.; Chen, G.; Ewing, N.P.; Ford, P.J.; Germann, U.A.; Green, J.; et al. Structure-guided design of potent and selective pyrimidylpyrrole inhibitors of extracellular signal-regulated kinase (ERK) using conformational control. J. Med. Chem. 2009, 52, 6362–6368. [Google Scholar] [CrossRef]
- Kalxdorf, M.; Gunthner, I.; Becher, I.; Kurzawa, N.; Knecht, S.; Savitski, M.M.; Eberl, H.C.; Bantscheff, M. Cell surface thermal proteome profiling tracks perturbations and drug targets on the plasma membrane. Nat. Methods 2021, 18, 84–91. [Google Scholar] [CrossRef]
- Seashore-Ludlow, B.; Axelsson, H.; Almqvist, H.; Dahlgren, B.; Jonsson, M.; Lundback, T. Quantitative Interpretation of Intracellular Drug Binding and Kinetics Using the Cellular Thermal Shift Assay. Biochemistry 2018, 57, 6715–6725. [Google Scholar] [CrossRef]
- Lee, C.R.; Park, Y.H.; Min, H.; Kim, Y.R.; Seok, Y.J. Determination of protein phosphorylation by polyacrylamide gel electrophoresis. J. Microbiol. 2019, 57, 93–100. [Google Scholar] [CrossRef]
- Heberle, A.M.; Razquin Navas, P.; Langelaar-Makkinje, M.; Kasack, K.; Sadik, A.; Faessler, E.; Hahn, U.; Marx-Stoelting, P.; Opitz, C.A.; Sers, C.; et al. The PI3K and MAPK/p38 pathways control stress granule assembly in a hierarchical manner. Life Sci. Alliance 2019, 2, e201800257. [Google Scholar] [CrossRef]
- Leech, C.M.; Flynn, M.J.; Arsenault, H.E.; Ou, J.; Liu, H.; Zhu, L.J.; Benanti, J.A. The coordinate actions of calcineurin and Hog1 mediate the stress response through multiple nodes of the cell cycle network. PLoS Genet. 2020, 16, e1008600. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, X.; Qin, X.; Wang, X.; Liu, F.; White, E.; Zheng, X.F. PP2AC Level Determines Differential Programming of p38-TSC-mTOR Signaling and Therapeutic Response to p38-Targeted Therapy in Colorectal Cancer. EBioMedicine 2015, 2, 1944–1956. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.; An, Z.; Zou, Z.; Sumpter, R.; Su, M.; Zang, X.; Sinha, S.; Gaestel, M.; Levine, B. The stress-responsive kinases MAPKAPK2/MAPKAPK3 activate starvation-induced autophagy through Beclin 1 phosphorylation. eLife 2015, 4, e05289. [Google Scholar] [CrossRef]
- Maayan, I.; Beenstock, J.; Marbach, I.; Tabachnick, S.; Livnah, O.; Engelberg, D. Osmostress induces autophosphorylation of Hog1 via a C-terminal regulatory region that is conserved in p38alpha. PLoS ONE 2012, 7, e44749. [Google Scholar] [CrossRef]
- Wu, Q.; Li, G.; Wen, C.; Zeng, T.; Fan, Y.; Liu, C.; Fu, G.F.; Xie, C.; Lin, Q.; Xie, L.; et al. Monoubiquitination of p120-catenin is essential for TGFbeta-induced epithelial-mesenchymal transition and tumor metastasis. Sci. Adv. 2020, 6, eaay9819. [Google Scholar] [CrossRef]
- Sreekanth, G.P.; Chuncharunee, A.; Sirimontaporn, A.; Panaampon, J.; Noisakran, S.; Yenchitsomanus, P.T.; Limjindaporn, T. SB203580 Modulates p38 MAPK Signaling and Dengue Virus-Induced Liver Injury by Reducing MAPKAPK2, HSP27, and ATF2 Phosphorylation. PLoS ONE 2016, 11, e0149486. [Google Scholar] [CrossRef]
- Huang, J.X.; Lee, G.; Cavanaugh, K.E.; Chang, J.W.; Gardel, M.L.; Moellering, R.E. High throughput discovery of functional protein modifications by Hotspot Thermal Profiling. Nat. Methods 2019, 16, 894–901. [Google Scholar] [CrossRef]
