
Postischemic Neuroprotection of Aminoethoxydiphenyl Borate Associates Shortening of Peri-Infarct Depolarizations

 ,
,  , and
, and
Abstract
:1. Introduction
2. Results
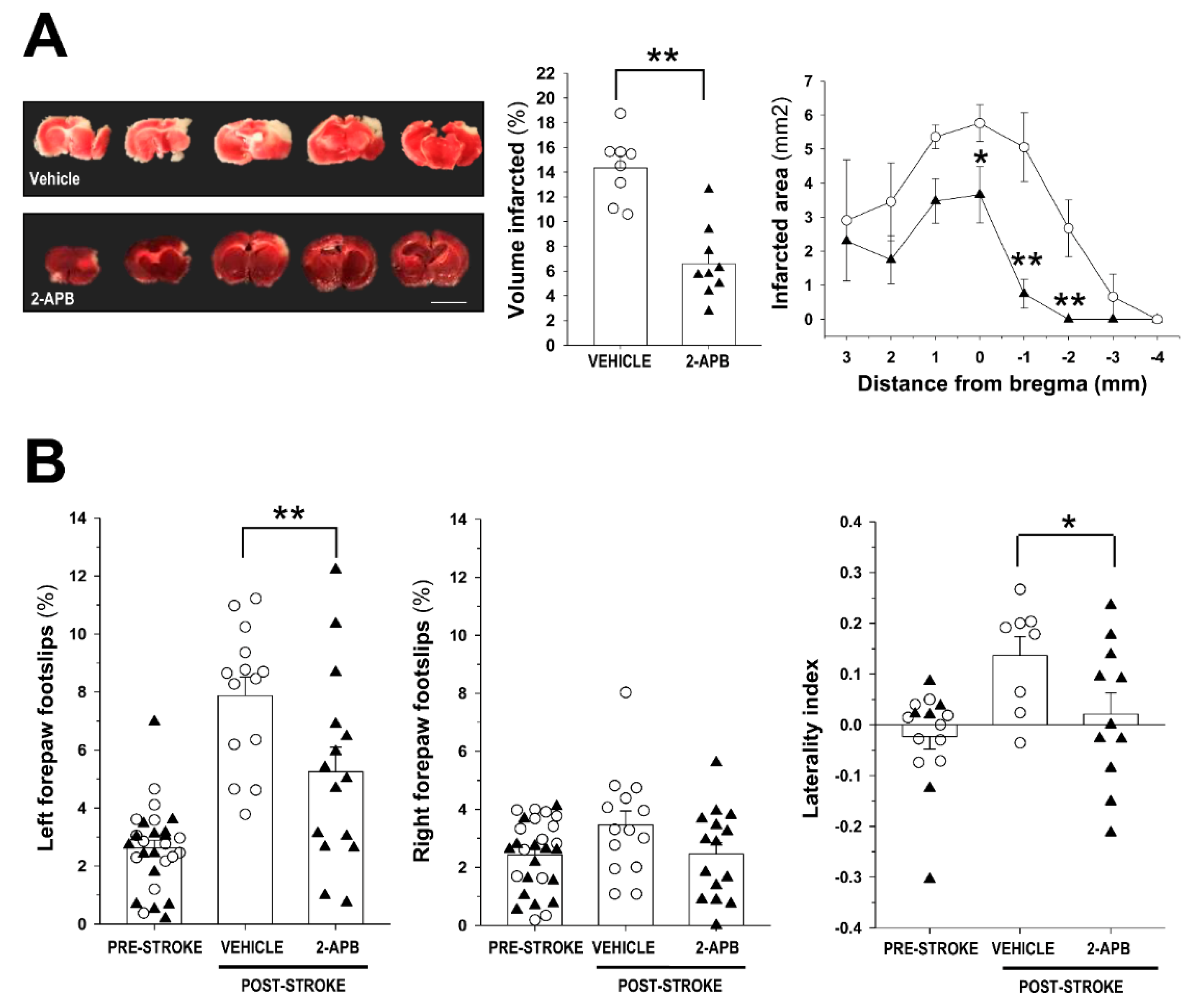
2.1. Evaluation of Infarct Size and Sensorimotor Coordination
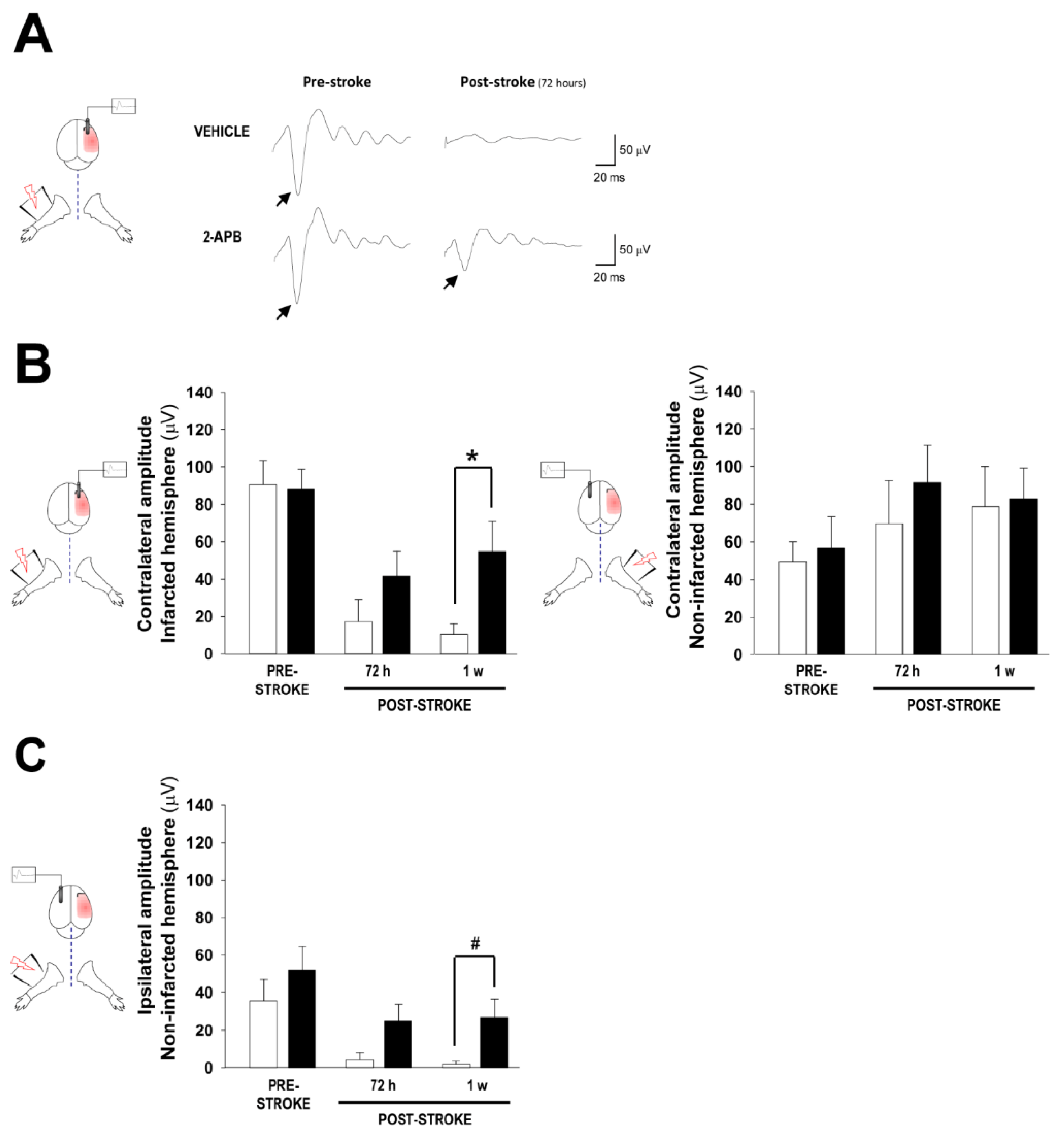
2.2. Assessment of Cortical Brain Function by Somatosensory Evoked Potentials
2.3. Evaluation of Secondary Injury: Oxidative Stress and Inflammatory Response
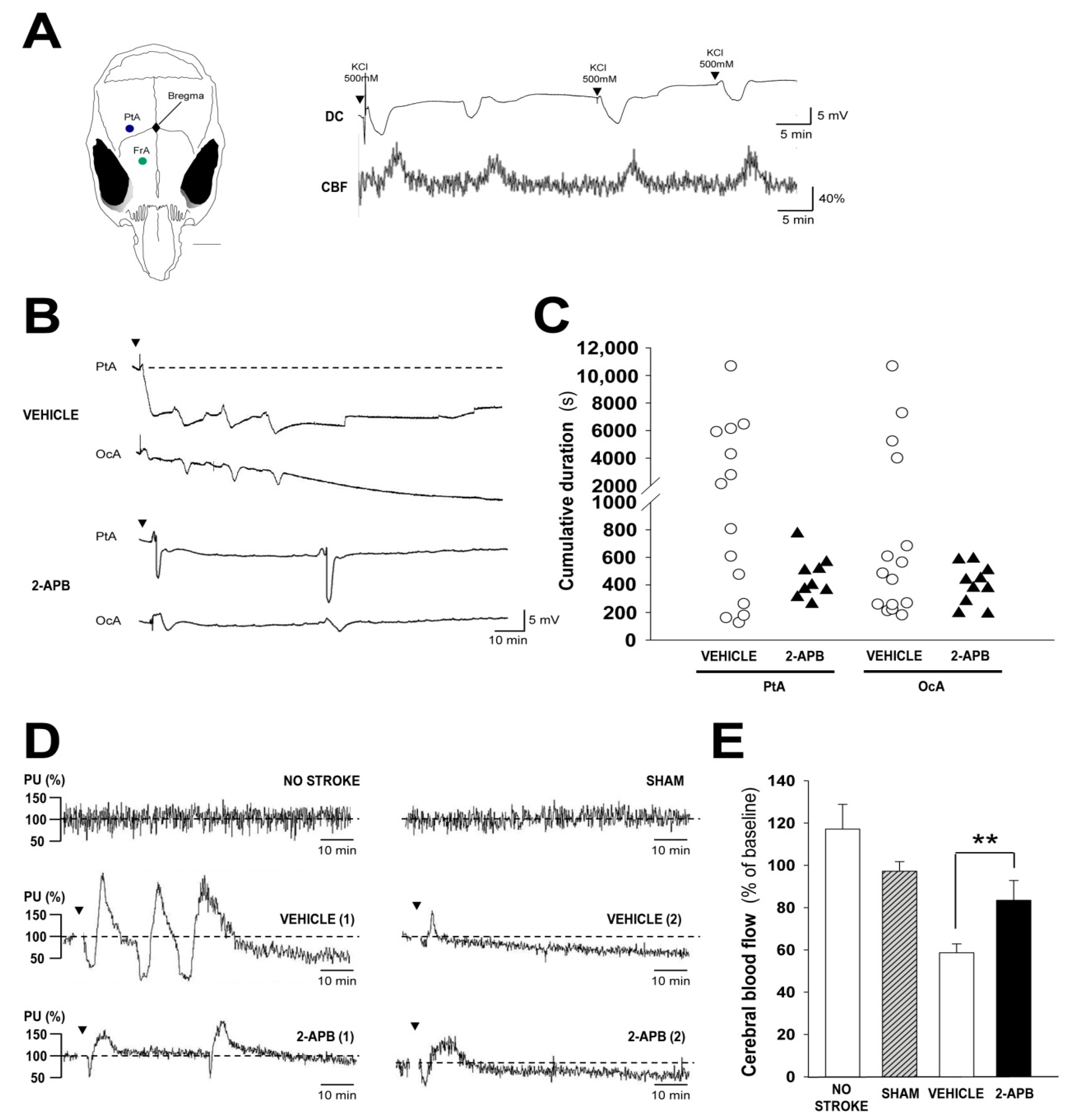
2.4. Evaluation of Secondary Injury: Peri-Infarct Depolarizations and Cerebral Blood Flow
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Permanent Middle Cerebral Artery Occlusion
4.3. Drug Administration
4.4. Infarct Volume and Area Measurement
4.5. Behavioral Tests
4.6. Evaluation of Functional Cortical Territory
4.7. Cortical Depolarization Recordings
4.8. Cerebral Blood Flow Assessment
4.9. Oxidative Stress Measurement
4.10. Immunohistochemistry
4.11. TUNEL Assay
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Donkor, E.S. Stroke in the 21(st) Century: A Snapshot of the Burden, Epidemiology, and Quality of Life. Stroke Res. Treat. 2018, 2018, 3238165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bejot, Y.; Bailly, H.; Durier, J.; Giroud, M. Epidemiology of stroke in Europe and trends for the 21st century. Presse Med. 2016, 45, e391–e398. [Google Scholar] [CrossRef] [PubMed]
- Feigin, V.L.; Roth, G.A.; Naghavi, M.; Parmar, P.; Krishnamurthi, R.; Chugh, S.; Mensah, G.A.; Norrving, B.; Shiue, I.; Ng, M.; et al. Global burden of stroke and risk factors in 188 countries, during 1990–2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet Neurol. 2016, 15, 913–924. [Google Scholar] [CrossRef] [Green Version]
- Wajngarten, M.; Silva, G.S. Hypertension and Stroke: Update on Treatment. Eur. Cardiol. 2019, 14, 111–115. [Google Scholar] [CrossRef] [Green Version]
- Kerwin, W.S. Carotid artery disease and stroke: Assessing risk with vessel wall MRI. ISRN Cardiol. 2012, 2012, 180710. [Google Scholar] [CrossRef] [Green Version]
- Essa, H.; Hill, A.M.; Lip, G.Y.H. Atrial Fibrillation and Stroke. Card. Electrophysiol. Clin. 2021, 13, 243–255. [Google Scholar] [CrossRef]
- Frost, L.; Vestergaard, P.; Mosekilde, L. Hyperthyroidism and risk of atrial fibrillation or flutter: A population-based study. Arch. Intern. Med. 2004, 164, 1675–1678. [Google Scholar] [CrossRef] [Green Version]
- Gencer, B.; Cappola, A.R.; Rodondi, N.; Collet, T.H. Challenges in the Management of Atrial Fibrillation With Subclinical Hyperthyroidism. Front. Endocrinol. 2021, 12, 795492. [Google Scholar] [CrossRef]
- Bielecka-Dabrowa, A.; Mikhailidis, D.P.; Rysz, J.; Banach, M. The mechanisms of atrial fibrillation in hyperthyroidism. Thyroid Res. 2009, 2, 4. [Google Scholar] [CrossRef] [Green Version]
- Mascia, G.; Della Bona, R.; Ameri, P.; Canepa, M.; Porto, I.; Brignole, M. Brugada syndrome and syncope: A systematic review. J. Cardiovasc. Electrophysiol. 2020, 31, 3334–3338. [Google Scholar] [CrossRef]
- Francis, J.; Antzelevitch, C. Atrial fibrillation and Brugada syndrome. J. Am. Coll. Cardiol. 2008, 51, 1149–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Platonov, P.G.; McNitt, S.; Polonsky, B.; Rosero, S.Z.; Zareba, W. Atrial Fibrillation in Long QT Syndrome by Genotype. Circ. Arrhythm. Electrophysiol. 2019, 12, e007213. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.D.; Folsom, A.R.; Blair, S.N. Physical activity and stroke risk: A meta-analysis. Stroke 2003, 34, 2475–2481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallanagh, S.; Quinn, T.J.; Alexander, J.; Walters, M.R. Physical activity in the prevention and treatment of stroke. ISRN Neurol. 2011, 2011, 953818. [Google Scholar] [CrossRef] [Green Version]
- Mont, L.; Elosua, R.; Brugada, J. Endurance sport practice as a risk factor for atrial fibrillation and atrial flutter. EP Eur. 2009, 11, 11–17. [Google Scholar] [CrossRef]
- Albers, G.W.; Goldstein, L.B.; Hess, D.C.; Wechsler, L.R.; Furie, K.L.; Gorelick, P.B.; Hurn, P.; Liebeskind, D.S.; Nogueira, R.G.; Saver, J.L. Stroke Treatment Academic Industry Roundtable (STAIR) recommendations for maximizing the use of intravenous thrombolytics and expanding treatment options with intra-arterial and neuroprotective therapies. Stroke 2011, 42, 2645–2650. [Google Scholar] [CrossRef]
- Hill, M.D.; Goyal, M.; Menon, B.K.; Nogueira, R.G.; McTaggart, R.A.; Demchuk, A.M.; Poppe, A.Y.; Buck, B.H.; Field, T.S.; Dowlatshahi, D.; et al. Efficacy and safety of nerinetide for the treatment of acute ischaemic stroke (ESCAPE-NA1): A multicentre, double-blind, randomised controlled trial. Lancet 2020, 395, 878–887. [Google Scholar] [CrossRef]
- Matsumoto, S.; Murozono, M.; Kanazawa, M.; Nara, T.; Ozawa, T.; Watanabe, Y. Edaravone and cyclosporine A as neuroprotective agents for acute ischemic stroke. Acute Med. Surg. 2018, 5, 213–221. [Google Scholar] [CrossRef] [Green Version]
- Bano, D.; Nicotera, P. Ca2+ signals and neuronal death in brain ischemia. Stroke 2007, 38, 674–676. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Chen, L.; Li, C.; Yang, X.; Zhang, J.; Wang, S.; Tan, S. Abstract TP286: The Study of the Role and Mechanism of TRPM7 in Acute Cerebral Ischemia-Reperfusion Injury. Stroke 2020, 51 (Suppl. S1), ATP286. [Google Scholar] [CrossRef]
- Nicoud, I.B.; Knox, C.D.; Jones, C.M.; Anderson, C.D.; Pierce, J.M.; Belous, A.E.; Earl, T.M.; Chari, R.S. 2-APB protects against liver ischemia-reperfusion injury by reducing cellular and mitochondrial calcium uptake. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 293, G623–G630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morihara, H.; Obana, M.; Tanaka, S.; Kawakatsu, I.; Tsuchiyama, D.; Mori, S.; Suizu, H.; Ishida, A.; Kimura, R.; Tsuchimochi, I.; et al. 2-aminoethoxydiphenyl borate provides an anti-oxidative effect and mediates cardioprotection during ischemia reperfusion in mice. PLoS ONE 2017, 12, e0189948. [Google Scholar] [CrossRef] [Green Version]
- Yildar, M.; Aksit, H.; Korkut, O.; Ozyigit, M.O.; Sunay, B.; Seyrek, K. Protective effect of 2-aminoethyl diphenylborinate on acute ischemia-reperfusion injury in the rat kidney. J. Surg. Res. 2014, 187, 683–689. [Google Scholar] [CrossRef]
- Bae, C.Y.; Sun, H.S. TRPM7 in cerebral ischemia and potential target for drug development in stroke. Acta Pharmacol. Sin. 2011, 32, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Verma, S.; Nakayama, S.; Quillinan, N.; Grafe, M.R.; Hurn, P.D.; Herson, P.S. Sex differences in neuroprotection provided by inhibition of TRPM2 channels following experimental stroke. J. Cereb. Blood Flow Metab. 2011, 31, 2160–2168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Kruchten, R.; Braun, A.; Feijge, M.A.; Kuijpers, M.J.; Rivera-Galdos, R.; Kraft, P.; Stoll, G.; Kleinschnitz, C.; Bevers, E.M.; Nieswandt, B.; et al. Antithrombotic potential of blockers of store-operated calcium channels in platelets. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1717–1723. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhao, Z.; Rege, S.V.; Wang, M.; Si, G.; Zhou, Y.; Wang, S.; Griffin, J.H.; Goldman, S.A.; Zlokovic, B.V. 3K3A-activated protein C stimulates postischemic neuronal repair by human neural stem cells in mice. Nat. Med. 2016, 22, 1050–1055. [Google Scholar] [CrossRef] [Green Version]
- Nih, L.R.; Gojgini, S.; Carmichael, S.T.; Segura, T. Dual-function injectable angiogenic biomaterial for the repair of brain tissue following stroke. Nat. Mater. 2018, 17, 642–651. [Google Scholar] [CrossRef]
- Allison, T.; McCarthy, G.; Wood, C.C.; Williamson, P.D.; Spencer, D.D. Human cortical potentials evoked by stimulation of the median nerve. II. Cytoarchitectonic areas generating long-latency activity. J. Neurophysiol. 1989, 62, 711–722. [Google Scholar] [CrossRef]
- Barios, J.A.; Pisarchyk, L.; Fernandez-Garcia, L.; Barrio, L.C.; Ramos, M.; Martinez-Murillo, R.; Gonzalez-Nieto, D. Long-term dynamics of somatosensory activity in a stroke model of distal middle cerebral artery oclussion. J. Cereb. Blood Flow Metab. 2016, 36, 606–620. [Google Scholar] [CrossRef] [Green Version]
- Peppiatt, C.M.; Collins, T.J.; Mackenzie, L.; Conway, S.J.; Holmes, A.B.; Bootman, M.D.; Berridge, M.J.; Seo, J.T.; Roderick, H.L. 2-Aminoethoxydiphenyl borate (2-APB) antagonises inositol 1,4,5-trisphosphate-induced calcium release, inhibits calcium pumps and has a use-dependent and slowly reversible action on store-operated calcium entry channels. Cell Calcium 2003, 34, 97–108. [Google Scholar] [CrossRef] [Green Version]
