
The Development of Novel Drug Treatments for Stroke Patients: A Review


Abstract
:1. Introduction
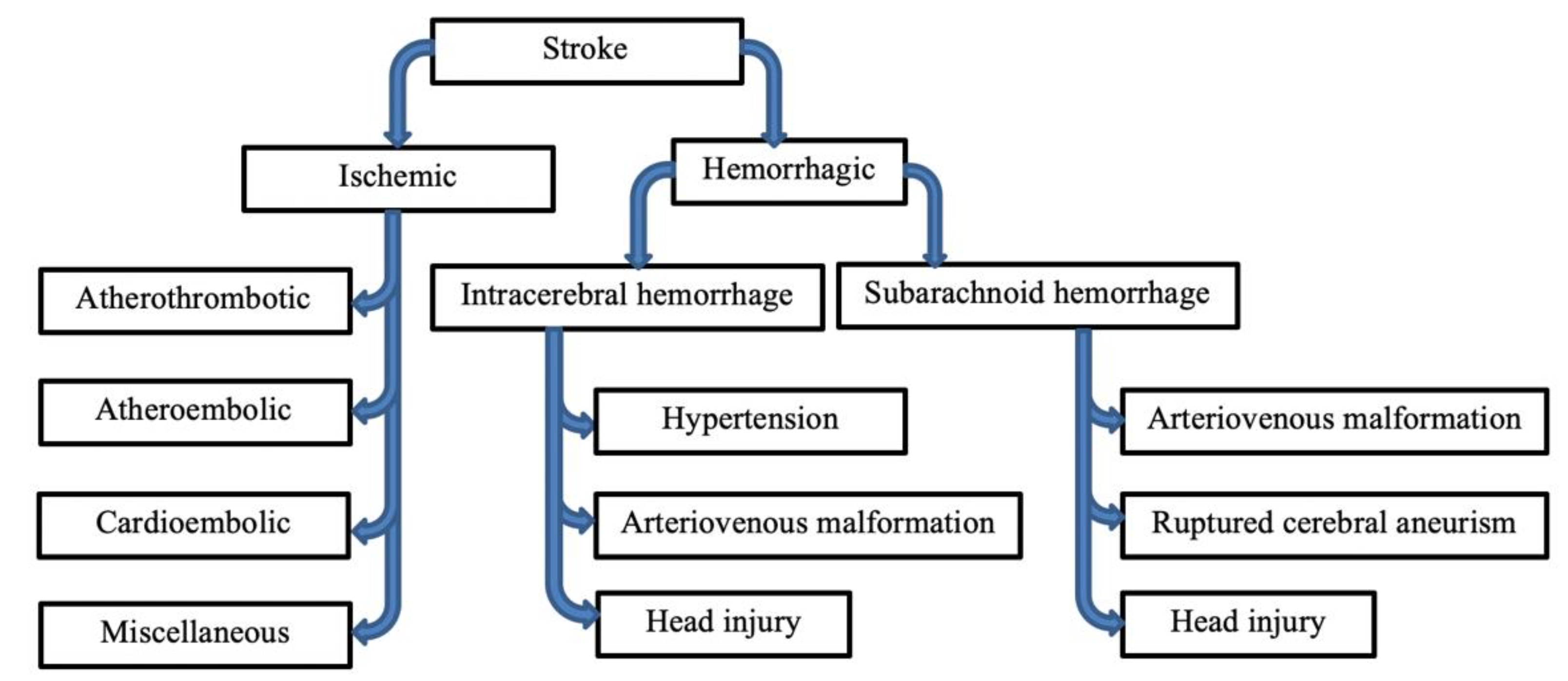
2. Etiology
3. Pathogenesis of Stroke
3.1. Ischemic and Hemorrhagic Stroke
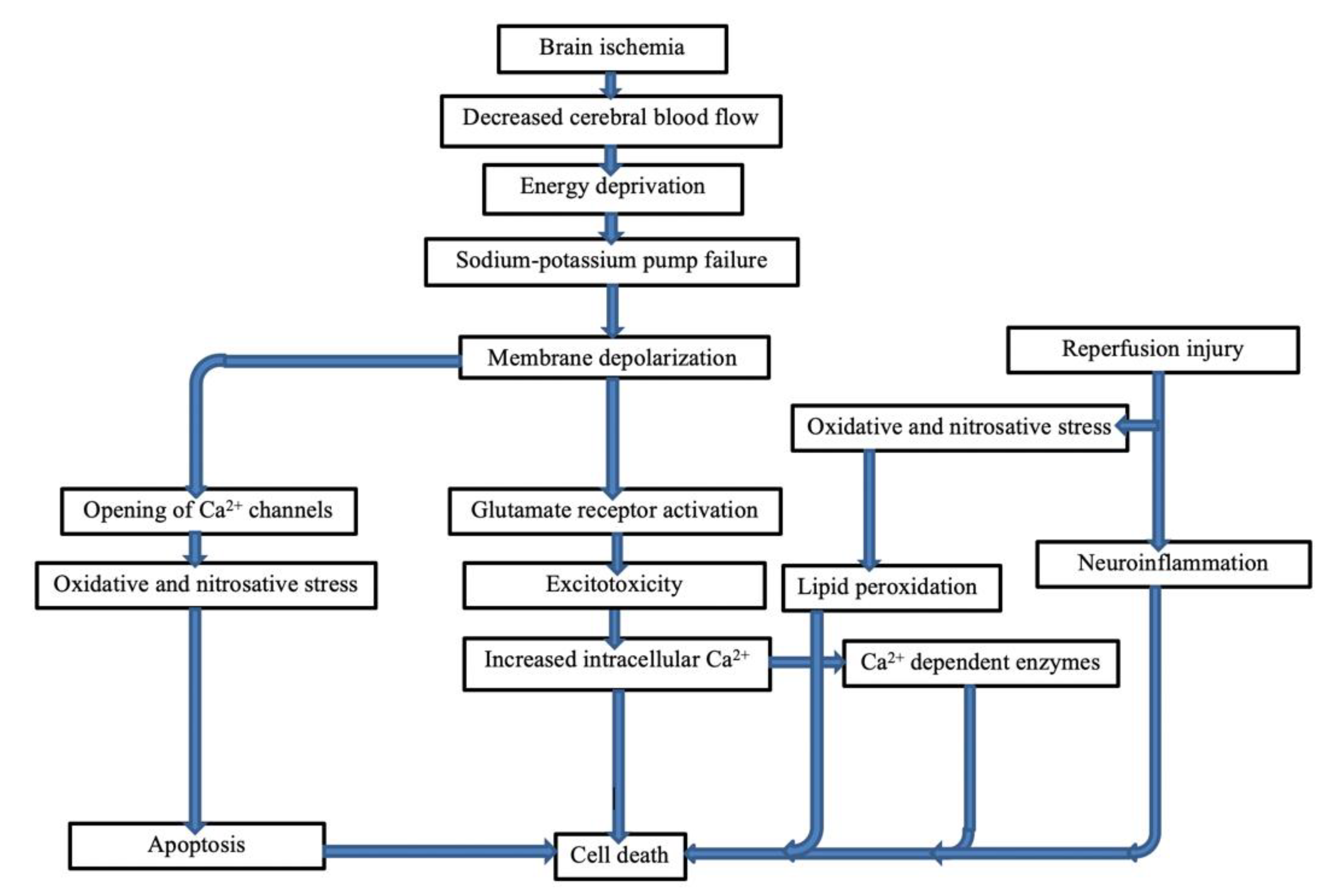
3.2. Excitotoxicity and Stroke
3.3. Excitotoxicity Mechanism
3.4. Ionic Imbalance
3.5. Calcium Homeostasis towards Cell Death
3.6. Activation of Calcium Dependent Enzymes
3.7. Mechanisms of Reperfusion Injury
3.8. Cell Damage during Ischemia–Reperfusion Injuries
3.9. Mechanisms of Cell Death Due to Ischemia–Reperfusion
3.10. Mechanisms of Oxidative Stress and Nitrosative Stress
3.11. Neuroinflammation
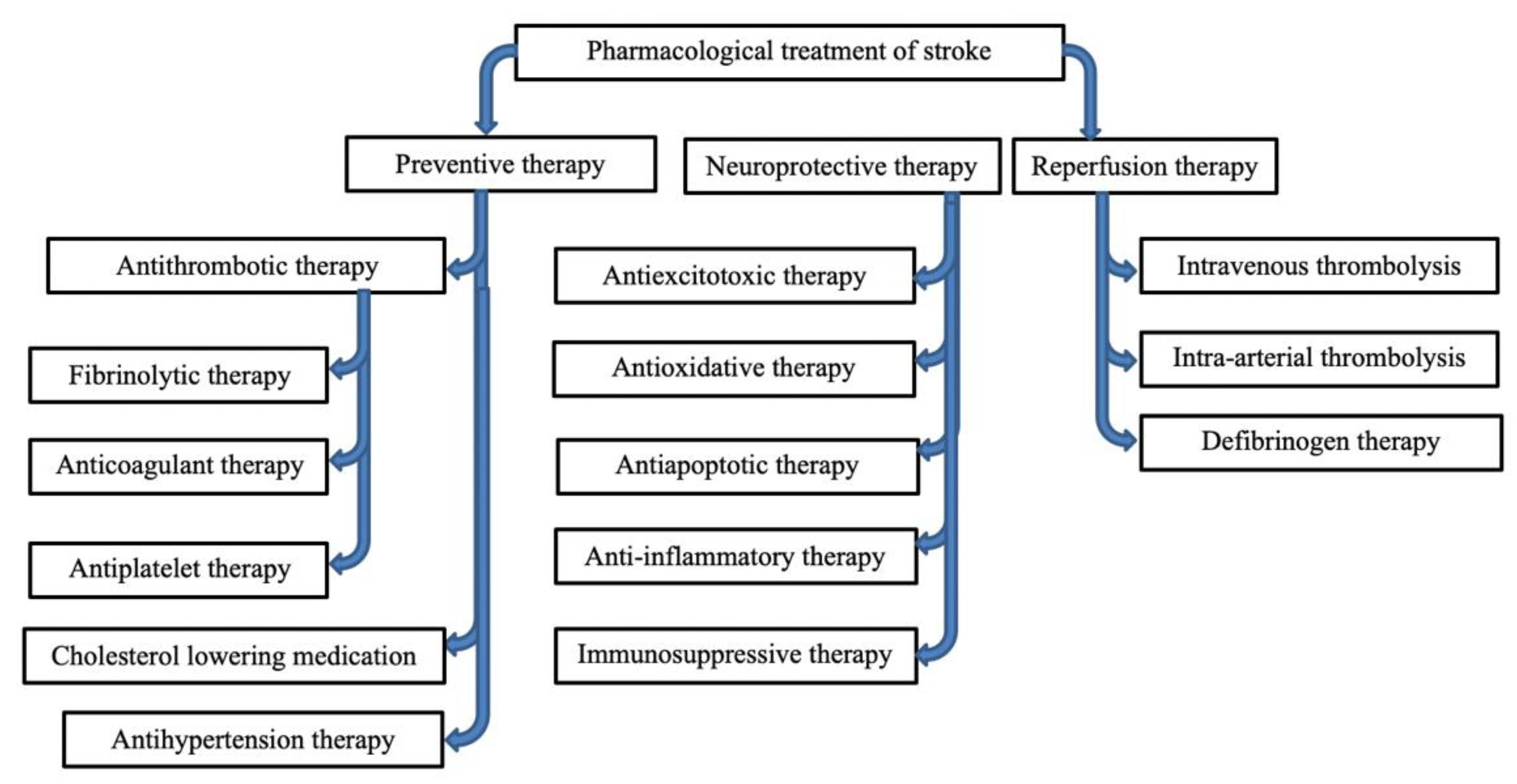
4. Drug Research
4.1. Thrombolytic
4.1.1. Intravenous Thrombolysis (IVT)
4.1.2. Mechanical Thrombectomy
4.1.3. IAT Intra-Arterial Thrombolysis Therapy
4.1.4. Fibrinogen Depleting Agents
4.2. Antithrombotic Drugs
4.3. Neuroprotective Therapy
4.3.1. Glutamate Receptors
4.3.2. Other Glutamate Targets
4.3.3. Magnesium Sulfate
4.3.4. Statins
4.3.5. Melatonin
4.3.6. Erythropoietin
4.3.7. Sodium Channel Blockers
4.3.8. Lubeluzole
4.3.9. Mexiletine
4.3.10. Calcium Channel Blockers and Chelators
4.3.11. GABA (Gamma Aminobutyric Acid Agonists)
4.3.12. Antioxidants
4.3.13. NXY-059
4.3.14. AEOL-10150
4.3.15. Deferoxamine
4.3.16. Pyruvate
4.3.17. C-Jun N Terminal Kinase (JNK) Inhibitor
4.3.18. Immunosuppressant Drugs
4.3.19. Antihypertension Therapy
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tomkins, A.J.; Hood, R.J.; Levi, C.R.; Spratt, N.J. Tissue Plasminogen Activator for preclinical stroke research: Neither “rat” nor “human” dose mimics clinical recanalization in a carotid occlusion model. Sci. Rep. 2015, 5, 16026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González, R.G.; Furie, K.L.; Goldmacher, G.V.; Smith, W.S.; Kamalian, S.; Payabvash, S.; Harris, G.J.; Halpern, E.F.; Koroshetz, W.J.; Camargo, E.C. Good outcome rate of 35% in IV-tPA–treated patients with computed tomography angiography confirmed severe anterior circulation occlusive stroke. Stroke 2013, 44, 3109–3113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fugate, J.E.; Rabinstein, A.A. Absolute and relative contraindications to IV rt-PA for acute ischemic stroke. Neurohospitalist 2015, 5, 110–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gianturco, L. Atherosclerosis: Yesterday, Today and Tomorrow; BoD–Books on Demand: Norderstedt, Germany, 2018. [Google Scholar]
- Pantoni, L.; Gorelick, P.B. Cerebral Small Vessel Disease; Cambridge University Press: Cambridge, UK, 2014. [Google Scholar]
- Johansson, B.B. Hypertension mechanisms causing stroke. Clin. Exp. Pharmacol. Physiol. 1999, 26, 563–565. [Google Scholar] [CrossRef]
- Creager, M.; Loscalzo, J.; Beckman, J.A. Vascular Medicine E-Book: A Companion to Braunwald’s Heart Disease; Elsevier Health Sciences: Amsterdam, The Netherlands, 2012. [Google Scholar]
- Hankey, G.J. Antithrombotic Therapy for Stroke Prevention: What’s New? Circulation 2019, 139, 1131–1133. [Google Scholar] [CrossRef] [PubMed]
- Boccardi, E.; Cenzato, M.; Curto, F.; Longoni, M.; Motto, C.; Oppo, V.; Perini, V.; Vidale, S. Hemorrhagic Stroke; Springer: Berlin/Heidelberg, Germany, 2017. [Google Scholar]
- Feliciano, D.V.; Mattox, K.L.; Moore, E.E. Trauma, 9th ed.; McGraw-Hill Education: New York, NY, USA, 2020. [Google Scholar]
- Abdu, H.; Tadese, F.; Seyoum, G. Comparison of Ischemic and Hemorrhagic Stroke in the Medical Ward of Dessie Referral Hospital, Northeast Ethiopia: A Retrospective Study. Neurol. Res. Int. 2021, 2021, 9996958. [Google Scholar] [CrossRef]
- Kazmi, Z.; Fatima, I.; Perveen, S.; Malik, S.S. Monosodium glutamate: Review on clinical reports. Int. J. Food Prop. 2017, 20, 1807–1815. [Google Scholar] [CrossRef] [Green Version]
- Zanfirescu, A.; Ungurianu, A.; Tsatsakis, A.M.; Nițulescu, G.M.; Kouretas, D.; Veskoukis, A.; Tsoukalas, D.; Engin, A.B.; Aschner, M.; Margină, D. A review of the alleged health hazards of monosodium glutamate. Compr. Rev. Food Sci. Food Saf. 2019, 18, 1111–1134. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Reddy, P.H. Role of glutamate and NMDA receptors in Alzheimer’s disease. J. Alzheimer’s Dis. 2017, 57, 1041–1048. [Google Scholar] [CrossRef] [Green Version]
- Dick, S.A.; Megeney, L.A. Cell death proteins: An evolutionary role in cellular adaptation before the advent of apoptosis. Bioessays 2013, 35, 974–983. [Google Scholar] [CrossRef]
- Reiner, A.; Levitz, J. Glutamatergic signaling in the central nervous system: Ionotropic and metabotropic receptors in concert. Neuron 2018, 98, 1080–1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terker, A.S.; Zhang, C.; McCormick, J.A.; Lazelle, R.A.; Zhang, C.; Meermeier, N.P.; Siler, D.A.; Park, H.J.; Fu, Y.; Cohen, D.M.; et al. Potassium modulates electrolyte balance and blood pressure through effects on distal cell voltage and chloride. Cell Metab. 2015, 21, 39–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abreu, G.E.A. Mechanisms of Neuroinflammation; BoD–Books on Demand: Norderstedt, Germany, 2017. [Google Scholar]
- Murphy, E.; Steenbergen, C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol. Rev. 2008, 88, 581–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Cheng, X.; Liu, X.; Wang, L.; Ha, J.; Gao, Z.; He, X.; Wu, Z.; Chen, A.; Sun, Y. Treatment of cerebral ischemia through NMDA receptors: Metabotropic signaling and future directions. Front. Pharmacol. 2022, 13, 831181. [Google Scholar] [CrossRef]
- Granzotto, A.; Canzoniero, L.M.; Sensi, S.L. A Neurotoxic Ménage-à-trois: Glutamate, calcium, and Zinc in the Excitotoxic cascade. Front. Mol. Neurosci. 2020, 13, 225. [Google Scholar] [CrossRef]
- Brazhe, A.R.; Verisokin, A.Y.; Verveyko, D.V.; Postnov, D.E. Sodium–calcium exchanger can account for regenerative Ca2+ entry in thin astrocyte processes. Front. Cell. Neurosci. 2018, 12, 250. [Google Scholar] [CrossRef]
- Luo, P.; Li, X.; Wu, X.; Dai, S.; Yang, Y.; Xu, H.; Jing, D.; Rao, W.; Xu, H.; Gao, X.; et al. Preso regulates NMDA receptor-mediated excitotoxicity via modulating nitric oxide and calcium responses after traumatic brain injury. Cell Death Dis. 2019, 10, 496. [Google Scholar] [CrossRef]
- Kaludercic, N.; Di Lisa, F. Mitochondrial ROS formation in the pathogenesis of diabetic cardiomyopathy. Front. Cardiovasc. Med. 2020, 7, 12. [Google Scholar] [CrossRef] [Green Version]
- Zemgulyte, G.; Tanaka, S.; Hide, I.; Sakai, N.; Pampuscenko, K.; Borutaite, V.; Rastenyte, D. Evaluation of the Effectiveness of Post-Stroke Metformin Treatment Using Permanent Middle Cerebral Artery Occlusion in Rats. Pharmaceuticals 2021, 14, 312. [Google Scholar] [CrossRef]
- Hagenston, A.M.; Bading, H. Calcium signaling in synapse-to-nucleus communication. Cold Spring Harb. Perspect. Biol. 2011, 3, a004564. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Nagata, S.; Nakano, H. Apoptotic and Non-Apoptotic Cell Death; Springer: Berlin/Heidelberg, Germany, 2017; Volume 403. [Google Scholar]
- Fuller, W.; Parmar, V.; Eaton, P.; Bell, J.R.; Shattock, M.J. Cardiac ischemia causes inhibition of the Na/K ATPase by a labile cytosolic compound whose production is linked to oxidant stress. Cardiovasc. Res. 2003, 57, 1044–1051. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.-Y.; Yiang, G.-T.; Liao, W.-T.; Tsai, A.P.-Y.; Cheng, Y.-L.; Cheng, P.-W.; Li, C.-Y.; Li, C.-J. Current mechanistic concepts in ischemia and reperfusion injury. Cell. Physiol. Biochem. 2018, 46, 1650–1667. [Google Scholar] [CrossRef] [PubMed]
- Curtin, N. Inhibiting the DNA damage response as a therapeutic manoeuvre in cancer. Br. J. Pharmacol. 2013, 169, 1745–1765. [Google Scholar] [CrossRef] [Green Version]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef]
- Panisello-Roselló, A.; Roselló-Catafau, J. Molecular mechanisms and pathophysiology of ischemia-reperfusion injury. Int. J. Mol. Sci. 2018, 19, 4093. [Google Scholar] [CrossRef] [Green Version]
