
An Insight into Recent Advances on Platelet Function in Health and Disease


Abstract
:1. Introduction
2. Principal Role of Platelet: Thrombosis and Hemostasis
3. Contribution of Platelets beyond Thrombosis and Hemostasis
3.1. Contribution of Platelet to Cardiovascular Diseases (CVDs)
3.2. Platelet in Diabetes Mellitus (DM)
3.3. Platelet in Wound Healing
3.4. Platelet in Inflammation and Immunity
3.4.1. General Role of Platelet in Inflammation and Immunity
3.4.2. Platelet in Sepsis
3.4.3. Role of Platelets in Neurovascular Inflammation
3.4.4. Platelet in Allergic Inflammation
3.5. Platelet in Malignancy
3.6. Platelet in Coronavirus Disease 2019 (COVID-19)
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| GF | Growth factor |
| PAMPs | Pathogen-associated molecular patterns |
| NO | Nitric oxide |
| PGI2 | Prostacyclin |
| vWF | Von Willebrand factor |
| GP | Glycoprotein |
| ADP | Adenosine diphosphate |
| TxA2 | Thromboxane A2 |
| VEGF | Vascular endothelial growth factor |
| FGF | Fibroblast growth factor |
| PDGF | Platelet derived growth factor |
| MMP | Matrix metalloproteinase |
| EMT | Epithelial-mesenchymal transition |
| PF | Platelet factor |
| RANTES | Regulated upon activation, normal T cell expressed and presumably secreted |
| IL | Interleukin |
| EGF | Epidermal growth factor |
| HGF | Hepatocyte growth factor |
| TGF-β | Transforming growth factor β |
| CXCL | C-X-C motif chemokine ligand |
| SDF-1α | Stromal derived factor 1α |
| CCL | C-C motif chemokine ligand |
| MCP | Monocyte chemoattractant protein |
| CD | Cluster of differentiation |
| TLT-1 | TREM-like transcript 1 |
| Ig | Immunoglobulin |
| PAI-1 | Plasminogen activator inhibitor 1 |
| TFPI | Tissue factor pathway inhibitor |
| ATP | Adenosine triphosphate |
| GTP | Guanosine triphosphate |
| GDP | Guanosine diphosphate |
| CVD | Cardiovascular disease |
| LDL | Low-density lipoprotein |
| ROS | Reactive oxygen species |
| WDR1 | WD repeat protein 1 |
| PDE3A | Phosphodiesterase 3A |
| cAMP | Cyclic adenosine monophosphate |
| SMC | Smooth muscle cell |
| SFKs | Src family kinases |
| MPs | Microparticles |
| MiRNA | MicroRNA |
| ACS | Acute coronary syndrome |
| VCAM-1 | Vascular cell adhesion molecule 1 |
| ICAM-1 | Intercellular adhesion molecule 1 |
| CXCR | C-X-C Motif Chemokine Receptor |
| MRP | Myeloid-related protein |
| PAFR | Platelet-activating factor receptor |
| LEPRL | Leptin activates long-form leptin receptor |
| JAK2 | Janus kinase 2 |
| PI3K | Phosphatidylinositol 3-kinase |
| PKB | Protein kinase B |
| IRS-1 | Insulin receptor substrate-1 |
| PLA | Platelet-leukocyte aggregation |
| ASA | Acetylsalicylic acid |
| DM | Diabetes mellitus |
| PKC | Protein kinase C |
| TNF-α | Tumor necrosis factor α |
| T2DM | Type 2 diabetes mellitus |
| Mac-1 | Macrophage antigen 1 |
| PRP | Platelet Rich Plasma |
| VEGFR | Vascular endothelial growth factor receptor |
| PEDF | Pigment epithelium-derived factor |
| DIC | Disseminated intravascular coagulation |
| NETs | Neutrophil extracellular traps |
| TLRs | Toll-like receptors |
| β-TG | β-thromboglobulin |
| SrpA | Serine-rich protein A |
| RGD | Arginine–glycine–aspartic acid |
| Clf | Clumping factors |
| TFPI | Cyclooxygenase-1 |