- Kostandy, B.B. The role of glutamate in neuronal ischemic injury: The role of spark in fire. Neurol. Sci. 2012, 33, 223–237. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Kondo, T.; Kawase, M.; Li, Y.; Sato, S.; Chen, S.F.; Chan, P.H. Mitochondrial susceptibility to oxidative stress exacerbates cerebral infarction that follows permanent focal cerebral ischemia in mutant mice with manganese superoxide dismutase deficiency. J. Neurosci. 1998, 18, 205–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayuso, M.I.; Chioua, M.; Martinez-Alonso, E.; Soriano, E.; Montaner, J.; Masjuan, J.; Hadjipavlou-Litina, D.J.; Marco-Contelles, J.; Alcazar, A. CholesteroNitrones for Stroke. J. Med. Chem. 2015, 58, 6704–6709. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Alonso, E.; Escobar-Peso, A.; Ayuso, M.I.; Gonzalo-Gobernado, R.; Chioua, M.; Montoya, J.J.; Montaner, J.; Fernandez, I.; Marco-Contelles, J.; Alcazar, A. Characterization of a CholesteroNitrone (ISQ-201), a Novel Drug Candidate for the Treatment of Ischemic Stroke. Antioxidants 2020, 9, 291. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Kearns, K.N.; Eli, I.; Sharifi, K.A.; Soldozy, S.; Carlson, E.W.; Scott, K.W.; Sluzewski, M.F.; Acton, S.T.; Stauderman, K.A.; et al. Microglial Calcium Waves during the Hyperacute Phase of Ischemic Stroke. Stroke 2021, 52, 274–283. [Google Scholar] [CrossRef]
- Dijkhuizen, R.M.; Beekwilder, J.P.; van der Worp, H.B.; Berkelbach van der Sprenkel, J.W.; Tulleken, K.A.; Nicolay, K. Correlation between tissue depolarizations and damage in focal ischemic rat brain. Brain Res. 1999, 840, 194–205. [Google Scholar] [CrossRef]
- Hartings, J.A.; Rolli, M.L.; Lu, X.C.; Tortella, F.C. Delayed secondary phase of peri-infarct depolarizations after focal cerebral ischemia: Relation to infarct growth and neuroprotection. J. Neurosci. 2003, 23, 11602–11610. [Google Scholar] [CrossRef] [Green Version]
- Mies, G.; Iijima, T.; Hossmann, K.A. Correlation between peri-infarct DC shifts and ischaemic neuronal damage in rat. Neuroreport 1993, 4, 709–711. [Google Scholar] [CrossRef]
- Shin, H.K.; Dunn, A.K.; Jones, P.B.; Boas, D.A.; Moskowitz, M.A.; Ayata, C. Vasoconstrictive neurovascular coupling during focal ischemic depolarizations. J. Cereb. Blood Flow Metab. 2006, 26, 1018–1030. [Google Scholar] [CrossRef] [Green Version]
- Chuquet, J.; Hollender, L.; Nimchinsky, E.A. High-resolution in vivo imaging of the neurovascular unit during spreading depression. J. Neurosci. 2007, 27, 4036–4044. [Google Scholar] [CrossRef] [PubMed]
- Dreier, J.P.; Lemale, C.L.; Kola, V.; Friedman, A.; Schoknecht, K. Spreading depolarization is not an epiphenomenon but the principal mechanism of the cytotoxic edema in various gray matter structures of the brain during stroke. Neuropharmacology 2018, 134, 189–207. [Google Scholar] [CrossRef] [PubMed]
- Dreier, J.P. The role of spreading depression, spreading depolarization and spreading ischemia in neurological disease. Nat. Med. 2011, 17, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Garcia, L.; Perez-Rigueiro, J.; Martinez-Murillo, R.; Panetsos, F.; Ramos, M.; Guinea, G.V.; Gonzalez-Nieto, D. Cortical Reshaping and Functional Recovery Induced by Silk Fibroin Hydrogels-Encapsulated Stem Cells Implanted in Stroke Animals. Front. Cell. Neurosci. 2018, 12, 296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabricius, M.; Fuhr, S.; Bhatia, R.; Boutelle, M.; Hashemi, P.; Strong, A.J.; Lauritzen, M. Cortical spreading depression and peri-infarct depolarization in acutely injured human cerebral cortex. Brain 2006, 129, 778–790. [Google Scholar] [CrossRef] [Green Version]
- Nedergaard, M.; Astrup, J. Infarct rim: Effect of hyperglycemia on direct current potential and [14C]2-deoxyglucose phosphorylation. J. Cereb. Blood Flow Metab. 1986, 6, 607–615. [Google Scholar] [CrossRef] [Green Version]
- Rother, J.; de Crespigny, A.J.; D’Arceuil, H.; Mosley, M.E. MR detection of cortical spreading depression immediately after focal ischemia in the rat. J. Cereb. Blood Flow Metab. 1996, 16, 214–220. [Google Scholar] [CrossRef] [Green Version]
- Zhou, N.; Gordon, G.R.; Feighan, D.; MacVicar, B.A. Transient swelling, acidification, and mitochondrial depolarization occurs in neurons but not astrocytes during spreading depression. Cereb. Cortex 2010, 20, 2614–2624. [Google Scholar] [CrossRef] [Green Version]
- Hartings, J.A.; Shuttleworth, C.W.; Kirov, S.A.; Ayata, C.; Hinzman, J.M.; Foreman, B.; Andrew, R.D.; Boutelle, M.G.; Brennan, K.C.; Carlson, A.P.; et al. The continuum of spreading depolarizations in acute cortical lesion development: Examining Leao’s legacy. J. Cereb. Blood Flow Metab. 2017, 37, 1571–1594. [Google Scholar] [CrossRef] [Green Version]
- Dreier, J.P.; Reiffurth, C. The stroke-migraine depolarization continuum. Neuron 2015, 86, 902–922. [Google Scholar] [CrossRef] [Green Version]
- Strong, A.J.; Anderson, P.J.; Watts, H.R.; Virley, D.J.; Lloyd, A.; Irving, E.A.; Nagafuji, T.; Ninomiya, M.; Nakamura, H.; Dunn, A.K.; et al. Peri-infarct depolarizations lead to loss of perfusion in ischaemic gyrencephalic cerebral cortex. Brain 2007, 130, 995–1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aldinger, K.A.; Sokoloff, G.; Rosenberg, D.M.; Palmer, A.A.; Millen, K.J. Genetic variation and population substructure in outbred CD-1 mice: Implications for genome-wide association studies. PLoS ONE 2009, 4, e4729. [Google Scholar] [CrossRef] [Green Version]