- Huang, J.X.; Wu, D.; Stein, B.D.; Moellering, R.E. Huang et al. reply. Nat. Methods 2021, 18, 763–767. [Google Scholar] [CrossRef]
- Potel, C.M.; Kurzawa, N.; Becher, I.; Typas, A.; Mateus, A.; Savitski, M.M. Impact of phosphorylation on thermal stability of proteins. Nat. Methods 2021, 18, 757–759. [Google Scholar] [CrossRef] [PubMed]
- Smith, I.R.; Hess, K.N.; Bakhtina, A.A.; Valente, A.S.; Rodríguez-Mias, R.A.; Villén, J. Identification of phosphosites that alter protein thermal stability. Nat. Methods 2021, 18, 760–762. [Google Scholar] [CrossRef] [PubMed]
- Sandtner, W.; Egwolf, B.; Khalili-Araghi, F.; Sanchez-Rodriguez, J.E.; Roux, B.; Bezanilla, F.; Holmgren, M. Ouabain binding site in a functioning Na+/K+ ATPase. J. Biol. Chem. 2011, 286, 38177–38183. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, H.; Shinoda, T.; Cornelius, F.; Toyoshima, C. Crystal structure of the sodium-potassium pump (Na+,K+-ATPase) with bound potassium and ouabain. Proc. Natl. Acad. Sci. USA 2009, 106, 13742–13747. [Google Scholar] [CrossRef] [Green Version]
- Yatime, L.; Laursen, M.; Morth, J.P.; Esmann, M.; Nissen, P.; Fedosova, N.U. Structural insights into the high affinity binding of cardiotonic steroids to the Na+,K+-ATPase. J. Struct. Biol. 2011, 174, 296–306. [Google Scholar] [CrossRef]
- Bantscheff, M.; Hopf, C.; Savitski, M.M.; Dittmann, A.; Grandi, P.; Michon, A.M.; Schlegl, J.; Abraham, Y.; Becher, I.; Bergamini, G.; et al. Chemoproteomics profiling of HDAC inhibitors reveals selective targeting of HDAC complexes. Nat. Biotechnol. 2011, 29, 255–265. [Google Scholar] [CrossRef]
- Perrin, J.; Werner, T.; Kurzawa, N.; Rutkowska, A.; Childs, D.D.; Kalxdorf, M.; Poeckel, D.; Stonehouse, E.; Strohmer, K.; Heller, B.; et al. Identifying drug targets in tissues and whole blood with thermal-shift profiling. Nat. Biotechnol. 2020, 38, 303–308. [Google Scholar] [CrossRef]
- Gao, Y.; Davies, S.P.; Augustin, M.; Woodward, A.; Patel, U.A.; Kovelman, R.; Harvey, K.J. A broad activity screen in support of a chemogenomic map for kinase signalling research and drug discovery. Biochem. J. 2013, 451, 313–328. [Google Scholar] [CrossRef]
- Davis, M.I.; Hunt, J.P.; Herrgard, S.; Ciceri, P.; Wodicka, L.M.; Pallares, G.; Hocker, M.; Treiber, D.K.; Zarrinkar, P.P. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29, 1046–1051. [Google Scholar] [CrossRef]
- Hillenmeyer, M.E.; Ericson, E.; Davis, R.W.; Nislow, C.; Koller, D.; Giaever, G. Systematic analysis of genome-wide fitness data in yeast reveals novel gene function and drug action. Genome Biol. 2010, 11, R30. [Google Scholar] [CrossRef] [Green Version]
- Ryan, C.J.; Krogan, N.J.; Cunningham, P.; Cagney, G. All or nothing: Protein complexes flip essentiality between distantly related eukaryotes. Genome Biol. Evol. 2013, 5, 1049–1059. [Google Scholar] [CrossRef] [Green Version]
- Schellman, J.A. The thermodynamics of solvent exchange. Biopolymers 1994, 34, 1015–1026. [Google Scholar] [CrossRef]
- Childs, D.; Bach, K.; Franken, H.; Anders, S.; Kurzawa, N.; Bantscheff, M.; Savitski, M.M.; Huber, W. Nonparametric Analysis of Thermal Proteome Profiles Reveals Novel Drug-binding Proteins. Mol. Cell. Proteom. 2019, 18, 2506–2515. [Google Scholar] [CrossRef] [Green Version]