| SIRS | Systemic inflammatory response syndrome |
| DAMPs | Damage-related molecular patterns |
| PSGL-1 | P-selectin glycoprotein ligand-1 |
| PAF | Platelet activating factor |
| MS | Multiple sclerosis |
| AD | Alzheimer’s disease |
| mTOR | Mammalian target of rapamycin |
| MAPK | Mitogen-activated protein kinase |
| CANTOS | Canakinumab anti-inflammatory thrombosis outcome study |
| DC | Dendritic cell |
| CLEC-2 | C-type lectin-like receptor II-type |
| MHC | Major histocompatibility complex |
| CNS | Central nervous system |
| BBB | Blood-brain barrier |
| EAE | Experimentally induced autoimmune encephalomyelitis |
| CSF | Cerebrospinal fluid |
| PSD95 | Postsynaptic density protein95 |
| CVT | Cerebral venous thrombosis |
| tMCAO | Transient middle cerebral artery occlusion |
| MPV | Mean platelet volume |
| DSCG | Disodium cromoglycate |
| G-CSF | Granulocyte colony-stimulating factor |
| GM-CSF | Granulocyte-macrophage colony-stimulating factors |
| RhoA | Ras homolog family member A |
| MYPT1 | Myosin phosphatase target subunit 1 |
| PP1 | Protein phosphatase 1 |
| YAP 1 | Yes-associated protein 1 |
| SMAD | Suppressor of Mothers Against Decapentaplegic |
| NF-kB | Nuclear factor kappa B |
| COVID-19 | Coronavirus disease 2019 |
| SARS-CoV-2 | severe acute respiratory syndrome coronavirus 2 |
| ITAM | Immunoreceptor tyrosine-based activation motif |
| ITP | Immune thrombocytopenia |
| IVIg | Gamma globulin intravenous infusion |
| rhTPO | Recombinant human thrombopoietin |
References
- Harvey, J.W. The feline blood film. J. Feline Med. Surg. 2017, 19, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Tanaka, S.; Hori, Y.; Hirayama, F.; Sato, E.; Inoue, M. Role of mitochondria in the maintenance of platelet function during in vitro storage. Transfus. Med. 2011, 21, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Melchinger, H.; Jain, K.; Tyagi, T.; Hwa, J. Role of platelet mitochondria: Life in a nucleus-free zone. Front. Cardiovasc. Med. 2019, 6, 153. [Google Scholar] [CrossRef] [PubMed]
- Senzel, L.; Gnatenko, D.V.; Bahou, W.F. The platelet proteome. Curr. Opin. Hematol. 2009, 16, 329–333. [Google Scholar] [CrossRef]
- White, J.G.; Key, N.S.; King, R.A.; Vercellotti, G.M. The White platelet syndrome: A new autosomal dominant platelet disorder. Platelets 2004, 15, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Amable, P.R.; Carias, R.B.; Teixeira, M.V.; da Cruz Pacheco, I.; Corrêa do Amaral, R.J.; Granjeiro, J.M.; Borojevic, R. Platelet-rich plasma preparation for regenerative medicine: Optimization and quantification of cytokines and growth factors. Stem Cell Res. Ther. 2013, 4, 67. [Google Scholar] [CrossRef] [Green Version]
- Bambace, N.M.; Holmes, C.E. The platelet contribution to cancer progression. J. Thromb. Haemost. 2011, 9, 237–249. [Google Scholar] [CrossRef]
- Rendu, F.; Brohard-Bohn, B. The platelet release reaction: Granules’ constituents, secretion and functions. Platelets 2001, 12, 261–273. [Google Scholar] [CrossRef]
- Thon, J.N.; Peters, C.G.; Aslam, R.; Rowley, J.; Weyrich, A.S.; Semple, J.W.; Flaumenhaft, R.C.; Italiano, J.E., Jr. The Functional Role of TLR9 in Human Platelets. Blood 2011, 118, 366. [Google Scholar] [CrossRef]
- Sharda, A.; