
Epigenetic Silencing of PTEN and Epi-Transcriptional Silencing of MDM2 Underlied Progression to Secondary Acute Myeloid Leukemia in Myelodysplastic Syndrome Treated with Hypomethylating Agents

 , , , , ,
, , , , , 
Abstract
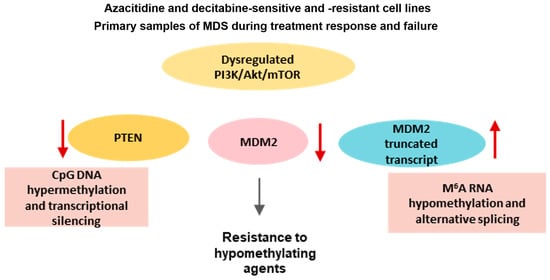
:
1. Introduction
2. Method
2.1. Cell Lines and Cell Cultures
2.2. Cell Viability Assays
2.3. DNA Whole Methylome Analysis
2.4. Whole Transcriptome Analysis
2.5. RNA Modifications and Alternative Splicing Detection
2.6. Pyrosequencing and Quantitative Polymerase Chain Reaction
2.7. Patients and Samples
2.8. Statistical Analyses and Data Sharing
3. Results
3.1. Establishment of AZA-R and DEC-R Sublines
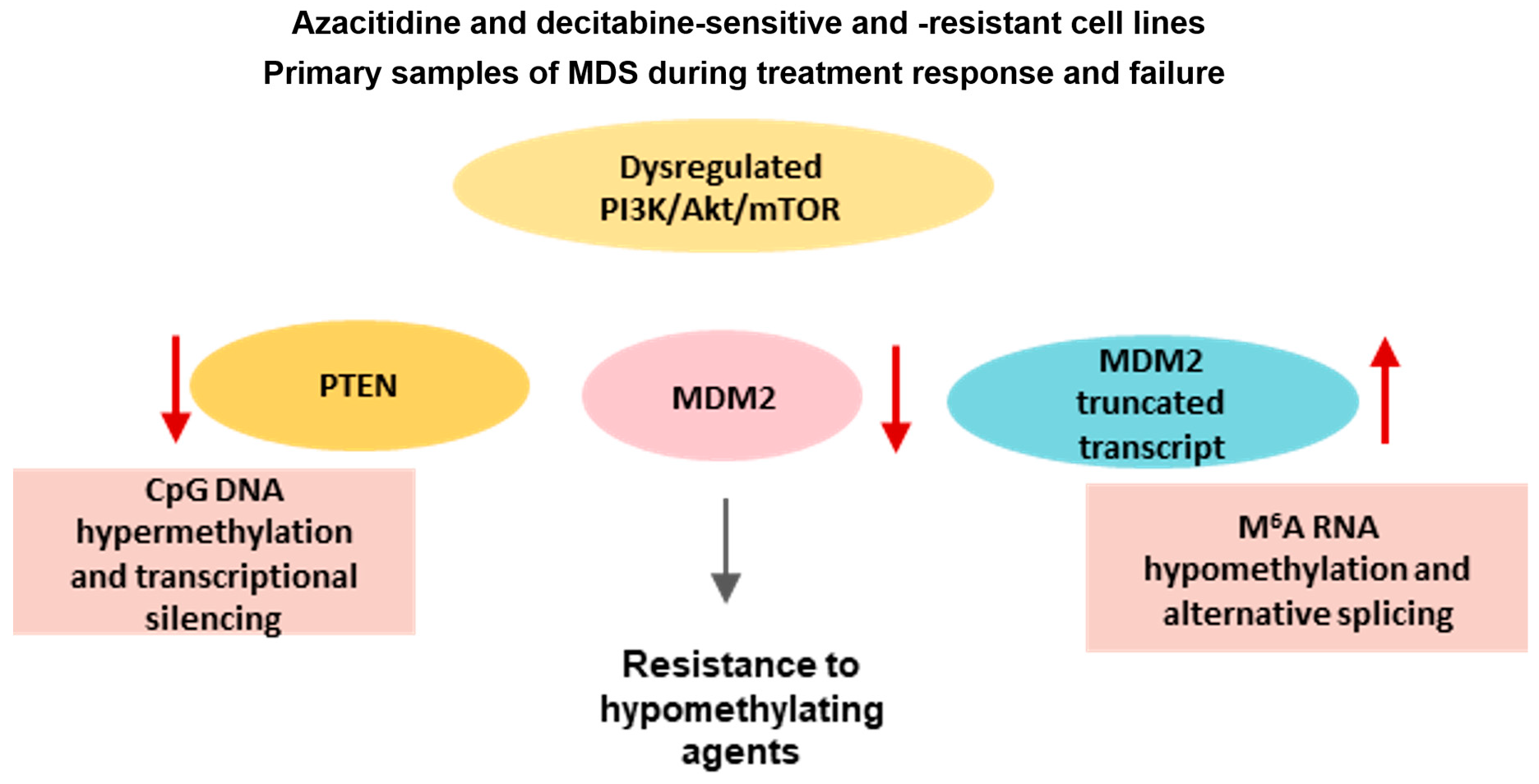
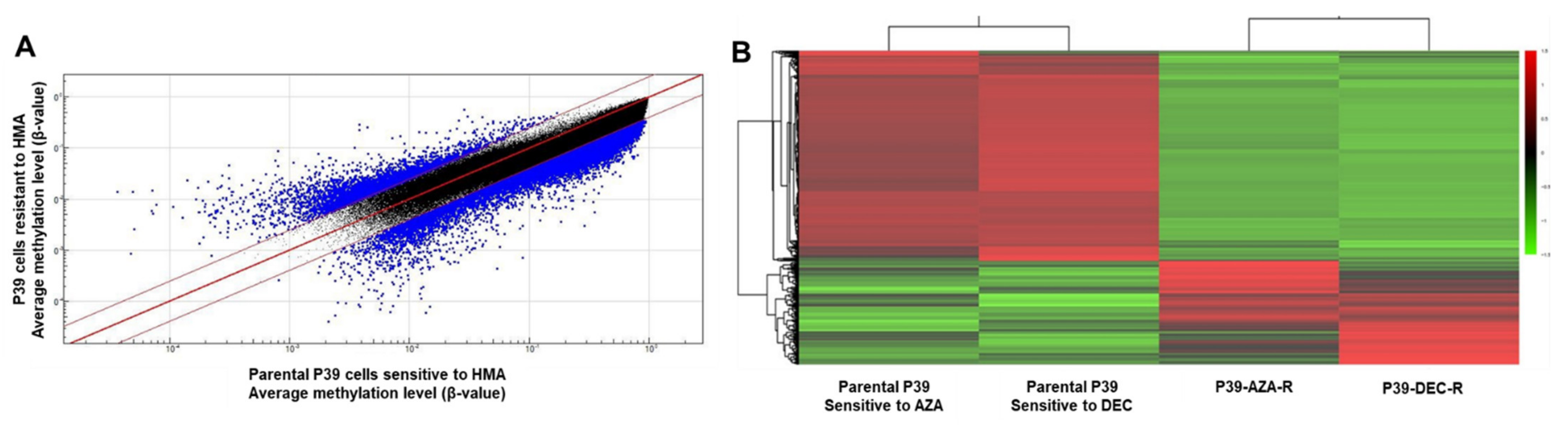
3.2. Methylome Dysregulation during HMA Resistance in Cell Lines
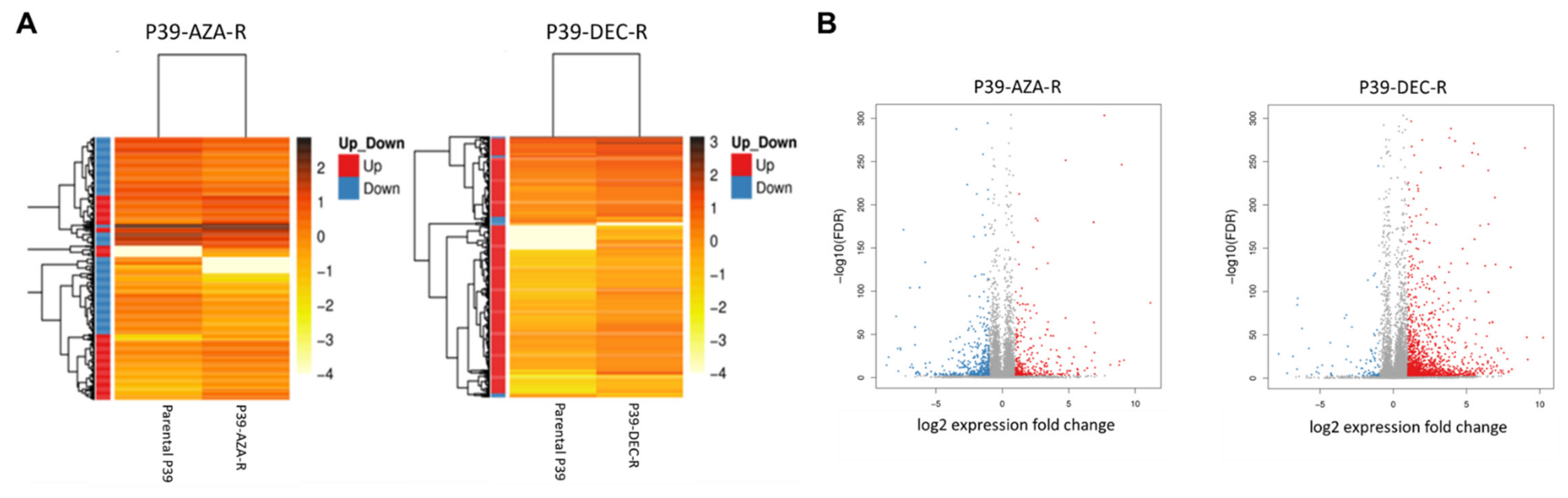
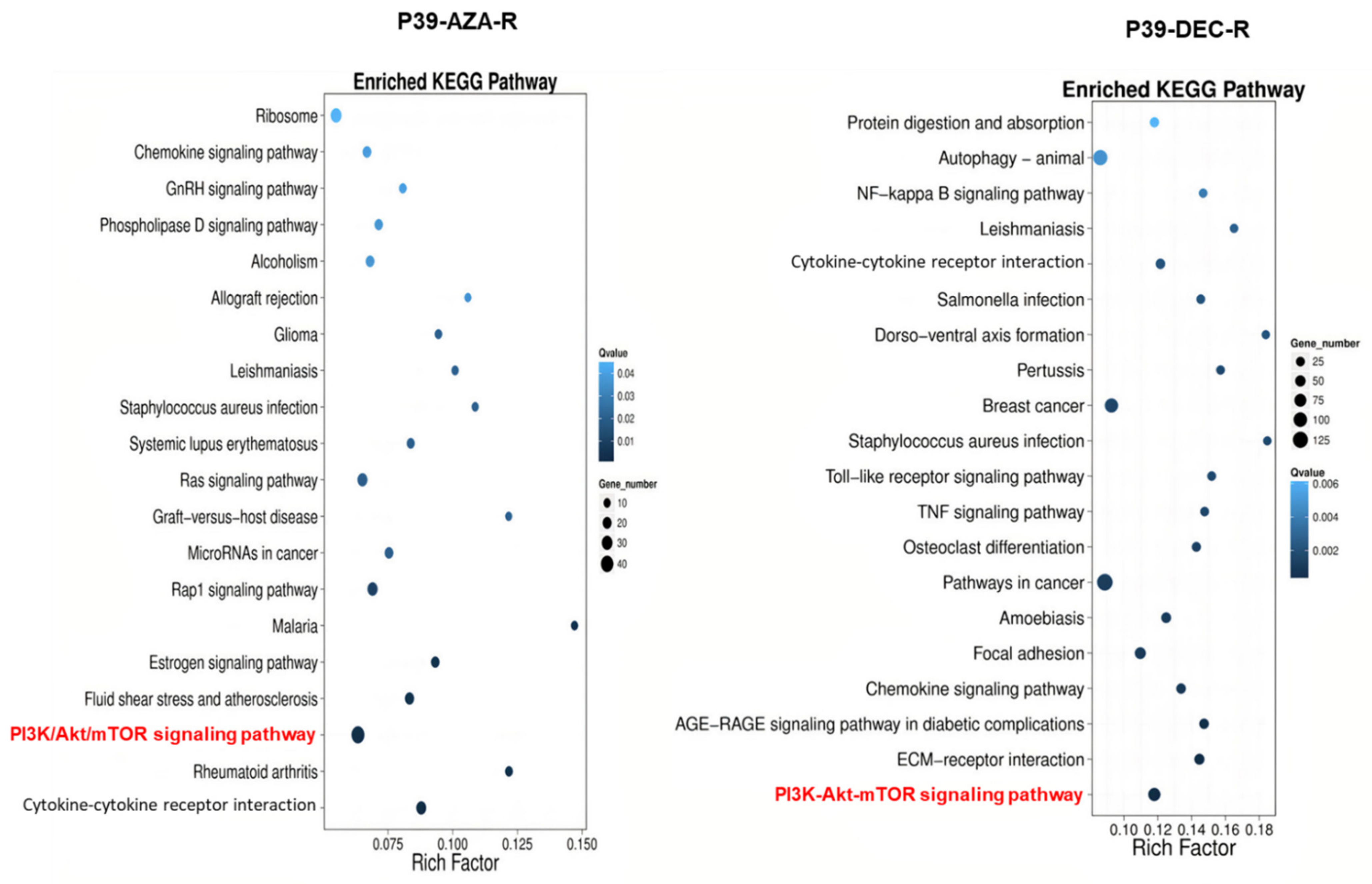
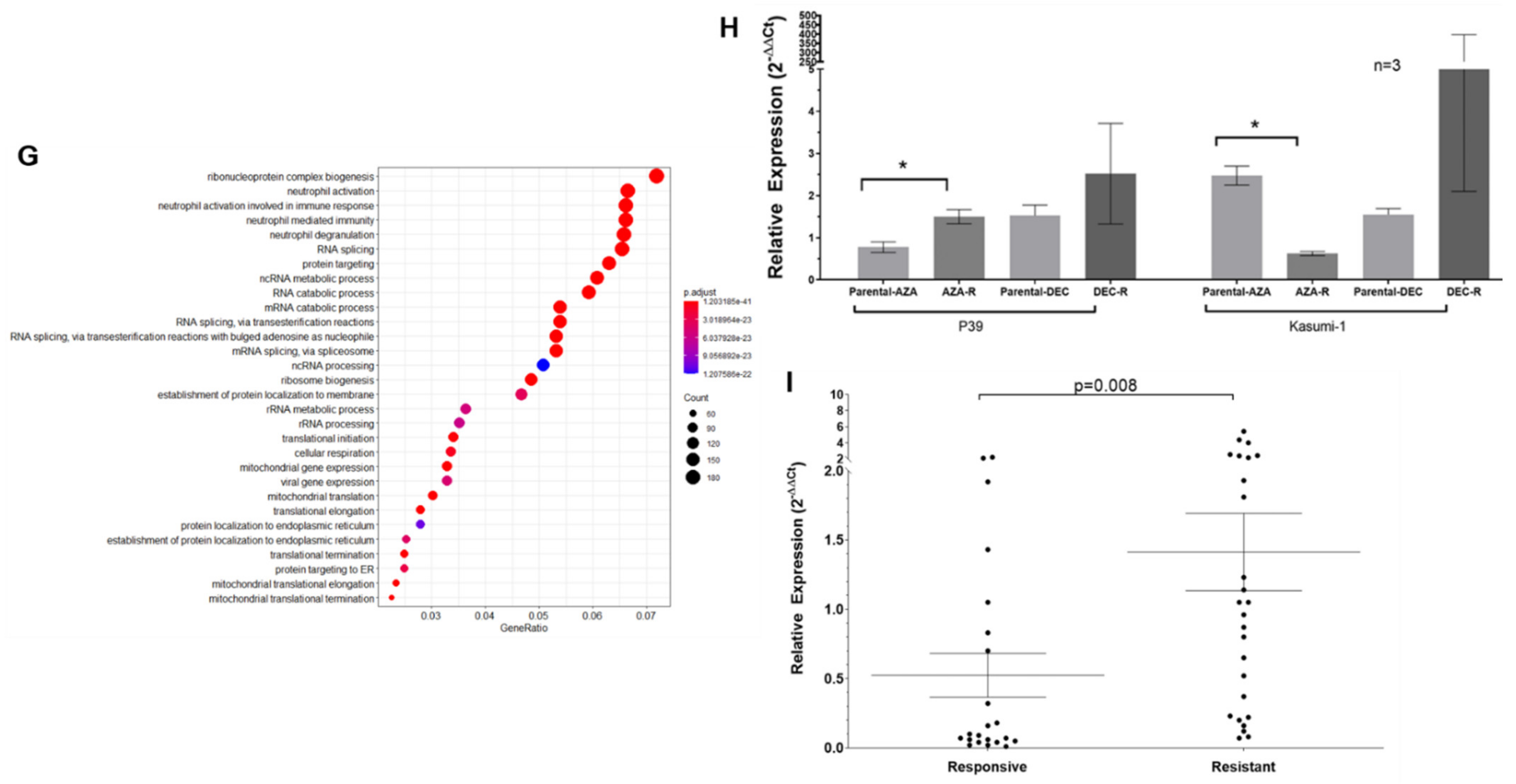
3.3. Transcriptomic Dysregulation and Corresponding DNA Methylomic Changes in HMA-Resistant Cell Lines
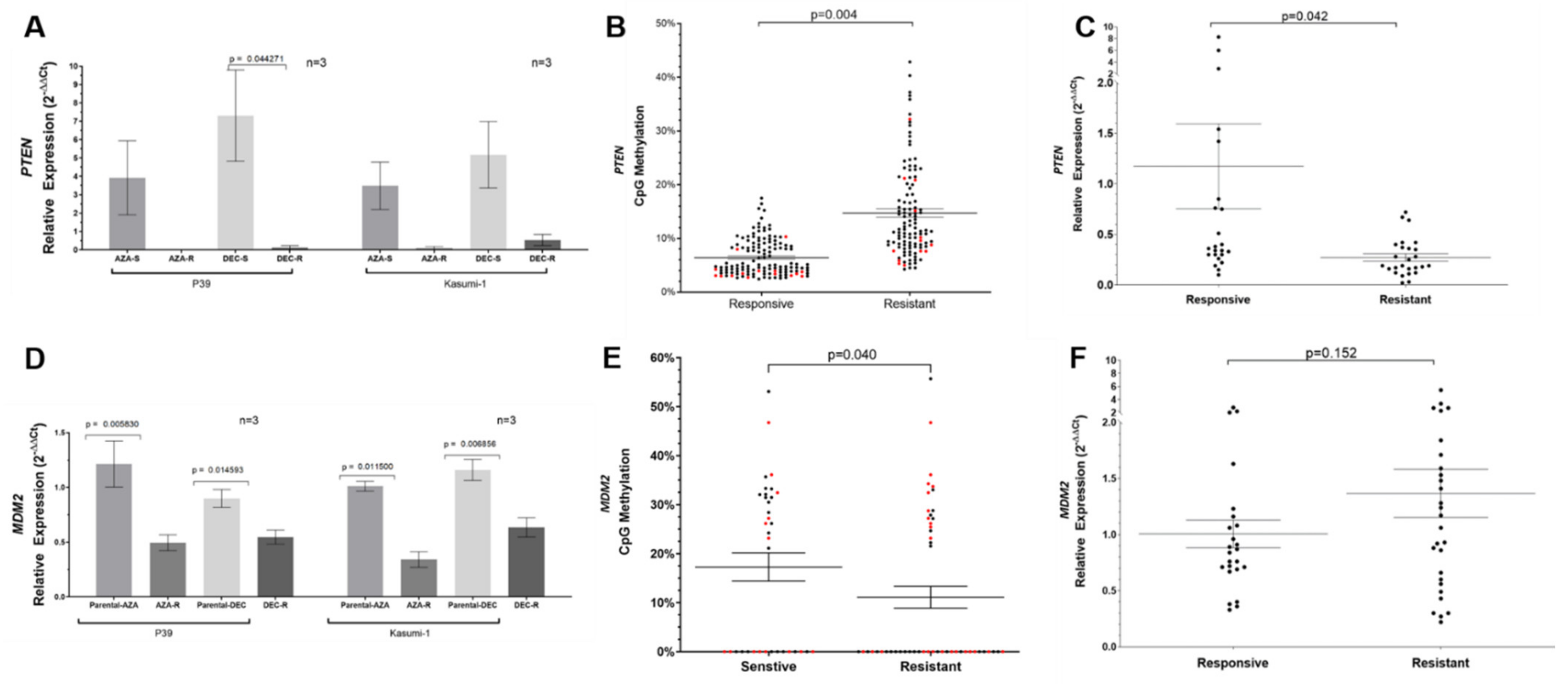
3.4. Aberrant DNA Methylation Associated with PTEN and MDM2 Dysregulation in HMA-Resistant Cell Lines
3.5. Epi-Transcriptomic Profiling of P39 and P39-AZA-R
3.6. Validation in Primary MDS Patient Samples
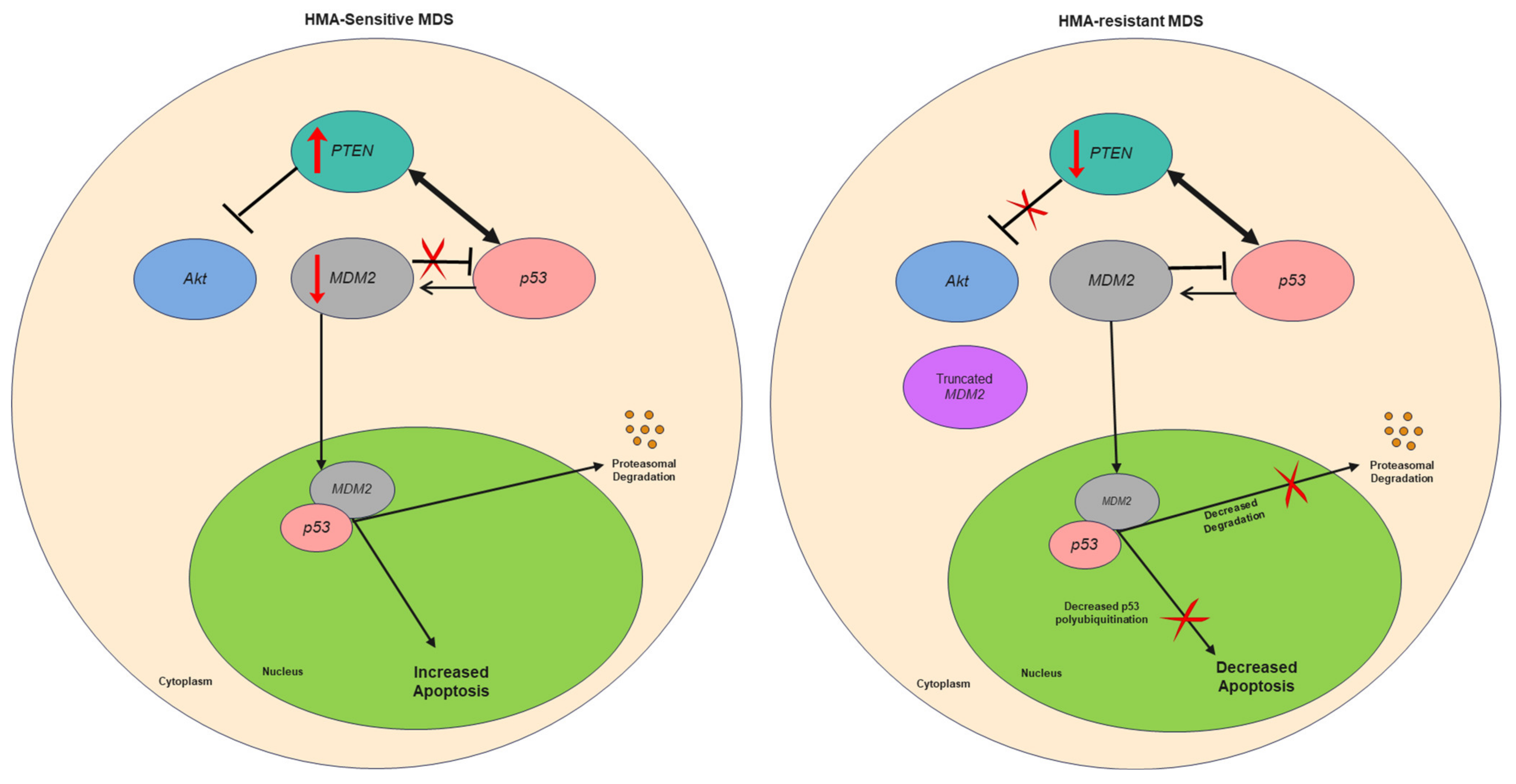
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Walter, M.J.; Shen, D.; Ding, L.; Shao, J.; Koboldt, D.C.; Chen, K.; Larson, D.E.; McLellan, M.D.; Dooling, D.; Abbott, R.; et al. Clonal architecture of secondary acute myeloid leukemia. N. Engl. J. Med. 2012, 366, 1090–1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woll, P.S.; Kjallquist, U.; Chowdhury, O.; Doolittle, H.; Wedge, D.C.; Thongjuea, S.; Erlandsson, R.; Ngara, M.; Anderson, K.; Deng, Q.; et al. Myelodysplastic syndromes are propagated by rare and distinct human cancer stem cells in vivo. Cancer Cell 2014, 25, 794–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gill, H.; Leung, A.Y.; Kwong, Y.L. Molecular and Cellular Mechanisms of Myelodysplastic Syndrome: Implications on Targeted Therapy. Int. J. Mol. Sci. 2016, 17, 440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Platzbecker, U. Treatment of MDS. Blood 2019, 133, 1096–1107. [Google Scholar] [CrossRef] [Green Version]
