
Structural Bases of Prion Variation in Yeast



{kind=link}
Abstract
:1. Amyloids and Prions—A General Description
2. Types of Amyloid Architecture
2.1. In-Register Arrangement
2.2. Helical, or Solenoid Type
2.3. Other Types
3. Yeast Prions, a Model for Prion/Amyloid Studies
4. [PSI+] Variants
4.1. Discrimination of [PSI+] Variants
4.2. [PSI+] Variants Obtained in Modified Backgrounds
5. Sup35 Variant Prion Structures
5.1. Methodological Note
5.2. Overall Prion Structure of the N and M Domains
5.3. Core 1 Structure
5.3.1. Amyloid β-Sheet Regions Can Be Smaller Than Those Protected from PK
5.3.2. Proline Residues Could Restrict Amyloid β-Sheet Structures
5.3.3. Probable Nested Structure of the Core 1 Variants
5.3.4. On the Importance of the Core 1
5.4. Contradictory Structures of the M Domain
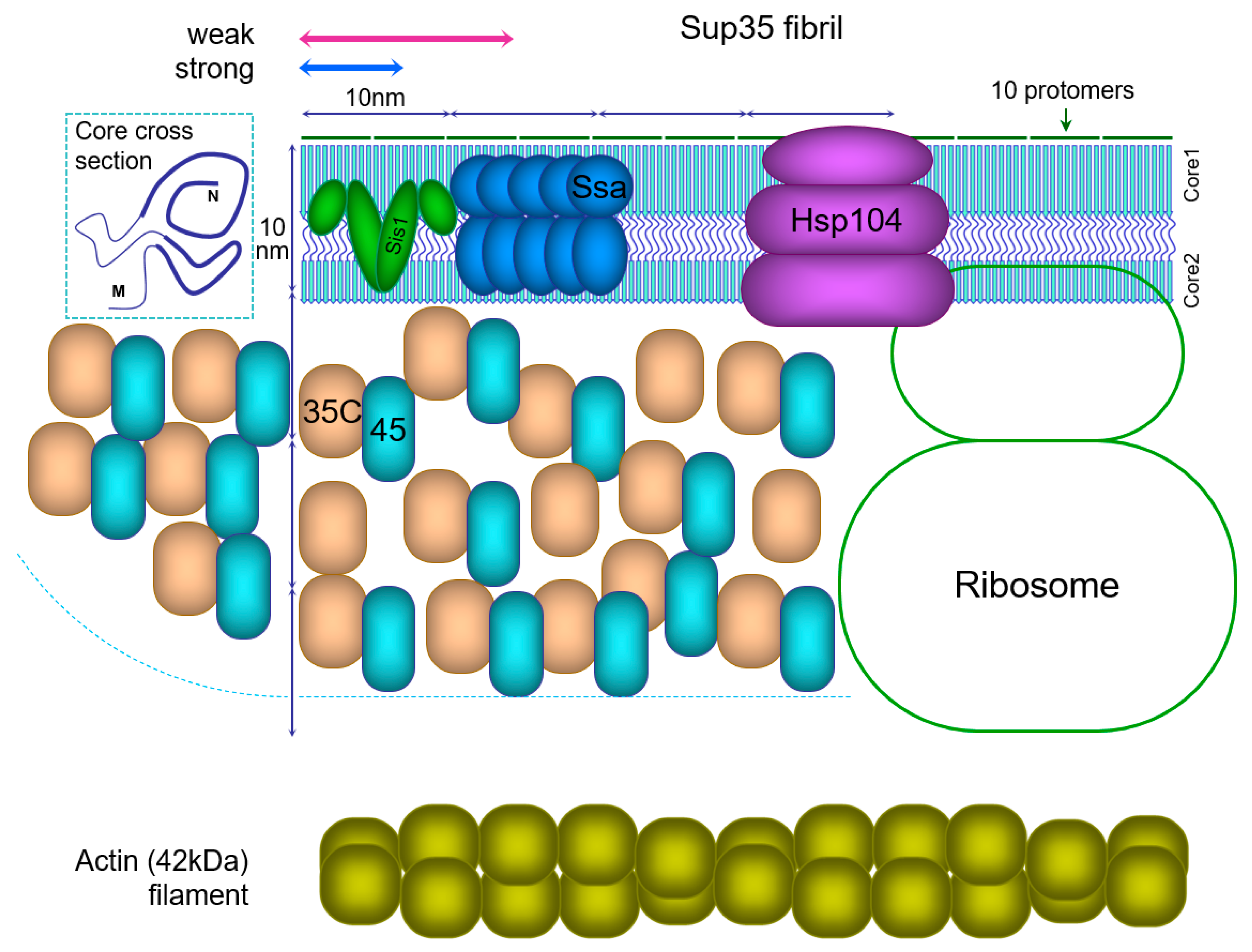
5.5. Structural Reconstruction of the Sup35 Prion Particle

6. The Mechanisms Relating Sup35 Prion Structure to Phenotype
6.1. The Balance of Soluble and Aggregated Sup35
6.2. Causes for the Reduced Translation Termination Activity of the Sup35 Prion Form
7. [URE3] and [RNQ+] Variants
7.1. [URE3] Prion Variants
7.2. Incorrect Use of PrD Hybrid Proteins
7.3. [RNQ+] Prion Variants
8. Conclusions
- Why are the terminal regions of prion domains the most important for prion properties and the phenotype? Do the interactions of unfolded regions with chaperones depend on the distance from the protein terminus?
- Why is Core 1 of the Sup35 prion so critical in yeast, but is not required for, or can even harm, prion propagation in mammalian cells?
- Why do prionogenic regions show much stronger prion properties when located in terminal positions, rather than inside the protein sequence, and do they acquire different structures in these two cases?
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chiti, F.; Dobson, C.M. Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress Over the Last Decade. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef]
- Prusiner, S.B. Biology and genetics of prions causing neurodegeneration. Annu. Rev. Genet. 2013, 47, 601–623. [Google Scholar] [CrossRef] [Green Version]
- Alyenbaawi, H.; Allison, W.T.; Mok, S.-A. Prion-Like Propagation Mechanisms in Tauopathies and Traumatic Brain Injury: Challenges and Prospects. Biomolecules 2020, 10, 1487. [Google Scholar] [CrossRef]
- Jo, M.; Lee, S.; Jeon, Y.-M.; Kim, S.; Kwon, Y.; Kim, H.-J. The role of TDP-43 propagation in neurodegenerative diseases: Integrating insights from clinical and experimental studies. Exp. Mol. Med. 2020, 52, 1652–1662. [Google Scholar] [CrossRef]
- Lam, S.; Petit, F.; Hérard, A.-S.; Boluda, S.; Eddarkaoui, S.; Guillermier, M.; Letournel, F.; Martin-Négrier, M.-L.; Faisant, M.; Godfraind, C.; et al. Transmission of amyloid-beta and tau pathologies is associated with cognitive impairments in a primate. Acta Neuropathol. Commun. 2021, 9, 165. [Google Scholar] [CrossRef]
- Purro, S.A.; Farrow, M.A.; Linehan, J.; Nazari, T.; Thomas, D.X.; Chen, Z.; Mengel, D.; Saito, T.; Saido, T.; Rudge, P.; et al. Transmission of amyloid-β protein pathology from cadaveric pituitary growth hormone. Nature 2018, 564, 415–419. [Google Scholar] [CrossRef]
- Kim, S.; Kwon, S.-H.; Kam, T.-I.; Panicker, N.; Karuppagounder, S.S.; Lee, S.; Lee, J.H.; Kim, W.R.; Kook, M.; Foss, C.A.; et al. Transneuronal Propagation of Pathologic α-Synuclein from the Gut to the Brain Models Parkinson’s Disease. Neuron 2019, 103, 627–641.e7. [Google Scholar] [CrossRef]
- Mukherjee, A.; Morales-Scheihing, D.; Salvadores, N.; Moreno-Gonzalez, I.; Gonzalez, C.; Taylor-Presse, K.; Mendez, N.; Shahnawaz, M.; Gaber, A.O.; Sabek, O.M.; et al. Induction of IAPP amyloid deposition and associated diabetic abnormalities by a prion-like mechanism. J. Exp. Med. 2017, 214, 2591–2610. [Google Scholar] [CrossRef]
- Otzen, D.; Riek, R. Functional Amyloids. Cold Spring Harb. Perspect. Biol. 2019, 11, a033860. [Google Scholar] [CrossRef]
- Keleman, K.; Krüttner, S.; Alenius, M.; Dickson, B.J. Function of the Drosophila CPEB protein Orb2 in long-term courtship memory. Nat. Neurosci. 2007, 10, 1587–1593. [Google Scholar] [CrossRef]
- Mastushita-Sakai, T.; White-Grindley, E.; Samuelson, J.; Seidel, C.; Si, K. Drosophila Orb2 targets genes involved in neuronal growth, synapse formation, and protein turnover. Proc. Natl. Acad. Sci. USA 2010, 107, 11987–11992. [Google Scholar] [CrossRef] [Green Version]
- Wickner, R.B. [URE3] as an altered URE2 protein: Evidence for a prion analog in Saccharomyces cerevisiae. Science 1994, 264, 566–569. [Google Scholar] [CrossRef] [Green Version]
- True, H.L.; Berlin, I.; Lindquist, S.L. Epigenetic regulation of translation reveals hidden genetic variation to produce complex traits. Nature 2004, 431, 184–187. [Google Scholar] [CrossRef]
- Oamen, H.P.; Lau, Y.; Caudron, F. Prion-like proteins as epigenetic devices of stress adaptation. Exp. Cell Res. 2020, 396, 112262. [Google Scholar] [CrossRef]
- Anfinsen, C.B. Principles that govern the folding of protein chains. Science 1973, 181, 223–230. [Google Scholar] [CrossRef] [Green Version]
- Ghaemmaghami, S. Biology and Genetics of PrP Prion Strains. Cold Spring Harb. Perspect. Med. 2017, 7, a026922. [Google Scholar] [CrossRef]
- Lau, H.H.C.; Ingelsson, M.; Watts, J.C. The existence of Aβ strains and their potential for driving phenotypic heterogeneity in Alzheimer’s disease. Acta Neuropathol. 2021, 142, 17–39. [Google Scholar] [CrossRef]
- Hoppe, S.O.; Uzunoğlu, G.; Nussbaum-Krammer, C. α-Synuclein Strains: Does Amyloid Conformation Explain the Heterogeneity of Synucleinopathies? Biomolecules 2021, 11, 931. [Google Scholar] [CrossRef]
- Wickner, R.B.; Son, M.; Edskes, H.K. Prion Variants of Yeast are Numerous, Mutable, and Segregate on Growth, Affecting Prion Pathogenesis, Transmission Barriers, and Sensitivity to Anti-Prion Systems. Viruses 2019, 11, 238. [Google Scholar] [CrossRef] [Green Version]
- Dergalev, A.A.; Alexandrov, A.I.; Ivannikov, R.I.; Ter-Avanesyan, M.D.; Kushnirov, V.V. Yeast Sup35 Prion Structure: Two Types, Four Parts, Many Variants. Int. J. Mol. Sci. 2019, 20, 2633. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.-W.; King, C.-Y. A complete catalog of wild-type Sup35 prion variants and their protein-only propagation. Curr. Genet. 2020, 66, 97–122. [Google Scholar] [CrossRef]
- Pezza, J.A.; Villali, J.; Sindi, S.S.; Serio, T.R. Amyloid-associated activity contributes to the severity and toxicity of a prion phenotype. Nat. Commun. 2014, 5, 4384. [Google Scholar] [CrossRef] [Green Version]
- Si, K.; Choi, Y.-B.; White-Grindley, E.; Majumdar, A.; Kandel, E.R. Aplysia CPEB can form prion-like multimers in sensory neurons that contribute to long-term facilitation. Cell 2010, 140, 421–435. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.R.; Li, L.; Pérez-Sánchez, C.; Saraf, A.; Florens, L.; Slaughter, B.D.; Unruh, J.R.; Si, K. Amyloidogenic Oligomerization Transforms Drosophila Orb2 from a Translation Repressor to an Activator. Cell 2015, 163, 1468–1483. [Google Scholar] [CrossRef] [Green Version]
- Malovichko, Y.V.; Antonets, K.S.; Maslova, A.R.; Andreeva, E.A.; Inge-Vechtomov, S.G.; Nizhnikov, A.A. RNA Sequencing Reveals Specific Transcriptomic Signatures Distinguishing Effects of the [SWI+] Prion and SWI1 Deletion in Yeast Saccharomyces cerevisiae. Genes 2019, 10, 212. [Google Scholar] [CrossRef] [Green Version]
- Dominguez, R.; Holmes, K.C. Actin structure and function. Annu. Rev. Biophys. 2011, 40, 169–186. [Google Scholar] [CrossRef] [Green Version]
- Wentink, A.S.; Nillegoda, N.B.; Feufel, J.; Ubartaitė, G.; Schneider, C.P.; De Los Rios, P.; Hennig, J.; Barducci, A.; Bukau, B. Molecular dissection of amyloid disaggregation by human HSP70. Nature 2020, 587, 483–488. [Google Scholar] [CrossRef]
- Benzinger, T.L.; Gregory, D.M.; Burkoth, T.S.; Miller-Auer, H.; Lynn, D.G.; Botto, R.E.; Meredith, S.C. Propagating structure of Alzheimer’s beta-amyloid(10-35) is parallel beta-sheet with residues in exact register. Proc. Natl. Acad. Sci. USA 1998, 95, 13407–13412. [Google Scholar] [CrossRef] [Green Version]
- Shewmaker, F.; Wickner, R.B.; Tycko, R. Amyloid of the prion domain of Sup35p has an in-register parallel beta-sheet structure. Proc. Natl. Acad. Sci. USA 2006, 103, 19754–19759. [Google Scholar] [CrossRef] [Green Version]
- Tuttle, M.D.; Comellas, G.; Nieuwkoop, A.J.; Covell, D.J.; Berthold, D.A.; Kloepper, K.D.; Courtney, J.M.; Kim, J.K.; Barclay, A.M.; Kendall, A.; et al. Solid-state NMR structure of a pathogenic fibril of full-length human α-synuclein. Nat. Struct. Mol. Biol. 2016, 23, 409–415. [Google Scholar] [CrossRef]
- Wasmer, C.; Lange, A.; Van Melckebeke, H.; Siemer, A.B.; Riek, R.; Meier, B.H. Amyloid fibrils of the HET-s(218-289) prion form a beta solenoid with a triangular hydrophobic core. Science 2008, 319, 1523–1526. [Google Scholar] [CrossRef]
- Daskalov, A.; Martinez, D.; Coustou, V.; El Mammeri, N.; Berbon, M.; Andreas, L.B.; Bardiaux, B.; Stanek, J.; Noubhani, A.; Kauffmann, B.; et al. Structural and molecular basis of cross-seeding barriers in amyloids. Proc. Natl. Acad. Sci. USA 2021, 118, e2014085118. [Google Scholar] [CrossRef]
- Shewmaker, F.; McGlinchey, R.P.; Thurber, K.R.; McPhie, P.; Dyda, F.; Tycko, R.; Wickner, R.B. The functional curli amyloid is not based on in-register parallel beta-sheet structure. J. Biol. Chem. 2009, 284, 25065–25076. [Google Scholar] [CrossRef] [Green Version]
- Peng, Z.; Peralta, M.D.R.; Cox, D.L.; Toney, M.D. Bottom-up synthesis of protein-based nanomaterials from engineered β-solenoid proteins. PLoS ONE 2020, 15, e0229319. [Google Scholar] [CrossRef]
- Peralta, M.D.R.; Karsai, A.; Ngo, A.; Sierra, C.; Fong, K.T.; Hayre, N.R.; Mirzaee, N.; Ravikumar, K.M.; Kluber, A.J.; Chen, X.; et al. Engineering amyloid fibrils from β-solenoid proteins for biomaterials applications. ACS Nano 2015, 9, 449–463. [Google Scholar] [CrossRef]
- Vázquez-Fernández, E.; Vos, M.R.; Afanasyev, P.; Cebey, L.; Sevillano, A.M.; Vidal, E.; Rosa, I.; Renault, L.; Ramos, A.; Peters, P.J.; et al. The Structural Architecture of an Infectious Mammalian Prion Using Electron Cryomicroscopy. PLoS Pathog. 2016, 12, e1005835. [Google Scholar] [CrossRef]
- Kraus, A.; Hoyt, F.; Schwartz, C.L.; Hansen, B.; Artikis, E.; Hughson, A.G.; Raymond, G.J.; Race, B.; Baron, G.S.; Caughey, B. High-resolution structure and strain comparison of infectious mammalian prions. Mol. Cell 2021, 81, 4540–4551.e6. [Google Scholar] [CrossRef]
- Wickner, R.B. Anti-prion systems in yeast. J. Biol. Chem. 2019, 294, 1729–1738. [Google Scholar] [CrossRef] [Green Version]
- Wickner, R.B.; Dyda, F.; Tycko, R. Amyloid of Rnq1p, the basis of the [PIN+] prion, has a parallel in-register beta-sheet structure. Proc. Natl. Acad. Sci. USA 2008, 105, 2403–2408. [Google Scholar] [CrossRef] [Green Version]
- Baxa, U.; Wickner, R.B.; Steven, A.C.; Anderson, D.E.; Marekov, L.N.; Yau, W.-M.; Tycko, R. Characterization of beta-sheet structure in Ure2p1-89 yeast prion fibrils by solid-state nuclear magnetic resonance. Biochemistry 2007, 46, 13149–13162. [Google Scholar] [CrossRef]
- Shewmaker, F.; Ross, E.D.; Tycko, R.; Wickner, R.B. Amyloids of shuffled prion domains that form prions have a parallel in-register beta-sheet structure. Biochemistry 2008, 47, 4000–4007. [Google Scholar] [CrossRef] [PubMed]
- Punihaole, D.; Workman, R.J.; Hong, Z.; Madura, J.D.; Asher, S.A. Polyglutamine Fibrils: New Insights into Antiparallel β-Sheet Conformational Preference and Side Chain Structure. J. Phys. Chem. B 2016, 120, 3012–3026. [Google Scholar] [CrossRef] [PubMed]
- Asakura, T.; Okonogi, M.; Naito, A. Toward Understanding the Silk Fiber Structure: 13C Solid-State NMR Studies of the Packing Structures of Alanine Oligomers before and after Trifluoroacetic Acid Treatment. J. Phys. Chem. B 2019, 123, 6716–6727. [Google Scholar] [CrossRef] [PubMed]
- Mo, Y.; Lu, Y.; Wei, G.; Derreumaux, P. Structural diversity of the soluble trimers of the human amylin(20–29) peptide revealed by molecular dynamics simulations. J. Chem. Phys. 2009, 130, 125101. [Google Scholar] [CrossRef] [PubMed]
- Soriaga, A.B.; Sangwan, S.; Macdonald, R.; Sawaya, M.R.; Eisenberg, D. Crystal Structures of IAPP Amyloidogenic Segments Reveal a Novel Packing Motif of Out-of-Register Beta Sheets. J. Phys. Chem. B 2016, 120, 5810–5816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Guo, C.; Watzlawik, J.O.; Randolph, P.S.; Lee, E.J.; Huang, D.; Stagg, S.M.; Zhou, H.-X.; Rosenberry, T.L.; Paravastu, A.K. Out-of-Register Parallel β-Sheets and Antiparallel β-Sheets Coexist in 150-kDa Oligomers Formed by Amyloid-β(1-42). J. Mol. Biol. 2020, 432, 4388–4407. [Google Scholar] [CrossRef]
- Alexandrov, A.I.; Polyanskaya, A.B.; Serpionov, G.V.; Ter-Avanesyan, M.D.; Kushnirov, V.V. The Effects of Amino Acid Composition of Glutamine-Rich Domains on Amyloid Formation and Fragmentation. PLoS ONE 2012, 7, 46458. [Google Scholar] [CrossRef]
- Dahlgren, P.R.; Karymov, M.A.; Bankston, J.; Holden, T.; Thumfort, P.; Ingram, V.M.; Lyubchenko, Y.L. Atomic force microscopy analysis of the Huntington protein nanofibril formation. Nanomed. Nanotechnol. Biol. Med. 2005, 1, 52–57. [Google Scholar] [CrossRef]
- Nekooki-Machida, Y.; Kurosawa, M.; Nukina, N.; Ito, K.; Oda, T.; Tanaka, M. Distinct conformations of in vitro and in vivo amyloids of huntingtin-exon1 show different cytotoxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 9679–9684. [Google Scholar] [CrossRef] [Green Version]
- Cox, B.S. Ψ, A cytoplasmic suppressor of super-suppressor in yeast. Heredity 1965, 20, 505–521. [Google Scholar] [CrossRef] [Green Version]
- Lacroute, F. Non-Mendelian mutation allowing ureidosuccinic acid uptake in yeast. J. Bacteriol. 1971, 106, 519–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patino, M.M.; Liu, J.J.; Glover, J.R.; Lindquist, S. Support for the prion hypothesis for inheritance of a phenotypic trait in yeast. Science 1996, 273, 622–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paushkin, S.V.; Kushnirov, V.V.; Smirnov, V.N.; Ter-Avanesyan, M.D. Propagation of the yeast prion-like [PSI+] determinant is mediated by oligomerization of the SUP35-encoded polypeptide chain release factor. EMBO J. 1996, 15, 3127–3134. [Google Scholar] [CrossRef] [PubMed]
- Masison, D.C.; Maddelein, M.L.; Wickner, R.B. The prion model for [URE3] of yeast: Spontaneous generation and requirements for propagation. Proc. Natl. Acad. Sci. USA 1997, 94, 12503–12508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ter-Avanesyan, M.D.; Kushnirov, V.V.; Dagkesamanskaya, A.R.; Didichenko, S.A.; Chernoff, Y.O.; Inge-Vechtomov, S.G.; Smirnov, V.N. Deletion analysis of the SUP35 gene of the yeast Saccharomyces cerevisiae reveals two non-overlapping functional regions in the encoded protein. Mol. Microbiol. 1993, 7, 683–692. [Google Scholar] [CrossRef]
- Alberti, S.; Halfmann, R.; King, O.; Kapila, A.; Lindquist, S. A systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cell 2009, 137, 146–158. [Google Scholar] [CrossRef] [Green Version]
- Si, K.; Lindquist, S.; Kandel, E.R. A neuronal isoform of the aplysia CPEB has prion-like properties. Cell 2003, 115, 879–891. [Google Scholar] [CrossRef] [Green Version]
- Paul, K.R.; Molliex, A.; Cascarina, S.; Boncella, A.E.; Taylor, J.P.; Ross, E.D. Effects of Mutations on the Aggregation Propensity of the Human Prion-Like Protein hnRNPA2B1. Mol. Cell. Biol. 2017, 37, e00652-16. [Google Scholar] [CrossRef] [Green Version]
- Chakrabortee, S.; Kayatekin, C.; Newby, G.A.; Mendillo, M.L.; Lancaster, A.; Lindquist, S. Luminidependens (LD) is an Arabidopsis protein with prion behavior. Proc. Natl. Acad. Sci. USA 2016, 113, 6065–6070. [Google Scholar] [CrossRef] [Green Version]
- Zajkowski, T.; Lee, M.D.; Mondal, S.S.; Carbajal, A.; Dec, R.; Brennock, P.D.; Piast, R.W.; Snyder, J.E.; Bense, N.B.; Dzwolak, W.; et al. The Hunt for Ancient Prions: Archaeal Prion-Like Domains Form Amyloid-Based Epigenetic Elements. Mol. Biol. Evol. 2021, 38, 2088–2103. [Google Scholar] [CrossRef]
- Nan, H.; Chen, H.; Tuite, M.F.; Xu, X. A viral expression factor behaves as a prion. Nat. Commun. 2019, 10, 359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coschigano, P.W.; Magasanik, B. The URE2 gene product of Saccharomyces cerevisiae plays an important role in the cellular response to the nitrogen source and has homology to glutathione s-transferases. Mol. Cell. Biol. 1991, 11, 822–832. [Google Scholar] [CrossRef] [PubMed]
- Derkatch, I.L.; Bradley, M.E.; Zhou, P.; Chernoff, Y.O.; Liebman, S.W. Genetic and environmental factors affecting the de novo appearance of the [PSI+] prion in Saccharomyces cerevisiae. Genetics 1997, 147, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Derkatch, I.L.; Bradley, M.E.; Hong, J.Y.; Liebman, S.W. Prions affect the appearance of other prions: The story of [PIN+]. Cell 2001, 106, 171–182. [Google Scholar] [CrossRef] [Green Version]
- Derkatch, I.L.; Uptain, S.M.; Outeiro, T.F.; Krishnan, R.; Lindquist, S.L.; Liebman, S.W. Effects of Q/N-rich, polyQ, and non-polyQ amyloids on the de novo formation of the [PSI+] prion in yeast and aggregation of Sup35 in vitro. Proc. Natl. Acad. Sci. USA 2004, 101, 12934–12939. [Google Scholar] [CrossRef] [Green Version]
- Bradley, M.E.; Edskes, H.K.; Hong, J.Y.; Wickner, R.B.; Liebman, S.W. Interactions among prions and prion “strains” in yeast. Proc. Natl. Acad. Sci. USA 2002, 99 (Suppl. S4), 16392–16399. [Google Scholar] [CrossRef] [Green Version]
- Du, Z.; Park, K.-W.; Yu, H.; Fan, Q.; Li, L. Newly identified prion linked to the chromatin-remodeling factor Swi1 in Saccharomyces cerevisiae. Nat. Genet. 2008, 40, 460–465. [Google Scholar] [CrossRef] [Green Version]
- Patel, B.K.; Gavin-Smyth, J.; Liebman, S.W. The yeast global transcriptional co-repressor protein Cyc8 can propagate as a prion. Nat. Cell Biol. 2009, 11, 344–349. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, G.; Shimazu, N.; Tanaka, M. A yeast prion, Mod5, promotes acquired drug resistance and cell survival under environmental stress. Science 2012, 336, 355–359. [Google Scholar] [CrossRef]
- Chernova, T.A.; Kiktev, D.A.; Romanyuk, A.V.; Shanks, J.R.; Laur, O.; Ali, M.; Ghosh, A.; Kim, D.; Yang, Z.; Mang, M.; et al. Yeast Short-Lived Actin-Associated Protein Forms a Metastable Prion in Response to Thermal Stress. Cell Rep. 2017, 18, 751–761. [Google Scholar] [CrossRef]
- Halfmann, R.; Wright, J.R.; Alberti, S.; Lindquist, S.; Rexach, M. Prion formation by a yeast GLFG nucleoporin. Prion 2012, 6, 391–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, J.C.S.; Lindquist, S. A heritable switch in carbon source utilization driven by an unusual yeast prion. Genes Dev. 2009, 23, 2320–2332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakrabortee, S.; Byers, J.S.; Jones, S.; Garcia, D.M.; Bhullar, B.; Chang, A.; She, R.; Lee, L.; Fremin, B.; Lindquist, S.; et al. Intrinsically Disordered Proteins Drive Emergence and Inheritance of Biological Traits. Cell 2016, 167, 369–381.e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derkatch, I.L.; Chernoff, Y.O.; Kushnirov, V.V.; Inge-Vechtomov, S.G.; Liebman, S.W. Genesis and variability of [PSI+] prion factors in Saccharomyces cerevisiae. Genetics 1996, 144, 1375–1386. [Google Scholar] [CrossRef]
- Chan, P.H.W.; Lee, L.; Kim, E.; Hui, T.; Stoynov, N.; Nassar, R.; Moksa, M.; Cameron, D.M.; Hirst, M.; Gsponer, J.; et al. The [PSI +] yeast prion does not wildly affect proteome composition whereas selective pressure exerted on [PSI +] cells can promote aneuploidy. Sci. Rep. 2017, 7, 8442. [Google Scholar] [CrossRef] [Green Version]
- Nizhnikov, A.A.; Ryzhova, T.A.; Volkov, K.V.; Zadorsky, S.P.; Sopova, J.V.; Inge-Vechtomov, S.G.; Galkin, A.P. Interaction of Prions Causes Heritable Traits in Saccharomyces cerevisiae. PLoS Genet. 2016, 12, e1006504. [Google Scholar] [CrossRef] [Green Version]
- Killian, A.N.; Miller, S.C.; Hines, J.K. Impact of Amyloid Polymorphism on Prion-Chaperone Interactions in Yeast. Viruses 2019, 11, 349. [Google Scholar] [CrossRef] [Green Version]
- Barbitoff, Y.A.; Matveenko, A.G.; Zhouravleva, G.A. Differential Interactions of Molecular Chaperones and Yeast Prions. J. Fungi 2022, 8, 122. [Google Scholar] [CrossRef]
- Kushnirov, V.V.; Kryndushkin, D.S.; Boguta, M.; Smirnov, V.N.; Ter-Avanesyan, M.D. Chaperones that cure yeast artificial [PSI+] and their prion-specific effects. Curr. Biol. 2000, 10, 1443–1446. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.-Y.; Lin, J.-Y.; Lee, H.-C.; Wang, H.-L.; King, C.-Y. Strain-specific sequences required for yeast [PSI+] prion propagation. Proc. Natl. Acad. Sci. USA 2008, 105, 13345–13350. [Google Scholar] [CrossRef] [Green Version]
- McGlinchey, R.P.; Kryndushkin, D.; Wickner, R.B. Suicidal [PSI+] is a lethal yeast prion. Proc. Natl. Acad. Sci. USA 2011, 108, 5337–5341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, M.; Wickner, R.B. Normal levels of ribosome-associated chaperones cure two groups of [PSI+] prion variants. Proc. Natl. Acad. Sci. USA 2020, 117, 26298–26306. [Google Scholar] [CrossRef] [PubMed]
- Gorkovskiy, A.; Reidy, M.; Masison, D.C.; Wickner, R.B. Hsp104 disaggregase at normal levels cures many [PSI +] prion variants in a process promoted by Sti1p, Hsp90, and Sis1p. Proc. Natl. Acad. Sci. USA 2017, 114, E4193–E4202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, M.; Wickner, R.B. Nonsense-mediated mRNA decay factors cure most [PSI+] prion variants. Proc. Natl. Acad. Sci. USA 2018, 115, E1184–E1193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wickner, R.B.; Kelly, A.C.; Bezsonov, E.E.; Edskes, H.K. [PSI+] prion propagation is controlled by inositol polyphosphates. Proc. Natl. Acad. Sci. USA 2017, 114, E8402–E8410. [Google Scholar] [CrossRef] [Green Version]
- Kryndushkin, D.S.; Shewmaker, F.; Wickner, R.B. Curing of the [URE3] prion by Btn2p, a Batten disease-related protein. EMBO J. 2008, 27, 2725–2735. [Google Scholar] [CrossRef] [Green Version]
- Wickner, R.B.; Bezsonov, E.E.; Son, M.; Ducatez, M.; Dewilde, M.; Edskes, H.K. Anti-Prion Systems in Yeast and Inositol Polyphosphates. Biochemistry 2018, 57, 1285–1292. [Google Scholar] [CrossRef]
- Wickner, R.B.; Edskes, H.K.; Son, M.; Wu, S.; Niznikiewicz, M. Innate immunity to prions: Anti-prion systems turn a tsunami of prions into a slow drip. Curr. Genet. 2021, 67, 833–847. [Google Scholar] [CrossRef]
- Huang, Y.-W.; Kushnirov, V.V.; King, C.-Y. Mutable yeast prion variants are stabilized by a defective Hsp104 chaperone. Mol. Microbiol. 2021, 115, 774–788. [Google Scholar] [CrossRef]
- Chernoff, Y.O.; Lindquist, S.L.; Ono, B.; Inge-Vechtomov, S.G.; Liebman, S.W. Role of the chaperone protein Hsp104 in propagation of the yeast prion-like factor [PSI+]. Science 1995, 268, 880–884. [Google Scholar] [CrossRef]
- Tipton, K.A.; Verges, K.J.; Weissman, J.S. In vivo monitoring of the prion replication cycle reveals a critical role for Sis1 in delivering substrates to Hsp104. Mol. Cell 2008, 32, 584–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urakov, V.N.; Mitkevich, O.V.; Dergalev, A.A.; Ter-Avanesyan, M.D. The Pub1 and Upf1 Proteins Act in Concert to Protect Yeast from Toxicity of the [PSI+] Prion. Int. J. Mol. Sci. 2018, 19, 3663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnan, R.; Lindquist, S.L. Structural insights into a yeast prion illuminate nucleation and strain diversity. Nature 2005, 435, 765–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toyama, B.H.; Kelly, M.J.S.; Gross, J.D.; Weissman, J.S. The structural basis of yeast prion strain variants. Nature 2007, 449, 233–237. [Google Scholar] [CrossRef] [Green Version]
- Frederick, K.K.; Debelouchina, G.T.; Kayatekin, C.; Dorminy, T.; Jacavone, A.C.; Griffin, R.G.; Lindquist, S. Distinct prion strains are defined by amyloid core structure and chaperone binding site dynamics. Chem. Biol. 2014, 21, 295–305. [Google Scholar] [CrossRef] [Green Version]
- Gorkovskiy, A.; Thurber, K.R.; Tycko, R.; Wickner, R.B. Locating folds of the in-register parallel beta-sheet of the Sup35p prion domain infectious amyloid. Proc. Natl. Acad. Sci. USA 2014, 111, E4615–E4622. [Google Scholar] [CrossRef] [Green Version]
- Ohhashi, Y.; Yamaguchi, Y.; Kurahashi, H.; Kamatari, Y.O.; Sugiyama, S.; Uluca, B.; Piechatzek, T.; Komi, Y.; Shida, T.; Müller, H.; et al. Molecular basis for diversification of yeast prion strain conformation. Proc. Natl. Acad. Sci. USA 2018, 115, 2389–2394. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, M.; Chien, P.; Naber, N.; Cooke, R.; Weissman, J.S. Conformational variations in an infectious protein determine prion strain differences. Nature 2004, 428, 323–328. [Google Scholar] [CrossRef]
- Frederick, K.K.; Michaelis, V.K.; Corzilius, B.; Ong, T.C.; Jacavone, A.C.; Griffin, R.G.; Lindquist, S. Sensitivity-Enhanced NMR Reveals Alterations in Protein Structure by Cellular Milieus. Cell 2015, 163, 620–628. [Google Scholar] [CrossRef] [Green Version]
- Hervas, R.; Rau, M.J.; Park, Y.; Zhang, W.; Murzin, A.G.; Fitzpatrick, J.A.J.; Scheres, S.H.W.; Si, K. Cryo-EM structure of a neuronal functional amyloid implicated in memory persistence in Drosophila. Science 2020, 367, 1230–1234. [Google Scholar] [CrossRef]