- Xu, B.; Strom, J.; Chen, Q.M. Dexamethasone induces transcriptional activation of Bcl-xL gene and inhibits cardiac injury by myocardial ischemia. Eur. J. Pharmacol. 2011, 668, 194–200. [Google Scholar] [CrossRef] [Green Version]
- Sekerdag, E.; Solaroglu, I.; Gursoy-Ozdemir, Y. Cell death mechanisms in stroke and novel molecular and cellular treatment options. Curr. Neuropharmacol. 2018, 16, 1396–1415. [Google Scholar] [CrossRef]
- Hameister, R.; Kaur, C.; Dheen, S.T.; Lohmann, C.H.; Singh, G. Reactive oxygen/nitrogen species (ROS/RNS) and oxidative stress in arthroplasty. J. Biomed. Mater. Res. Part B Appl. Biomater. 2020, 108, 2073–2087. [Google Scholar] [CrossRef]
- Li, R.; Jia, Z.; Trush, M.A. Defining ROS in biology and medicine. React. Oxyg. Species 2016, 1, 9–27. [Google Scholar] [CrossRef] [Green Version]
- Granger, D.N.; Kvietys, P.R. Reperfusion injury and reactive oxygen species: The evolution of a concept. Redox Biol. 2015, 6, 524–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McIntyre, A.; Adekayode, C.; Kim, H.; Woodley, J.; Dittakavi, T.; Finnegan, M.; Heron, J.; Amuquandoh, K.; James, I.; Benjamin, I.; et al. Protein Kinase C Epsilon Peptide Inhibitor Exerts Cardioprotective Effects in Myocardial Ischemia/Reperfusion Injury. J. Cardiobiol. 2018, 5, 6. [Google Scholar] [CrossRef]
- Wu, L.; Xiong, X.; Wu, X.; Ye, Y.; Jian, Z.; Zhi, Z.; Gu, L. Targeting oxidative stress and inflammation to prevent ischemia-reperfusion injury. Front. Mol. Neurosci. 2020, 13, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayaraj, R.L.; Azimullah, S.; Beiram, R.; Jalal, F.Y.; Rosenberg, G.A. Neuroinflammation: Friend and foe for ischemic stroke. J. Neuroinflamm. 2019, 16, 142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.; Pan, M.-X.; Tang, J.-C.; Zhang, Y.; Liao, H.-B.; Zhuang, Y.; Zhao, D.; Wan, Q. Role of neuroinflammation in ischemic stroke. Neuroimmunol. Neuroinflamm. 2017, 4, 158–166. [Google Scholar] [CrossRef] [Green Version]
- Kwon, H.S.; Koh, S.-H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 42. [Google Scholar] [CrossRef]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef] [Green Version]
- Mokin, M.; Jauch, E.C.; Linfante, I.; Siddiqui, A.; Levy, E. Acute Stroke Management in the First 24 Hours: A Practical Guide for Clinicians; Oxford University Press: Oxford, UK, 2018. [Google Scholar]
- Seifert, H.A.; Pennypacker, K.R. Molecular and cellular immune responses to ischemic brain injury. Transl. Stroke Res. 2014, 5, 543–553. [Google Scholar] [CrossRef]
- Balami, J.S.; Chen, R.; Sutherland, B.A.; Buchan, A.M. Thrombolytic agents for acute ischaemic stroke treatment: The past, present and future. In CNS & Neurological Disorders-Drug Targets (Formerly Current Drug Targets-CNS & Neurological Disorders); Bentham Science Publishers: Sharjah, United Arab Emirates, 2013; Volume 12, pp. 145–154. [Google Scholar]
- Carhart-Harris, R.L.; Goodwin, G.M. The therapeutic potential of psychedelic drugs: Past, present, and future. Neuropsychopharmacology 2017, 42, 2105–2113. [Google Scholar] [CrossRef]
- Park, J. Acute Ischemic Stroke: Medical, Endovascular, and Surgical Techniques; Springer: Berlin/Heidelberg, Germany, 2017. [Google Scholar]
- Adams, H.P.; Brott, T.G.; Furlan, A.J.; Gomez, C.R.; Grotta, J.; Helgason, C.M.; Kwiatkowski, T.; Lyden, P.D.; Marler, J.R.; Torner, J.; et al. Guidelines for thrombolytic therapy for acute stroke: A supplement to the guidelines for the management of patients with acute ischemic stroke: A statement for healthcare professionals from a Special Writing Group of the Stroke Council, American Heart Association. Circulation 1996, 94, 1167–1174. [Google Scholar]
- Paz, J.C.; West, M.P. Acute Care Handbook for Physical Therapists E-Book; Elsevier Health Sciences: Amsterdam, The Netherlands, 2019. [Google Scholar]
- Lee, S.-H. Stroke Revisited: Diagnosis and Treatment of Ischemic Stroke; Springer: Berlin/Heidelberg, Germany, 2017. [Google Scholar]