- Xiao, J.; Liang, D.; Zhao, H.; Liu, Y.; Zhang, H.; Lu, X.; Li, J.; Peng, L.; Chen, Y.H. 2-Aminoethoxydiphenyl borate, a inositol 1,4,5-triphosphate receptor inhibitor, prevents atrial fibrillation. Exp. Biol. Med. 2010, 235, 862–868. [Google Scholar] [CrossRef] [PubMed]
- Bootman, M.D.; Collins, T.J.; Mackenzie, L.; Roderick, H.L.; Berridge, M.J.; Peppiatt, C.M. 2-aminoethoxydiphenyl borate (2-APB) is a reliable blocker of store-operated Ca2+ entry but an inconsistent inhibitor of InsP3-induced Ca2+ release. FASEB J. 2002, 16, 1145–1150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobrydneva, Y.; Abelt, C.J.; Dovel, B.; Thadigiri, C.M.; Williams, R.L.; Blackmore, P.F. 2-aminoethoxydiphenyl borate as a prototype drug for a group of structurally related calcium channel blockers in human platelets. Mol. Pharmacol. 2006, 69, 247–256. [Google Scholar] [CrossRef] [Green Version]
- Colton, C.K.; Zhu, M.X. 2-Aminoethoxydiphenyl borate as a common activator of TRPV1, TRPV2, and TRPV3 channels. Transient Recept. Potential (TRP) Channels 2007, 179, 173–187. [Google Scholar] [CrossRef] [Green Version]
- Rakers, C.; Petzold, G.C. Astrocytic calcium release mediates peri-infarct depolarizations in a rodent stroke model. J. Clin. Investig. 2017, 127, 511–516. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.C.; Chen, R.Y.; Cheng, T.C.; Chiang, Y.C.; Shen, M.L.; Hsu, L.L.; Zhou, N. Spreading Depression Promotes Astrocytic Calcium Oscillations and Enhances Gliotransmission to Hippocampal Neurons. Cereb. Cortex 2018, 28, 3204–3216. [Google Scholar] [CrossRef]
- Bai, D.; del Corsso, C.; Srinivas, M.; Spray, D.C. Block of specific gap junction channel subtypes by 2-aminoethoxydiphenyl borate (2-APB). J. Pharmacol. Exp. Ther. 2006, 319, 1452–1458. [Google Scholar] [CrossRef] [Green Version]
- Bargiotas, P.; Muhammad, S.; Rahman, M.; Jakob, N.; Trabold, R.; Fuchs, E.; Schilling, L.; Plesnila, N.; Monyer, H.; Schwaninger, M. Connexin 36 promotes cortical spreading depolarization and ischemic brain damage. Brain Res. 2012, 1479, 80–85. [Google Scholar] [CrossRef]
- Herreras, O.; Largo, C.; Ibarz, J.M.; Somjen, G.G.; Martin del Rio, R. Role of neuronal synchronizing mechanisms in the propagation of spreading depression in the in vivo hippocampus. J. Neurosci. 1994, 14, 7087–7098. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Denisova, J.V.; Kang, K.S.; Fontes, J.D.; Zhu, B.T.; Belousov, A.B. Neuronal gap junctions are required for NMDA receptor-mediated excitotoxicity: Implications in ischemic stroke. J. Neurophysiol. 2010, 104, 3551–3556. [Google Scholar] [CrossRef] [PubMed]