- Shkundina, I.S.; Kushnirov, V.V.; Tuite, M.F.; Ter-Avanesyan, M.D. The role of the N-terminal oligopeptide repeats of the yeast Sup35 prion protein in propagation and transmission of prion variants. Genetics 2006, 172, 827–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, R.; Dong, J.; Wall, J.; Frederick, K.K. Amyloid fibrils embodying distinctive yeast prion phenotypes exhibit diverse morphologies. FEMS Yeast Res. 2018, 18, foy059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balguerie, A.; Dos Reis, S.; Ritter, C.; Chaignepain, S.; Coulary-Salin, B.; Forge, V.; Bathany, K.; Lascu, I.; Schmitter, J.-M.; Riek, R.; et al. Domain organization and structure-function relationship of the HET-s prion protein of Podospora anserina. EMBO J. 2003, 22, 2071–2081. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.L.; Covell, D.J.; Daniels, J.P.; Iba, M.; Stieber, A.; Zhang, B.; Riddle, D.M.; Kwong, L.K.; Xu, Y.; Trojanowski, J.Q.; et al. Distinct α-synuclein strains differentially promote tau inclusions in neurons. Cell 2013, 154, 103–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miake, H.; Mizusawa, H.; Iwatsubo, T.; Hasegawa, M. Biochemical characterization of the core structure of alpha-synuclein filaments. J. Biol. Chem. 2002, 277, 19213–19219. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Ge, P.; Murray, K.A.; Sheth, P.; Zhang, M.; Nair, G.; Sawaya, M.R.; Shin, W.S.; Boyer, D.R.; Ye, S.; et al. Cryo-EM of full-length α-synuclein reveals fibril polymorphs with a common structural kernel. Nat. Commun. 2018, 9, 3609. [Google Scholar] [CrossRef]
- Daskalov, A.; Gantner, M.; Wälti, M.A.; Schmidlin, T.; Chi, C.N.; Wasmer, C.; Schütz, A.; Ceschin, J.; Clavé, C.; Cescau, S.; et al. Contribution of specific residues of the β-solenoid fold to HET-s prion function, amyloid structure and stability. PLoS Pathog. 2014, 10, e1004158. [Google Scholar] [CrossRef] [Green Version]
- Duernberger, Y.; Liu, S.; Riemschoss, K.; Paulsen, L.; Bester, R.; Kuhn, P.-H.; Schölling, M.; Lichtenthaler, S.F.; Vorberg, I. Prion replication in the mammalian cytosol: Functional regions within a prion domain driving induction, propagation and inheritance. Mol. Cell. Biol. 2018, 38, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Dagkesamanskaia, A.R.; Kushnirov, V.V.; Paushkin, S.V.; Ter-Avanesian, M.D. Fusion of glutathione S-transferase with the N-terminus of yeast Sup35p protein inhibits its prion-like properties. Genetika 1997, 33, 610–615. [Google Scholar]
- Dergalev, A.A.; Urakov, V.N.; Agaphonov, M.O.; Alexandrov, A.I.; Kushnirov, V.V. Dangerous Stops: Nonsense Mutations Can Dramatically Increase Frequency of Prion Conversion. Int. J. Mol. Sci. 2021, 22, 1542. [Google Scholar] [CrossRef]
- Antonets, K.S.; Sargsyan, H.M.; Nizhnikov, A.A. A Glutamine/Asparagine-Rich Fragment of Gln3, but not the Full-Length Protein, Aggregates in Saccharomyces cerevisiae. Biochemistry 2016, 81, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Bagriantsev, S.N.; Gracheva, E.O.; Richmond, J.E.; Liebman, S.W. Variant-specific [PSI+] infection is transmitted by Sup35 polymers within [PSI+] aggregates with heterogeneous protein composition. Mol. Biol. Cell 2008, 19, 2433–2443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagawa, Y.; Shen, H.C.-H.; Komi, Y.; Sugiyama, S.; Kurinomaru, T.; Tomabechi, Y.; Krayukhina, E.; Okamoto, K.; Yokoyama, T.; Shirouzu, M.; et al. Amyloid conformation-dependent disaggregation in a reconstituted yeast prion system. Nat. Chem. Biol. 2022, 18, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Kryndushkin, D.S.; Alexandrov, I.M.; Ter-Avanesyan, M.D.; Kushnirov, V.V. Yeast [PSI+] prion aggregates are formed by small Sup35 polymers fragmented by Hsp104. J. Biol. Chem. 2003, 278, 49636–49643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baxa, U.; Keller, P.W.; Cheng, N.; Wall, J.S.; Steven, A.C. In Sup35p filaments (the [PSI+] prion), the globular C-terminal domains are widely offset from the amyloid fibril backbone. Mol. Microbiol. 2011, 79, 523–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salnikova, A.B.; Kryndushkin, D.S.; Smirnov, V.N.; Kushnirov, V.V.; Ter-Avanesyan, M.D. Nonsense suppression in yeast cells overproducing Sup35 (eRF3) is caused by its non-heritable amyloids. J. Biol. Chem. 2005, 280, 8808–8812. [Google Scholar] [CrossRef] [Green Version]
- Uptain, S.M.; Sawicki, G.J.; Caughey, B.; Lindquist, S. Strains of [PSI(+)] are distinguished by their efficiencies of prion-mediated conformational conversion. EMBO J. 2001, 20, 6236–6245. [Google Scholar] [CrossRef] [Green Version]
- Chabelskaya, S.; Kiktev, D.; Inge-Vechtomov, S.; Philippe, M.; Zhouravleva, G. Nonsense mutations in the essential gene SUP35 of Saccharomyces cerevisiae are non-lethal. Mol. Genet. Genom. 2004, 272, 297–307. [Google Scholar] [CrossRef]
- Park, S.; Wang, X.; Xi, W.; Richardson, R.; Laue, T.M.; Denis, C.L. The non-prion SUP35 preexists in large chaperone-containing molecular complexes. Proteins 2021, 90, 869–880. [Google Scholar] [CrossRef]
- Kushnirov, V.V.; Alexandrov, I.M.; Mitkevich, O.V.; Shkundina, I.S.; Ter-Avanesyan, M.D. Purification and analysis of prion and amyloid aggregates. Methods 2006, 39, 50–55. [Google Scholar] [CrossRef]