- Jabbour, E.; Short, N.J.; Montalban-Bravo, G.; Huang, X.; Bueso-Ramos, C.; Qiao, W.; Yang, H.; Zhao, C.; Kadia, T.; Borthakur, G.; et al. Randomized phase 2 study of low-dose decitabine vs low-dose azacitidine in lower-risk MDS and MDS/MPN. Blood 2017, 130, 1514–1522. [Google Scholar] [CrossRef] [Green Version]
- Tobiasson, M.; Abdulkadir, H.; Lennartsson, A.; Katayama, S.; Marabita, F.; De Paepe, A.; Karimi, M.; Krjutskov, K.; Einarsdottir, E.; Grovdal, M.; et al. Comprehensive mapping of the effects of azacitidine on DNA methylation, repressive/permissive histone marks and gene expression in primary cells from patients with MDS and MDS-related disease. Oncotarget 2017, 8, 28812–28825. [Google Scholar] [CrossRef]
- Makishima, H.; Yoshizato, T.; Yoshida, K.; Sekeres, M.A.; Radivoyevitch, T.; Suzuki, H.; Przychodzen, B.; Nagata, Y.; Meggendorfer, M.; Sanada, M.; et al. Dynamics of clonal evolution in myelodysplastic syndromes. Nat. Genet. 2017, 49, 204–212. [Google Scholar] [CrossRef]
- Itzykson, R.; Kosmider, O.; Cluzeau, T.; Mansat-De Mas, V.; Dreyfus, F.; Beyne-Rauzy, O.; Quesnel, B.; Vey, N.; Gelsi-Boyer, V.; Raynaud, S.; et al. Impact of TET2 mutations on response rate to azacitidine in myelodysplastic syndromes and low blast count acute myeloid leukemias. Leukemia 2011, 25, 1147–1152. [Google Scholar] [CrossRef] [Green Version]
- Traina, F.; Visconte, V.; Elson, P.; Tabarroki, A.; Jankowska, A.M.; Hasrouni, E.; Sugimoto, Y.; Szpurka, H.; Makishima, H.; O’Keefe, C.L.; et al. Impact of molecular mutations on treatment response to DNMT inhibitors in myelodysplasia and related neoplasms. Leukemia 2014, 28, 78–87. [Google Scholar] [CrossRef]
- Valencia, A.; Masala, E.; Rossi, A.; Martino, A.; Sanna, A.; Buchi, F.; Canzian, F.; Cilloni, D.; Gaidano, V.; Voso, M.T.; et al. Expression of nucleoside-metabolizing enzymes in myelodysplastic syndromes and modulation of response to azacitidine. Leukemia 2014, 28, 621–628. [Google Scholar] [CrossRef] [Green Version]
- Qin, T.; Castoro, R.; El Ahdab, S.; Jelinek, J.; Wang, X.; Si, J.; Shu, J.; He, R.; Zhang, N.; Chung, W.; et al. Mechanisms of resistance to decitabine in the myelodysplastic syndrome. PLoS ONE 2011, 6, e23372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santini, V. How I treat MDS after hypomethylating agent failure. Blood 2019, 133, 521–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meldi, K.; Qin, T.; Buchi, F.; Droin, N.; Sotzen, J.; Micol, J.B.; Selimoglu-Buet, D.; Masala, E.; Allione, B.; Gioia, D.; et al. Specific molecular signatures predict decitabine response in chronic myelomonocytic leukemia. J. Clin. Investig. 2015, 125, 1857–1872. [Google Scholar] [CrossRef] [PubMed]
- The Gene Ontology. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 2019, 47, D330–D338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, Y.; Shi, C.; Huang, Z.; Zhang, Y.; Li, S.; Li, Y.; Ye, J.; Yu, C.; Li, Z.; et al. SOAPnuke: A MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience 2018, 7, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; Van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Team, R.C. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013. [Google Scholar]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mi, H.; Muruganujan, A.; Ebert, D.; Huang, X.; Thomas, P.D. PANTHER version 14: More genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 2019, 47, D419–D426. [Google Scholar] [CrossRef] [PubMed]
- Jia, W.; Qiu, K.; He, M.; Song, P.; Zhou, Q.; Zhou, F.; Yu, Y.; Zhu, D.; Nickerson, M.L.; Wan, S.; et al. SOAPfuse: An algorithm for identifying fusion transcripts from paired-end RNA-Seq data. Genome Biol. 2013, 14, R12. [Google Scholar] [CrossRef] [Green Version]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [Green Version]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Loman, N.J.; Quick, J.; Simpson, J.T. A complete bacterial genome assembled de novo using only nanopore sequencing data. Nat. Methods 2015, 12, 733–735. [Google Scholar] [CrossRef]