- Lyden, P.; Levy, H.; Weymer, S.; Pryor, K.; Kramer, W.; Griffin, J.H.; Davis, T.P.; Zlokovic, B. Phase 1 safety, tolerability and pharmacokinetics of 3K3A-APC in healthy adult volunteers. Curr. Pharm. Des. 2013, 19, 7479–7485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qureshi, A.I.; Harris-Lane, P.; Kirmani, J.F.; Janjua, N.; Divani, A.A.; Mohammad, Y.M.; Suarez, J.I.; Montgomery, M.O. Intra-arterial reteplase and intravenous abciximab in patients with acute ischemic stroke: An open-label, dose-ranging, phase I study. Neurosurgery 2006, 59, 789–797. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, C. Randomized Assessment of Rapid Endovascular Treatment of Ischemic Stroke. J. Emerg. Med. 2015, 49, 258–259. [Google Scholar] [CrossRef]
- Warach, S.J.; Dula, A.N.; Milling Jr, T.J. Tenecteplase thrombolysis for acute ischemic stroke. Stroke 2020, 51, 3440–3451. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Cheripelli, B.K.; Lloyd, S.M.; Kalladka, D.; Moreton, F.C.; Siddiqui, A.; Ford, I.; Muir, K.W. Alteplase versus tenecteplase for thrombolysis after ischaemic stroke (ATTEST): A phase 2, randomised, open-label, blinded endpoint study. Lancet Neurol. 2015, 14, 368–376. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Fang, X.; Wang, D.; Xiao, Y. Effect of intravenous thrombolysis with alteplase on clinical efficacy, inflammatory factors, and neurological function in patients with acute cerebral infarction. Braz. J. Med. Biol. Res. 2021, 54. [Google Scholar] [CrossRef] [PubMed]
- Page, P.S.; Khattar, N.K.; White, A.C.; Cambon, A.C.; Brock, G.N.; Rai, S.N.; James, R.F. Intra-arterial thrombolysis for acute central retinal artery occlusion: A systematic review and meta-analysis. Front. Neurol. 2018, 9, 76. [Google Scholar] [CrossRef] [Green Version]
- Hao, Z.; Liu, M.; Counsell, C.; Wardlaw, J.M.; Lin, S.; Zhao, X. Fibrinogen depleting agents for acute ischaemic stroke. Cochrane Database Syst. Rev. 2012. [Google Scholar] [CrossRef]
- Liu, S.; Marder, V.J.; Levy, D.E.; Wang, S.-J.; Yang, F.; Paganini-Hill, A.; Fisher, M.J. Ancrod and fibrin formation: Perspectives on mechanisms of action. Stroke 2011, 42, 3277–3280. [Google Scholar] [CrossRef]
- Lippi, G.; Montagnana, M.; Danese, E.; Favaloro, E.J.; Franchini, M. Glycoprotein IIb/IIIa inhibitors: An update on the mechanism of action and use of functional testing methods to assess antiplatelet efficacy. Biomark. Med. 2011, 5, 63–70. [Google Scholar] [CrossRef]
- Medcalf, R.L.; Lawrence, D.A. The role of the plasminogen activating system in neurobiology. Front. Cell. Neurosci. 2016, 10, 222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takado, Y.; Sato, N.; Kanbe, Y.; Tomiyasu, M.; Xin, L.; Near, J.; Yoshikawa, K.; Sahara, N.; Higashi, T.; Suhara, T.; et al. Association between brain and plasma glutamine levels in healthy young subjects investigated by MRS and LC/MS. Nutrients 2019, 11, 1649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scalise, M.; Pochini, L.; Galluccio, M.; Console, L.; Indiveri, C. Glutamine transport and mitochondrial metabolism in cancer cell growth. Front. Oncol. 2017, 7, 306. [Google Scholar] [CrossRef] [Green Version]
- Goodwani, S.; Saternos, H.; Alasmari, F.; Sari, Y. Metabotropic and ionotropic glutamate receptors as potential targets for the treatment of alcohol use disorder. Neurosci. Biobehav. Rev. 2017, 77, 14–31. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yan, H.; Zhang, S.; Wei, X.; Zheng, J.; Li, J. The GluN3A subunit exerts a neuroprotective effect in brain ischemia and the hypoxia process. ASN Neuro 2013, 5, AN20130009. [Google Scholar] [CrossRef] [PubMed]
- Choo, A.M.; Geddes-Klein, D.M.; Hockenberry, A.; Scarsella, D.; Mesfin, M.N.; Singh, P.; Patel, T.P.; Meaney, D.F. NR2A and NR2B subunits differentially mediate MAP kinase signaling and mitochondrial morphology following excitotoxic insult. Neurochem. Int. 2012, 60, 506–516. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Ding, Q.; Chen, Z.; Yun, H.; Wang, H. Involvement of the GluN2A and GluN2B subunits in synaptic and extrasynaptic N-methyl-D-aspartate receptor function and neuronal excitotoxicity. J. Biol. Chem. 2013, 288, 24151–24159. [Google Scholar] [CrossRef] [Green Version]