- Tanaka, M.; Collins, S.R.; Toyama, B.H.; Weissman, J.S. The physical basis of how prion conformations determine strain phenotypes. Nature 2006, 442, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Zhouravleva, G.; Frolova, L.; Le Goff, X.; Le Guellec, R.; Inge-Vechtomov, S.; Kisselev, L.; Philippe, M. Termination of translation in eukaryotes is governed by two interacting polypeptide chain release factors, eRF1 and eRF3. EMBO J. 1995, 14, 4065–4072. [Google Scholar] [CrossRef] [PubMed]
- Kiktev, D.; Moskalenko, S.; Murina, O.; Rousset, A.B.J. The paradox of viable sup45 STOP mutations: A necessary equilibrium between translational readthrough, activity and stability of the protein. Mol. Genet. Genom. 2009, 282, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Stansfield, I.; Jones, K.M.; Kushnirov, V.V.; Dagkesamanskaya, A.R.; Poznyakovski, A.I.; Paushkin, S.V.; Nierras, C.R.; Cox, B.S.; Ter-Avanesyan, M.D.; Tuite, M.F. The products of the SUP45 (eRF1) and SUP35 genes interact to mediate translation termination in Saccharomyces cerevisiae. EMBO J. 1995, 14, 4365–4373. [Google Scholar] [CrossRef]
- Vishveshwara, N.; Bradley, M.E.; Liebman, S.W. Sequestration of essential proteins causes prion associated toxicity in yeast. Mol. Microbiol. 2009, 73, 1101–1114. [Google Scholar] [CrossRef] [Green Version]
- Schlumpberger, M.; Prusiner, S.B.; Herskowitz, I. Induction of distinct [URE3] yeast prion strains. Mol. Cell. Biol. 2001, 21, 7035–7046. [Google Scholar] [CrossRef] [Green Version]
- Kochneva-Pervukhova, N.V.; Chechenova, M.B.; Valouev, I.A.; Kushnirov, V.V.; Smirnov, V.N.; Ter-Avanesyan, M.D. [Psi(+)] prion generation in yeast: Characterization of the “strain” difference. Yeast 2001, 18, 489–497. [Google Scholar] [CrossRef]
- Bai, M.; Zhou, J.-M.; Perrett, S. The yeast prion protein Ure2 shows glutathione peroxidase activity in both native and fibrillar forms. J. Biol. Chem. 2004, 279, 50025–50030. [Google Scholar] [CrossRef] [Green Version]
- Baxa, U.; Speransky, V.; Steven, A.C.; Wickner, R.B. Mechanism of inactivation on prion conversion of the Saccharomyces cerevisiae Ure2 protein. Proc. Natl. Acad. Sci. USA 2002, 99, 5253–5260. [Google Scholar] [CrossRef] [Green Version]
- Crow, E.T.; Du, Z.; Li, L. A Small, Glutamine-Free Domain Propagates the [SWI+] Prion in Budding Yeast. Mol. Cell. Biol. 2011, 31, 3436–3444. [Google Scholar] [CrossRef] [Green Version]
- Bardill, J.P.; True, H.L. Heterologous prion interactions are altered by mutations in the prion protein Rnq1p. J. Mol. Biol. 2009, 388, 583–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sondheimer, N.; Lindquist, S. Rnq1: An Epigenetic Modifier of Protein Function in Yeast. Yeast 2000, 5, 163–172. [Google Scholar]
- Stein, K.C.; True, H.L. Extensive Diversity of Prion Strains Is Defined by Differential Chaperone Interactions and Distinct Amyloidogenic Regions. PLoS Genet. 2014, 10, e1004337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothe, S.; Prakash, A.; Tyedmers, J. The Insoluble Protein Deposit (IPOD) in Yeast. Front. Mol. Neurosci. 2018, 11, 237. [Google Scholar] [CrossRef] [PubMed]
- Dorweiler, J.E.; Oddo, M.J.; Lyke, D.R.; Reilly, J.A.; Wisniewski, B.T.; Davis, E.E.; Kuborn, A.M.; Merrill, S.J.; Manogaran, A.L. The actin cytoskeletal network plays a role in yeast prion transmission and contributes to prion stability. Mol. Microbiol. 2020, 114, 480–494. [Google Scholar] [CrossRef]
- Sharma, J.; Liebman, S.W. Exploring the basis of [PIN(+)] variant differences in [PSI(+)] induction. J. Mol. Biol. 2013, 425, 3046–3059. [Google Scholar] [CrossRef] [Green Version]
- Huang, V.J.; Stein, K.C.; True, H.L. Spontaneous variants of the [RNQ+] prion in yeast demonstrate the extensive conformational diversity possible with prion proteins. PLoS ONE 2013, 8, e79582. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kushnirov, V.V.; Dergalev, A.A.; Alieva, M.K.; Alexandrov, A.I. Structural Bases of Prion Variation in Yeast. Int. J. Mol. Sci. 2022, 23, 5738. https://doi.org/10.3390/ijms23105738
Kushnirov VV, Dergalev AA, Alieva MK, Alexandrov AI. Structural Bases of Prion Variation in Yeast. International Journal of Molecular Sciences. 2022; 23(10):5738. https://doi.org/10.3390/ijms23105738
Chicago/Turabian StyleKushnirov, Vitaly V., Alexander A. Dergalev, Maya K. Alieva, and Alexander I. Alexandrov. 2022. "Structural Bases of Prion Variation in Yeast" International Journal of Molecular Sciences 23, no. 10: 5738. https://doi.org/10.3390/ijms23105738
APA StyleKushnirov, V. V., Dergalev, A. A., Alieva, M. K., & Alexandrov, A. I. (2022). Structural Bases of Prion Variation in Yeast. International Journal of Molecular Sciences, 23(10), 5738. https://doi.org/10.3390/ijms23105738