- Pratanwanich, P.N.; Yao, F.; Chen, Y.; Koh, C.W.Q.; Hendra, C.; Poon, P.; Goh, Y.T.; Yap, P.M.L.; Yuan, C.J.; Chng, W.J.; et al. Detection of Differential RNA Modifications from Direct RNA Sequencing of Human Cell Lines; Cold Spring Harbor Laboratory: LongIsland, NY, USA, 2020. [Google Scholar]
- Steensma, D.P. P39/Tsugane cells are a false cell line contaminated with HL-60 cells and are not suitable for mechanistic studies in myelodysplastic syndromes. Haematologica 2010, 95, 1229–1230. [Google Scholar] [CrossRef] [Green Version]
- Drexler, H.G.; Dirks, W.G.; Macleod, R.A. Many are called MDS cell lines: One is chosen. Leuk. Res. 2009, 33, 1011–1016. [Google Scholar] [CrossRef]
- Khan, R.; Aggerholm, A.; Hokland, P.; Hassan, M.; Hellstrom-Lindberg, E. A pharmacodynamic study of 5-azacytidine in the P39 cell line. Exp. Hematol. 2006, 34, 35–43. [Google Scholar] [CrossRef]
- Tsujioka, T.; Yokoi, A.; Uesugi, M.; Kishimoto, M.; Tochigi, A.; Suemori, S.; Tohyama, Y.; Tohyama, K. Effects of DNA methyltransferase inhibitors (DNMTIs) on MDS-derived cell lines. Exp. Hematol. 2013, 41, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, E.C.; Grand, R.S.; Perry, J.K.; Vickers, M.H.; Olins, A.L.; Olins, D.E.; O’Sullivan, J.M. Hi-C detects novel structural variants in HL-60 and HL-60/S4 cell lines. Genomics 2020, 112, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Trotman, L.C.; Shaffer, D.; Lin, H.K.; Dotan, Z.A.; Niki, M.; Koutcher, J.A.; Scher, H.I.; Ludwig, T.; Gerald, W.; et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 2005, 436, 725–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, T.; Meng, X.; Wang, J.; Chen, X.; Yin, D.; Liang, Y.; Song, X.; Pan, S.; Jiang, H.; Liu, L. PTEN- and p53-mediated apoptosis and cell cycle arrest by FTY720 in gastric cancer cells and nude mice. J. Cell. Biochem. 2010, 111, 218–228. [Google Scholar] [CrossRef]
- Morotti, A.; Panuzzo, C.; Crivellaro, S.; Carra, G.; Torti, D.; Guerrasio, A.; Saglio, G. The Role of PTEN in Myeloid Malignancies. Hematol. Rep. 2015, 7, 5844. [Google Scholar] [CrossRef] [Green Version]