- Neubig, R.R.; Spedding, M.; Kenakin, T.; Christopoulos, A. International Union of Pharmacology Committee on Receptor Nomenclature and Drug Classification. XXXVIII. Update on terms and symbols in quantitative pharmacology. Pharmacol. Rev. 2003, 55, 597–606. [Google Scholar] [CrossRef] [Green Version]
- McCracken, N.A.; Peck Justice, S.A.; Wijeratne, A.B.; Mosley, A.L. Inflect: Optimizing Computational Workflows for Thermal Proteome Profiling Data Analysis. J. Proteome Res. 2021, 20, 1874–1888. [Google Scholar] [CrossRef]
- Spiess, A.N.; Neumeyer, N. An evaluation of R2 as an inadequate measure for nonlinear models in pharmacological and biochemical research: A Monte Carlo approach. BMC Pharmacol. 2010, 10, 6. [Google Scholar] [CrossRef] [Green Version]
- Schabenberger, O.P.F.J. Contemporary Statistical Models for the Plant and Soil Sciences; CRC Press: Boca Raton, FL, USA, 2002. [Google Scholar]
- Cui, Y.; Zhang, X.; Yu, M.; Zhu, Y.; Xing, J.; Lin, J. Techniques for detecting protein-protein interactions in living cells: Principles, limitations, and recent progress. Sci. China Life Sci. 2019, 62, 619–632. [Google Scholar] [CrossRef]
- O’Reilly, F.J.; Rappsilber, J. Cross-linking mass spectrometry: Methods and applications in structural, molecular and systems biology. Nat. Struct. Mol. Biol. 2018, 25, 1000–1008. [Google Scholar] [CrossRef] [Green Version]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; Garcia-Seisdedos, D.; Hewapathirana, S.; Kamatchinathan, S.; Kundu, D.J.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE database resources in 2022: A hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022, 50, D543–D552. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LIVING CELLS | Cell Extract a | ΔTm to DMSO in Cell Extract Compared to Living Cells/°C | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Protein | Inhibitor | n | Tm/°C | Std. Error | ΔTm to DMSO/°C | p-Value (ANOVA, to DMSO) | Tm/°C | Std. Error | ΔTm to DMSO/°C | p-Value (ANOVA, to DMSO) | |
| MAPK14 | AMG-548 | 7 | 60.58 | 0.52 | 14.76 | 4.1 × 10−46 ***,b | 59.15 | 0.57 | 13.60 | 5.8 × 10−22 *** | −1.16 |
| SB203580 | 4 | 52.86 | 0.35 | 7.04 | 4.3 × 10−25 *** | 56.86 | 0.45 | 11.32 | 2.7 × 10−26 *** | 4.28 | |
| ERK 11e | 4 | 45.56 | 0.43 | −0.26 | 0.17 | 45.15 | 0.53 | −0.39 | 0.64 | −0.13 | |
| DMSO | 4 | 45.82 | 0.36 | 45.54 | 0.52 | ||||||
| MAPKAPK3 | AMG-548 | 6 | 54.68 | 0.36 | 6.62 | 3.7 × 10−15 *** | 49.31 | 0.36 | 2.63 | 9.0 × 10−7 *** | −3.99 |
| SB203580 | 3 | 51.56 | 0.48 | 3.50 | 2.2 × 10−7 *** | 47.42 | 0.34 | 0.74 | 0.21 | −2.76 | |
| ERK 11e | 3 | 48.32 | 0.46 | 0.26 | 0.57 | 46.13 | 0.39 | −0.55 | 0.24 | −0.81 | |
| DMSO | 3 | 48.06 | 0.40 | 46.68 | 0.36 | ||||||
| MAPKAPK2 | AMG-548 | 6 | 54.39 | 0.29 | 5.18 | 5.2 × 10−7 *** | 51.06 | 0.48 | 2.96 | 3.5 × 10−4 *** | −2.22 |
| SB203580 | 3 | 50.96 | 0.56 | 1.75 | 0.18 | 49.35 | 0.46 | 1.24 | 0.13 | −0.51 | |
| ERK 11e | 3 | 49.19 | 1.03 | −0.02 | 0.99 | 48.33 | 0.49 | 0.23 | 0.91 | 0.25 | |
| DMSO | 3 | 49.20 | 1.40 | 48.1 | 0.48 | ||||||
| MAPKAPK2p | AMG-548 | n.d. c | 50.95 | 0.6 | 2.82 | 0.014 * | |||||