- Yan, J.-Z.; Liu, Y.; Zong, Y.-Y.; Zhang, G.-Y. Knock-down of postsynaptic density protein 95 expression by antisense oligonucleotides protects against apoptosis-like cell death induced by oxygen-glucose deprivation in vitro. Neurosci. Bull. 2012, 28, 69–76. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Cheng, X.; Hu, J.; Gao, Z. The role of GluN2A in cerebral ischemia: Promoting neuron death and survival in the early stage and thereafter. Mol. Neurobiol. 2018, 55, 1208–1216. [Google Scholar] [CrossRef]
- Buonarati, O.R.; Cook, S.G.; Goodell, D.J.; Chalmers, N.E.; Rumian, N.L.; Tullis, J.E.; Restrepo, S.; Coultrap, S.J.; Quillinan, N.; Herson, P.S.; et al. CaMKII versus DAPK1 binding to GluN2B in ischemic neuronal cell death after resuscitation from cardiac arrest. Cell Rep. 2020, 30, 1–8.e4. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, M.E.; Dong, Y.; Lu, Y.; Tucker, D.; Wang, R.; Zhang, Q. Beneficial effects of a CaMKIIα inhibitor TatCN21 peptide in global cerebral ischemia. J. Mol. Neurosci. 2017, 61, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Milani, D.; Cross, J.L.; Anderton, R.S.; Blacker, D.J.; Knuckey, N.W.; Meloni, B.P. Neuroprotective efficacy of poly-arginine R18 and NA-1 (TAT-NR2B9c) peptides following transient middle cerebral artery occlusion in the rat. Neurosci. Res. 2017, 114, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Boyko, M.; Melamed, I.; Gruenbaum, B.F.; Gruenbaum, S.E.; Ohayon, S.; Leibowitz, A.; Brotfain, E.; Shapira, Y.; Zlotnik, A. The effect of blood glutamate scavengers oxaloacetate and pyruvate on neurological outcome in a rat model of subarachnoid hemorrhage. Neurotherapeutics 2012, 9, 649–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gruenbaum, B.F.; Kutz, R.; Zlotnik, A.; Boyko, M. Blood glutamate scavenging as a novel glutamate-based therapeutic approach for post-stroke depression. Ther. Adv. Psychopharmacol. 2020, 10, 2045125320903951. [Google Scholar] [CrossRef] [Green Version]
- Boyko, M.; Zlotnik, A.; Gruenbaum, B.F.; Gruenbaum, S.E.; Ohayon, S.; Kuts, R.; Melamed, I.; Regev, A.; Shapira, Y.; Teichberg, V.I. Pyruvate’s blood glutamate scavenging activity contributes to the spectrum of its neuroprotective mechanisms in a rat model of stroke. Eur. J. Neurosci. 2011, 34, 1432–1441. [Google Scholar] [CrossRef]
- Frank, D.; Kuts, R.; Tsenter, P.; Gruenbaum, B.F.; Grinshpun, Y.; Zvenigorodsky, V.; Shelef, I.; Natanel, D.; Brotfain, E.; Zlotnik, A.; et al. The effect of pyruvate on the development and progression of post-stroke depression: A new therapeutic approach. Neuropharmacology 2019, 155, 173–184. [Google Scholar] [CrossRef]
- Gragossian, A.; Bashir, K.; Friede, R. Hypomagnesemia; StatPearls Publishing: Treasure Island, FL, USA, 2018. [Google Scholar]
- Muir, K. Magnesium in stroke treatment. Postgrad. Med. J. 2002, 78, 641–645. [Google Scholar] [CrossRef] [Green Version]
- Vink, R.; Nechifor, M. Magnesium in the Central Nervous System; University of Adelaide Press: Adelaide, Australia, 2011. [Google Scholar]
- Kaur, P.; Sharma, S. Recent advances in pathophysiology of traumatic brain injury. Curr. Neuropharmacol. 2018, 16, 1224–1238. [Google Scholar] [CrossRef]
- Kirkland, A.E.; Sarlo, G.L.; Holton, K.F. The role of magnesium in neurological disorders. Nutrients 2018, 10, 730. [Google Scholar] [CrossRef] [Green Version]
- Saver, J.L.; Starkman, S.; Eckstein, M.; Stratton, S.J.; Pratt, F.D.; Hamilton, S.; Conwit, R.; Liebeskind, D.S.; Sung, G.; Kramer, I.; et al. Prehospital use of magnesium sulfate as neuroprotection in acute stroke. N. Engl. J. Med. 2015, 372, 528–536. [Google Scholar] [CrossRef] [Green Version]
- Wong, G.K.; Poon, W.S.; Chan, M.T.; Boet, R.; Gin, T.; Ng, S.C.; Zee, B.C. Plasma magnesium concentrations and clinical outcomes in aneurysmal subarachnoid hemorrhage patients: Post hoc analysis of intravenous magnesium sulphate for aneurysmal subarachnoid hemorrhage trial. Stroke 2010, 41, 1841–1844. [Google Scholar] [CrossRef] [PubMed]