- Moody, J.L.; Xu, L.; Helgason, C.D.; Jirik, F.R. Anemia, thrombocytopenia, leukocytosis, extramedullary hematopoiesis, and impaired progenitor function in Pten+/-SHIP-/- mice: A novel model of myelodysplasia. Blood 2004, 103, 4503–4510. [Google Scholar] [CrossRef]
- Kim, J.; Eltoum, I.E.A.; Roh, M.; Wang, J.; Abdulkadir, S.A. Interactions between Cells with Distinct Mutations in c-MYC and Pten in Prostate Cancer. PLoS Genet. 2009, 5, e1000542. [Google Scholar] [CrossRef]
- Li, A.G.; Piluso, L.G.; Cai, X.; Wei, G.; Sellers, W.R.; Liu, X. Mechanistic insights into maintenance of high p53 acetylation by PTEN. Mol. Cell 2006, 23, 575–587. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research, N.; Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.J.; Robertson, A.; Hoadley, K.; Triche, T.J., Jr.; Laird, P.W.; et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J.Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Gao, L.; Luo, X.; Wang, L.; Gao, X.; Wang, W.; Sun, J.; Dou, L.; Li, J.; Xu, C.; et al. Epigenetic silencing of microRNA-193a contributes to leukemogenesis in t(8;21) acute myeloid leukemia by activating the PTEN/PI3K signal pathway. Blood 2013, 121, 499–509. [Google Scholar] [CrossRef]
- Liu, G.; Terzian, T.; Xiong, S.; Van Pelt, C.S.; Audiffred, A.; Box, N.F.; Lozano, G. The p53-Mdm2 network in progenitor cell expansion during mouse postnatal development. J. Pathol. 2007, 213, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Cumbo, C.; Tota, G.; Anelli, L.; Zagaria, A.; Specchia, G.; Albano, F. TP53 in Myelodysplastic Syndromes: Recent Biological and Clinical Findings. Int. J. Mol. Sci. 2020, 21, 3432. [Google Scholar] [CrossRef] [PubMed]
- Hunt, S.E.; McLaren, W.; Gil, L.; Thormann, A.; Schuilenburg, H.; Sheppard, D.; Parton, A.; Armean, I.M.; Trevanion, S.J.; Flicek, P.; et al. Ensembl variation resources. Database 2018, 2018, bay119. [Google Scholar] [CrossRef] [PubMed]
- Manfredi, J.J. The Mdm2-p53 relationship evolves: Mdm2 swings both ways as an oncogene and a tumor suppressor. Genes Dev. 2010, 24, 1580–1589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez-Monge, J.; Rousset-Roman, A.B.; Medina-Medina, I.; Olivares-Illana, V. Dual function of MDM2 and MDMX toward the tumor suppressors p53 and RB. Genes Cancer 2016, 7, 278–287. [Google Scholar] [CrossRef]
- Hu, B.; Gilkes, D.M.; Chen, J. Efficient p53 activation and apoptosis by simultaneous disruption of binding to MDM2 and MDMX. Cancer Res. 2007, 67, 8810–8817. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.; Liang, R.; Bai, T.; Lin, Y.; Mai, R.; Wei, M.; Ye, X.; Li, L.; Wu, F. RBM38 plays a tumor-suppressor role via stabilizing the p53-mdm2 loop function in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2018, 37, 212. [Google Scholar] [CrossRef]
- McGraw, K.L.; Cluzeau, T.; Sallman, D.A.; Basiorka, A.A.; Irvine, B.A.; Zhang, L.; Epling-Burnette, P.K.; Rollison, D.E.; Mallo, M.; Sokol, L.; et al. TP53 and MDM2 single nucleotide polymorphisms influence survival in non-del(5q) myelodysplastic syndromes. Oncotarget 2015, 6, 34437–34445. [Google Scholar] [CrossRef]
- Falk, I.J.; Willander, K.; Chaireti, R.; Lund, J.; Nahi, H.; Hermanson, M.; Green, H.; Lotfi, K.; Soderkvist, P. TP53 mutations and MDM2(SNP309) identify subgroups of AML patients with impaired outcome. Eur. J. Haematol. 2015, 94, 355–362. [Google Scholar] [CrossRef] [Green Version]
- Wei, S.; Chen, X.; McGraw, K.; Zhang, L.; Komrokji, R.; Clark, J.; Caceres, G.; Billingsley, D.; Sokol, L.; Lancet, J.; et al. Lenalidomide promotes p53 degradation by inhibiting MDM2 auto-ubiquitination in myelodysplastic syndrome with chromosome 5q deletion. Oncogene 2013, 32, 1110–1120. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, S. Genetics of MDS. Blood 2019, 133, 1049–1059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pleyer, L.; Valent, P.; Greil, R. Mesenchymal Stem and Progenitor Cells in Normal and Dysplastic Hematopoiesis-Masters of Survival and Clonality? Int. J. Mol. Sci. 2016, 17, 1009. [Google Scholar] [CrossRef] [PubMed]
- Mercer, T.R.; Dinger, M.E.; Mattick, J.S. Long non-coding RNAs: Insights into functions. Nat. Rev. Genet. 2009, 10, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Kong, X.; Zhang, Y.; Sun, W.; Xu, E.; Chen, X. Mdm2 is a target and mediator of IRP2 in cell growth control. FASEB J. 2020, 34, 2301–2311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCubrey, J.A.; Steelman, L.S.; Abrams, S.L.; Bertrand, F.E.; Ludwig, D.E.; Basecke, J.; Libra, M.; Stivala, F.; Milella, M.; Tafuri, A.; et al. Targeting survival cascades induced by activation of Ras/Raf/MEK/ERK, PI3K/PTEN/Akt/mTOR and Jak/STAT pathways for effective leukemia therapy. Leukemia 2008, 22, 708–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steelman, L.S.; Abrams, S.L.; Whelan, J.; Bertrand, F.E.; Ludwig, D.E.; Basecke, J.; Libra, M.; Stivala, F.; Milella, M.; Tafuri, A.; et al. Contributions of the Raf/MEK/ERK, PI3K/PTEN/Akt/mTOR and Jak/STAT pathways to leukemia. Leukemia 2008, 22, 686–707. [Google Scholar] [CrossRef] [Green Version]
- Morris, L.G.; Chan, T.A. Therapeutic targeting of tumor suppressor genes. Cancer 2015, 121, 1357–1368. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line * | CpG 1 (%) | CpG 2 (%) | CpG 3 (%) | CpG 4 (%) | CpG 5 (%) | CpG 6# (%) | CpG 7 (%) | CpG 8 (%) | CpG 9 (%) | Mean (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| Parental P39 sensitive to AZA ** | 5.95 | 7.78 | 7.41 | 11.91 | 7.23 | 3.96 | 3.54 | 6.04 | 14.1 | 7.55 |
| Parental P39 sensitive to DEC ** | 4.91 | 6.5 | 6.91 | 12.37 | 7.57 | 4.39 | 3.8 | 6.7 | 13.7 | 7.43 |
| P39-AZA-R | 4.87 | 7.46 | 6.82 | 13.07 | 8.69 | 4.67 | 5.01 | 7.22 | 15.16 | 8.11 |
| P39-DEC-R | 5.47 | 7.88 | 8.07 | 12.93 | 8.4 | 4.35 | 3.94 | 6.87 | 15.16 | 8.12 |
| Parental Kasumi-1 sensitive to AZA ** | 5.86 | 7.98 | 5.61 | 11.57 | 7.56 | 4.53 | 4.22 | 6.09 | 13.48 | 7.43 |
| Parental Kasumi-1 sensitive to DEC ** | 4.86 | 7.69 | 5.35 | 11.75 | 6.66 | 4.31 | 4.54 | 6.89 | 14.45 | 7.39 |
| Kasumi-1-AZA-R | 6.56 | 8.52 | 7.35 | 15.41 | 7.77 | 5.67 | 6.53 | 8.21 | 18.82 | 9.43 |
| Kasumi-1-DEC-R | 4.61 | 7.26 | 5.74 | 11.18 | 6.07 | 4.19 | 3.74 | 5.52 | 12.34 | 6.74 |
| Cell Line * | CpG 1 (%) | CpG 2 (%) # | Mean (%) |
|---|---|---|---|
| Parental P39 sensitive to AZA ** | 45.92 | 28.05 | 36.99 |