- Margineanu, D.G.; Klitgaard, H. The connexin 36 blockers quinine, quinidine and mefloquine inhibit cortical spreading depression in a rat neocortical slice model in vitro. Brain Res. Bull. 2006, 71, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Nieto, D.; Gomez-Hernandez, J.M.; Larrosa, B.; Gutierrez, C.; Munoz, M.D.; Fasciani, I.; O’Brien, J.; Zappala, A.; Cicirata, F.; Barrio, L.C. Regulation of neuronal connexin-36 channels by pH. Proc. Natl. Acad. Sci. USA 2008, 105, 17169–17174. [Google Scholar] [CrossRef] [Green Version]
- Ixmatlahua, D.J.; Vizcarra, B.; Gomez-Lira, G.; Romero-Maldonado, I.; Ortiz, F.; Rojas-Piloni, G.; Gutierrez, R. Neuronal Glutamatergic Network Electrically Wired with Silent But Activatable Gap Junctions. J. Neurosci. 2020, 40, 4661–4672. [Google Scholar] [CrossRef]
- Yuzawa, I.; Sakadzic, S.; Srinivasan, V.J.; Shin, H.K.; Eikermann-Haerter, K.; Boas, D.A.; Ayata, C. Cortical spreading depression impairs oxygen delivery and metabolism in mice. J. Cereb. Blood Flow Metab. 2012, 32, 376–386. [Google Scholar] [CrossRef] [Green Version]
- Brennan, K.C.; Romero Reyes, M.; Lopez Valdes, H.E.; Arnold, A.P.; Charles, A.C. Reduced threshold for cortical spreading depression in female mice. Ann. Neurol. 2007, 61, 603–606. [Google Scholar] [CrossRef]
- Krizman, J.; Rotondo, E.K.; Nicol, T.; Kraus, N.; Bieszczad, K.M. Sex differences in auditory processing vary across estrous cycle. Sci. Rep. 2021, 11, 22898. [Google Scholar] [CrossRef]
- Lauritzen, M.; Dreier, J.P.; Fabricius, M.; Hartings, J.A.; Graf, R.; Strong, A.J. Clinical relevance of cortical spreading depression in neurological disorders: Migraine, malignant stroke, subarachnoid and intracranial hemorrhage, and traumatic brain injury. J. Cereb. Blood Flow Metab. 2011, 31, 17–35. [Google Scholar] [CrossRef]
- Klass, A.; Sanchez-Porras, R.; Santos, E. Systematic review of the pharmacological agents that have been tested against spreading depolarizations. J. Cereb. Blood Flow Metab. 2018, 38, 1149–1179. [Google Scholar] [CrossRef]
- Lapchak, P.A.; Zhang, J.H.; Noble-Haeusslein, L.J. RIGOR guidelines: Escalating STAIR and STEPS for effective translational research. Transl. Stroke Res. 2013, 4, 279–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alim, I.; Teves, L.; Li, R.; Mori, Y.; Tymianski, M. Modulation of NMDAR subunit expression by TRPM2 channels regulates neuronal vulnerability to ischemic cell death. J. Neurosci. 2013, 33, 17264–17277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, H.S.; Jackson, M.F.; Martin, L.J.; Jansen, K.; Teves, L.; Cui, H.; Kiyonaka, S.; Mori, Y.; Jones, M.; Forder, J.P.; et al. Suppression of hippocampal TRPM7 protein prevents delayed neuronal death in brain ischemia. Nat. Neurosci. 2009, 12, 1300–1307. [Google Scholar] [CrossRef] [PubMed]