- Habgood, M.; Bye, N.; Dziegielewska, K.; Ek, C.; Lane, M.; Potter, A.; Morganti-Kossmann, C.; Saunders, N. Changes in blood–brain barrier permeability to large and small molecules following traumatic brain injury in mice. Eur. J. Neurosci. 2007, 25, 231–238. [Google Scholar] [CrossRef]
- Kinjo, T.; Tomita, H. Atrial fibrillation newly diagnosed after a stroke: Which came first, atrial fibrillation or stroke? J. Xiangya Med. 2018, 3, 34. [Google Scholar] [CrossRef]
- Unit, E.S. Efficacy and safety of cholesterol-lowering treatment: Prospective meta-analysis of data from 90 056 participants in 14 randomised trials of statins. Lancet 2005, 366, 1267–1278. [Google Scholar]
- Parizadeh, S.M.; Azarpazhooh, M.R.; Moohebati, M.; Nematy, M.; Ghayour-Mobarhan, M.; Tavallaie, S.; Rahsepar, A.A.; Amini, M.; Sahebkar, A.; Mohammadi, M.; et al. Simvastatin therapy reduces prooxidant-antioxidant balance: Results of a placebo-controlled cross-over trial. Lipids 2011, 46, 333–340. [Google Scholar] [CrossRef]
- Montaner, J.; Chacón, P.; Krupinski, J.; Rubio, F.; Millán, M.; Molina, C.; Hereu, P.; Quintana, M.; Alvarez-Sabín, J. Simvastatin in the acute phase of ischemic stroke: A safety and efficacy pilot trial. Eur. J. Neurol. 2008, 15, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Parker, R.A.; Huang, Q.; Tesfamariam, B. Influence of 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) reductase inhibitors on endothelial nitric oxide synthase and the formation of oxidants in the vasculature. Atherosclerosis 2003, 169, 19–29. [Google Scholar] [CrossRef]
- Kaneko, Y.; Hayashi, T.; Yu, S.; Tajiri, N.; Bae, E.C.; Solomita, M.A.; Chheda, S.H.; Weinbren, N.L.; Parolini, O.; Borlongan, C.V. Human amniotic epithelial cells express melatonin receptor MT1, but not melatonin receptor MT2: A new perspective to neuroprotection. J. Pineal Res. 2011, 50, 272–280. [Google Scholar] [CrossRef]
- Lee, M.Y.; Kuan, Y.H.; Chen, H.Y.; Chen, T.Y.; Chen, S.T.; Huang, C.C.; Yang, I.P.; Hsu, Y.S.; Wu, T.S.; Lee, E.J. Intravenous administration of melatonin reduces the intracerebral cellular inflammatory response following transient focal cerebral ischemia in rats. J. Pineal Res. 2007, 42, 297–309. [Google Scholar] [CrossRef]
- Shinozuka, K.; Staples, M.; Borlongan, C.V. Melatonin-based therapeutics for neuroprotection in stroke. Int. J. Mol. Sci. 2013, 14, 8924–8947. [Google Scholar] [CrossRef]
- Kovermann, P.; Untiet, V.; Kolobkova, Y.; Engels, M.; Baader, S.; Schilling, K.; Fahlke, C. Increased glutamate transporter-associated anion currents cause glial apoptosis in episodic ataxia 6. Brain Commun. 2020, 2, fcaa022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, F.; Sandhu, A.F.; Rungratanawanich, W.; Williams, G.E.; Akbar, M.; Zhou, S.; Song, B.-J.; Wang, X. Melatonin and autophagy in aging-related neurodegenerative diseases. Int. J. Mol. Sci. 2020, 21, 7174. [Google Scholar] [CrossRef] [PubMed]
- Grasso, G.; Sfacteria, A.; Cerami, A.; Brines, M. Erythropoietin as a tissue-protective cytokine in brain injury: What do we know and where do we go? Neuroscientist 2004, 10, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Wang, J.; Li, Q.-Y.; Yu, J.-Z.; Ma, C.-G.; Wang, X.; Lu, C.-Z.; Xiao, B.-G. Neuroprotection and CD131/GDNF/AKT pathway of carbamylated erythropoietin in hypoxic neurons. Mol. Neurobiol. 2017, 54, 5051–5060. [Google Scholar] [CrossRef]
- Priest, B.T.; Kaczorowski, G.J. Subtype-selective sodium channel blockers promise a new era of pain research. Proc. Natl. Acad. Sci. USA 2007, 104, 8205–8206. [Google Scholar] [CrossRef] [Green Version]
- Gandolfo, C.; Sandercock, P.A.; Conti, M. Lubeluzole for acute ischaemic stroke. Cochrane Database Syst. Rev. 2002. [Google Scholar] [CrossRef]
- Grotta, J. Lubeluzole treatment of acute ischemic stroke. Stroke 1997, 28, 2338–2346. [Google Scholar] [CrossRef]
- Mani, V.E. Arbinda Mukherjee: IAN Textbook of Neurology. Neurol. India 2018, 66, 1536. [Google Scholar] [CrossRef]