| SB203580 | n.d. c | 49.03 | 0.65 | 0.89 | 0.63 | ||||||
| ERK11e | 1 | 47.18 | 0.90 | −1.07 | 0.13 | 48.34 | 0.58 | 0.21 | 0.65 | 1.16 | |
| DMSO | 1 | 48.25 | 0.49 | 48.13 | 0.70 | −0.12 | |||||
| GSK-3α | AMG-548 | 7 | 53.26 | 0.25 | −0.71 | 0.24 | 48.26 | 0.45 | −0.76 | 0.31 | −0.05 |
| SB203580 | 4 | 54.03 | 0.33 | 0.05 | 0.82 | 49.77 | 0.44 | 0.75 | 0.45 | 0.70 | |
| ERK 11e | 4 | 54.11 | 0.30 | 0.14 | 0.94 | 50.25 | 0.43 | 1.24 | 0.11 | 1.10 | |
| DMSO | 4 | 53.97 | 0.31 | 49.02 | 0.45 | ||||||
| Inhibitor | Protein | Living Cells a | Cell Extract a | ||
|---|---|---|---|---|---|
| pEC50 | Std. Error | pEC50 | Std. Error | ||
| AMG-548 | MAPK14 | 8.93 | 0.15 | 7.90 | 0.05 |
| MAPKAPK3 | 8.97 | 0.16 | 8.06 | 0.13 | |
| MAPKAPK2 | 8.78 | 0.17 | 8.03 | 0.17 | |
| MAPKAPK2p | 8.80 | 0.12 | 7.78 | 0.18 | |
| SB203580 | MAPK14 | 5.34 | 0.13 | 5.85 | 0.08 |
| MAPKAPK3 | 4.98 | 0.29 | n.d.b | ||
| MAPKAPK2 | 5.20 | 0.21 | n.d.b | ||
| MAPKAPK2p | 6.39 | 0.22 | n.d.b | ||
| Inhibitor | Protein | pEC50 | Std. Error | pseudo R2 | n a |
|---|---|---|---|---|---|
| AMG-548 | MAPK14 | 8.33 | 0.04 | 0.98 | 5 |
| MAPKAPK3 | 8.60 | 0.05 | 0.96 | 5 | |
| MAPKAPK2 | 8.60 | 0.05 | 0.95 | 5 | |
| MYLK | 8.22 | 0.15 | 0.77 | 5 | |
| USP24 | 8.57 | 0.13 | 0.75 | 5 | |
| STX4 | 6.88 | 0.30 | 0.71 | 2 | |
| HN1L | 6.41 | 0.17 | 0.69 | 5 | |
| MARS | 6.68 | 0.22 | 0.64 | 4 | |
| NUDT1 | 5.64 | 0.13 | 0.62 | 5 | |
| MAPK8 | 7.91 | 0.46 | 0.58 | 2 | |
| TRAPPC4 | 6.39 | 0.21 | 0.57 | 5 | |
| MRPS34 | 7.22 | 0.44 | 0.55 | 2 | |
| MTMR1 | 6.98 | 0.39 | 0.55 | 2 | |
| SB203580 | MAPKAPK2 | 6.15 | 0.06 | 0.94 | 4 |
| MAPKAPK3 | 6.05 | 0.06 | 0.94 | 4 | |
| MAPK14 | 5.71 | 0.06 | 0.93 | 4 | |
| NAPG | 8.71 | 0.44 | 0.73 | 2 | |
| WDR46 | 7.49 | 0.42 | 0.61 | 2 | |
| MAPK8 | 6.50 | 0.33 | 0.58 | 2 | |
| MYLK | 6.02 | 0.30 | 0.58 | 2 | |
| C14orf166 | 6.39 | 0.29 | 0.52 | 4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lenz, T.; Stühler, K. Small Molecule Arranged Thermal Proximity Coaggregation (smarTPCA)—A Novel Approach to Characterize Protein–Protein Interactions in Living Cells by Similar Isothermal Dose–Responses. Int. J. Mol. Sci. 2022, 23, 5605. https://doi.org/10.3390/ijms23105605
Lenz T, Stühler K. Small Molecule Arranged Thermal Proximity Coaggregation (smarTPCA)—A Novel Approach to Characterize Protein–Protein Interactions in Living Cells by Similar Isothermal Dose–Responses. International Journal of Molecular Sciences. 2022; 23(10):5605. https://doi.org/10.3390/ijms23105605
Chicago/Turabian StyleLenz, Thomas, and Kai Stühler. 2022. "Small Molecule Arranged Thermal Proximity Coaggregation (smarTPCA)—A Novel Approach to Characterize Protein–Protein Interactions in Living Cells by Similar Isothermal Dose–Responses" International Journal of Molecular Sciences 23, no. 10: 5605. https://doi.org/10.3390/ijms23105605
APA StyleLenz, T., & Stühler, K. (2022). Small Molecule Arranged Thermal Proximity Coaggregation (smarTPCA)—A Novel Approach to Characterize Protein–Protein Interactions in Living Cells by Similar Isothermal Dose–Responses. International Journal of Molecular Sciences, 23(10), 5605. https://doi.org/10.3390/ijms23105605