- Eroglu, E.; Unel, C.C.; Harmanci, N.; Erol, K.; Ari, N.S.; Ozatik, O. 2-Aminoethoxydiphenyl borate ameliorates functional and structural abnormalities in cisplatin-induced peripheral neuropathy. J. Trace Elem. Med. Biol. 2022, 70, 126909. [Google Scholar] [CrossRef] [PubMed]
- Koehn, D.; Meyer, K.J.; Syed, N.A.; Anderson, M.G. Ketamine/Xylazine-Induced Corneal Damage in Mice. PLoS ONE 2015, 10, e0132804. [Google Scholar] [CrossRef]
- Jin, Q.; Cai, Y.; Li, S.; Liu, H.; Zhou, X.; Lu, C.; Gao, X.; Qian, J.; Zhang, J.; Ju, S.; et al. Edaravone-Encapsulated Agonistic Micelles Rescue Ischemic Brain Tissue by Tuning Blood-Brain Barrier Permeability. Theranostics 2017, 7, 884–898. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Studies (*) | Groups | Animals (n) |
|---|---|---|
| Infarct volume and area determination | Vehicle | 8 |
| 2-APB | 9 | |
| Behavioral testing | Vehicle | 14 (gwt), 8 (ct) |
| 2-APB | 15 (gwt), 11 (ct) | |
| Somatosensory evoked potentials recordings | Vehicle | 7 |
| 2-APB | 11 | |
| Oxidative stress, inflammation, and neuronal death studies | Vehicle | 8 (os), 7 (inf and nd) |
| 2-APB | 8 (os), 7 (inf and nd) | |
| F2 | 5 (os), 3 (nd) | |
| Peri-infarct depolarizations recordings | Vehicle | 16 |
| 2-APB | 11 | |
| Cerebral blood flow assessments | No stroke | 3 |
| Sham | 5 | |
| Vehicle | 8 | |
| 2-APB | 8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernández-Serra, R.; Martínez-Alonso, E.; Alcázar, A.; Chioua, M.; Marco-Contelles, J.; Martínez-Murillo, R.; Ramos, M.; Guinea, G.V.; González-Nieto, D. Postischemic Neuroprotection of Aminoethoxydiphenyl Borate Associates Shortening of Peri-Infarct Depolarizations. Int. J. Mol. Sci. 2022, 23, 7449. https://doi.org/10.3390/ijms23137449
Fernández-Serra R, Martínez-Alonso E, Alcázar A, Chioua M, Marco-Contelles J, Martínez-Murillo R, Ramos M, Guinea GV, González-Nieto D. Postischemic Neuroprotection of Aminoethoxydiphenyl Borate Associates Shortening of Peri-Infarct Depolarizations. International Journal of Molecular Sciences. 2022; 23(13):7449. https://doi.org/10.3390/ijms23137449
Chicago/Turabian StyleFernández-Serra, Rocío, Emma Martínez-Alonso, Alberto Alcázar, Mourad Chioua, José Marco-Contelles, Ricardo Martínez-Murillo, Milagros Ramos, Gustavo V. Guinea, and Daniel González-Nieto. 2022. "Postischemic Neuroprotection of Aminoethoxydiphenyl Borate Associates Shortening of Peri-Infarct Depolarizations" International Journal of Molecular Sciences 23, no. 13: 7449. https://doi.org/10.3390/ijms23137449
APA StyleFernández-Serra, R., Martínez-Alonso, E., Alcázar, A., Chioua, M., Marco-Contelles, J., Martínez-Murillo, R., Ramos, M., Guinea, G. V., & González-Nieto, D. (2022). Postischemic Neuroprotection of Aminoethoxydiphenyl Borate Associates Shortening of Peri-Infarct Depolarizations. International Journal of Molecular Sciences, 23(13), 7449. https://doi.org/10.3390/ijms23137449