- Aronson, J.K. Side Effects of Drugs Annual: A World-Wide Yearly Survey of New Data and Trends in Adverse Drug Reactions; Elsevier: Amsterdam, The Netherlands, 2003. [Google Scholar]
- Lodder, J.; Van Raak, L.; Hilton, A.; Hardy, E.; Kessels, A. Diazepam to improve acute stroke outcome: Results of the early GABA-ergic activation study in stroke trial. Cerebrovasc. Dis. 2006, 21, 120–127. [Google Scholar] [CrossRef]
- Lyden, P.D. Thrombolytic Therapy for Acute Stroke; Springer: Berlin/Heidelberg, Germany, 2005. [Google Scholar]
- Diener, H.-C.; Lees, K.R.; Lyden, P.; Grotta, J.; Davalos, A.; Davis, S.M.; Shuaib, A.; Ashwood, T.; Wasiewski, W.; Alderfer, V. NXY-059 for the treatment of acute stroke: Pooled analysis of the SAINT I and II Trials. Stroke 2008, 39, 1751–1758. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.-r.; Zhou, W.-x.; Zhang, Y.-x. Improvements in SOD mimic AEOL-10150, a potent broad-spectrum antioxidant. Mil. Med. Res. 2018, 5, 30. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Tan, L.; Li, H.; Zhang, Q.; Li, Y.; Guo, J. Deferoxamine therapy for intracerebral hemorrhage: A systematic review. PLoS ONE 2018, 13, e0193615. [Google Scholar] [CrossRef] [PubMed]
- Kontoghiorghe, C.N.; Kontoghiorghes, G.J. Efficacy and safety of iron-chelation therapy with deferoxamine, deferiprone, and deferasirox for the treatment of iron-loaded patients with non-transfusion-dependent thalassemia syndromes. Drug Des. Dev. Ther. 2016, 10, 465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, B. Metabolomics in Neurodegenerative Disease; MDPI: Basel, Switzerland, 2020. [Google Scholar]
- Zilberter, Y.; Gubkina, O.; Ivanov, A.I. A unique array of neuroprotective effects of pyruvate in neuropathology. Front. Neurosci. 2015, 9, 17. [Google Scholar] [CrossRef] [Green Version]
- Andrabi, S.A.; Umanah, G.K.; Chang, C.; Stevens, D.A.; Karuppagounder, S.S.; Gagné, J.-P.; Poirier, G.G.; Dawson, V.L.; Dawson, T.M. Poly (ADP-ribose) polymerase-dependent energy depletion occurs through inhibition of glycolysis. Proc. Natl. Acad. Sci. USA 2014, 111, 10209–10214. [Google Scholar] [CrossRef] [Green Version]
- Ying, W.; Chen, Y.; Alano, C.C.; Swanson, R.A. Tricarboxylic acid cycle substrates prevent PARP-mediated death of neurons and astrocytes. J. Cereb. Blood Flow Metab. 2002, 22, 774–779. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; van Hoecke, M.; Tang, X.N.; Lee, H.; Zheng, Z.; Swanson, R.A.; Yenari, M.A. Pyruvate protects against experimental stroke via an anti-inflammatory mechanism. Neurobiol. Dis. 2009, 36, 223–231. [Google Scholar] [CrossRef] [Green Version]
- Plotnikov, M.B.; Chernysheva, G.A.; Aliev, O.I.; Smol’iakova, V.I.; Fomina, T.I.; Osipenko, A.N.; Rydchenko, V.S.; Anfinogenova, Y.J.; Khlebnikov, A.I.; Schepetkin, I.A.; et al. Protective effects of a new C-Jun N-terminal kinase inhibitor in the model of global cerebral ischemia in rats. Molecules 2019, 24, 1722. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.S.; Khan, A.; Ahmad, S.; Ahmad, R.; Rehman, I.U.; Ikram, M.; Kim, M.O. Inhibition of JNK alleviates chronic hypoperfusion-related ischemia induces oxidative stress and brain degeneration via Nrf2/HO-1 and NF-κB signaling. Oxidative Med. Cell. Longev. 2020, 2020, 5291852. [Google Scholar] [CrossRef]
- Zawadzka, M.; Kaminska, B. A novel mechanism of FK506-mediated neuroprotection: Downregulation of cytokine expression in glial cells. Glia 2005, 49, 36–51. [Google Scholar] [CrossRef]
- Pillans, P. Immunosuppressants-mechanisms of action and monitoring. Aust. Prescr. 2006, 29, 99–101. [Google Scholar] [CrossRef] [Green Version]
- Szydlowska, K.; Gozdz, A.; Dabrowski, M.; Zawadzka, M.; Kaminska, B. Prolonged activation of ERK triggers glutamate-induced apoptosis of astrocytes: Neuroprotective effect of FK506. J. Neurochem. 2010, 113, 904–918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edmunds, M.W.; Mayhew, M.S. Pharmacology for the Primary Care Provider-E-Book; Elsevier Health Sciences: Amsterdam, The Netherlands, 2013. [Google Scholar]