| Parental P39 sensitive to DEC ** | 56.31 | 32.24 | 44.28 |
| P39-AZA-R | 26.37 | 16.77 | 21.57 |
| P39-DEC-R | 31.44 | 21.71 | 26.58 |
| Parental Kasumi-1 sensitive to AZA ** | 57.97 | 35.89 | 46.93 |
| Parental Kasumi-1 sensitive to DEC ** | 66.45 | 42.91 | 54.68 |
| Kasumi-1-AZA-R | 37.49 | 19.56 | 28.53 |
| Kasumi-1-DEC-R | 44.07 | 26.95 | 35.51 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, P.; Yim, R.; Miu, K.-K.; Fung, S.-H.; Liao, J.J.; Wang, Z.; Li, J.; Yung, Y.; Chu, H.-T.; Yip, P.-K.; et al. Epigenetic Silencing of PTEN and Epi-Transcriptional Silencing of MDM2 Underlied Progression to Secondary Acute Myeloid Leukemia in Myelodysplastic Syndrome Treated with Hypomethylating Agents. Int. J. Mol. Sci. 2022, 23, 5670. https://doi.org/10.3390/ijms23105670
Lee P, Yim R, Miu K-K, Fung S-H, Liao JJ, Wang Z, Li J, Yung Y, Chu H-T, Yip P-K, et al. Epigenetic Silencing of PTEN and Epi-Transcriptional Silencing of MDM2 Underlied Progression to Secondary Acute Myeloid Leukemia in Myelodysplastic Syndrome Treated with Hypomethylating Agents. International Journal of Molecular Sciences. 2022; 23(10):5670. https://doi.org/10.3390/ijms23105670
Chicago/Turabian StyleLee, Paul, Rita Yim, Kai-Kei Miu, Sin-Hang Fung, Jason Jinyue Liao, Zhangting Wang, Jun Li, Yammy Yung, Hiu-Tung Chu, Pui-Kwan Yip, and et al. 2022. "Epigenetic Silencing of PTEN and Epi-Transcriptional Silencing of MDM2 Underlied Progression to Secondary Acute Myeloid Leukemia in Myelodysplastic Syndrome Treated with Hypomethylating Agents" International Journal of Molecular Sciences 23, no. 10: 5670. https://doi.org/10.3390/ijms23105670
APA StyleLee, P., Yim, R., Miu, K. -K., Fung, S. -H., Liao, J. J., Wang, Z., Li, J., Yung, Y., Chu, H. -T., Yip, P. -K., Lee, E., Tse, E., Kwong, Y. -L., & Gill, H. (2022). Epigenetic Silencing of PTEN and Epi-Transcriptional Silencing of MDM2 Underlied Progression to Secondary Acute Myeloid Leukemia in Myelodysplastic Syndrome Treated with Hypomethylating Agents. International Journal of Molecular Sciences, 23(10), 5670. https://doi.org/10.3390/ijms23105670