- Boncoraglio, G.B.; Del Giovane, C.; Tramacere, I. Antihypertensive Drugs for Secondary Prevention After Ischemic Stroke or Transient Ischemic Attack: A Systematic Review and Meta-Analysis. Stroke 2021, 52, 1974–1982. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Neuroprotectors | Mechanism of Action |
|---|---|
| NV-AAMO77 | Antiexcitotoxic, antagonizes GluN2A receptors |
| Zn+ | Antiexcitotoxic, antagonizes GluN2A receptors |
| Tat-CN21 | Antiexcitotoxic, minimizes brain damage by stopping GluN2B from binding with CaMKII |
| Poly-arginine R18 | Antiexcitotoxic, inhibits proprotein convertases, reducing calcium ion influx |
| NA-1 | Antiexcitotoxic, inhibits proprotein convertases, reducing calcium ion influx |
| MgSO4 | Antiexcitotoxic, noncompetitive NMDAR blocking, regulation of calcium channels |
| Simvastatin, Mevastatin, Rovostatin, Atorvastatin | Anti-inflammatory, reduces oxidized LDL and malondialdehyde and synthesizes endothelial nitric oxide type III, alters inflammatory gene expression in molecules such as ICAM-1, VCAM-1, interleukins, and E-selection |
| Melatonin | Antioxidant; antiapoptotic; alters the expression of antioxidant enzyme genes such as glutathione peroxidase, catalase, and superoxide dismutase; attenuates AP-1 and NF-kB downregulating cyclooxygenase 2, tumour necrosis factor-alpha, and interleukin-1-B; protects cells from dying by inhibiting JKN 1 and suppressing apoptotic factors; detoxes free radicals and protects DNA |
| IQ-IS | Antiapoptotic, C-Jun N Terminal Kinase Inhibiting |
| SP600125 | Antiapoptotic, C-Jun N Terminal Kinase Inhibiting |
| AEOL 10150 | Antioxidant |
| Deferoxamine | Antioxidant |
| NXY 059 | Antioxidant |
| Glutamate-oxaloacetate transaminase | Antiexcitotoxic. blood glutamate scavenging |
| Glutamate-pyruvate transaminase | Antiexcitotoxic. blood glutamate scavenging |
| Oxaloacetate | Antiexcitotoxic. blood glutamate scavenging |
| Pyruvate | Antiexcitotoxic, antioxidant, blood glutamate scavenging, anti-inflammatory |
| Erythropoietin | Antiapoptotic. stimulates the Janus tyrosine kinase by signalling pathways, expressing of extracellular-regulated kinase, Bcl-2, nuclear factor-kappa, and protein kinase |
| Lubeluzole | Sodium channel blocking |
| Mexiletine | Sodium channel blocking |
| DP b99 | Calcium channel blocking |
| Clomethiasole | Gamma aminobutyric acid agonist |
| Diazepam | Gamma aminobutyric acid agonist |
| Cyclosporin | Immunosuppressing, antiapoptotic, suppresses cells and interleukin-2 production, blocks ERK 1/2 |
| Tacrolimus | Immunosuppressant, antiapoptotic, suppresses cells and interleukin-2 production, blocks ERK 1/2, inhibits nitric oxide and calcineurin production, reduces tumour-necrosis factor-alpha |
| Metformin | Antioxidant, antiapoptotic, anti-inflammatory, hinders mitochondrial respiratory-chain-complex 1, enhances the mitochondria’s calcium capacity and prevents the opening of its permeability pore |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frank, D.; Zlotnik, A.; Boyko, M.; Gruenbaum, B.F. The Development of Novel Drug Treatments for Stroke Patients: A Review. Int. J. Mol. Sci. 2022, 23, 5796. https://doi.org/10.3390/ijms23105796
Frank D, Zlotnik A, Boyko M, Gruenbaum BF. The Development of Novel Drug Treatments for Stroke Patients: A Review. International Journal of Molecular Sciences. 2022; 23(10):5796. https://doi.org/10.3390/ijms23105796
Chicago/Turabian StyleFrank, Dmitry, Alexander Zlotnik, Matthew Boyko, and Benjamin Fredrick Gruenbaum. 2022. "The Development of Novel Drug Treatments for Stroke Patients: A Review" International Journal of Molecular Sciences 23, no. 10: 5796. https://doi.org/10.3390/ijms23105796
APA StyleFrank, D., Zlotnik, A., Boyko, M., & Gruenbaum, B. F. (2022). The Development of Novel Drug Treatments for Stroke Patients: A Review. International Journal of Molecular Sciences, 23(10), 5796. https://doi.org/10.3390/ijms23105796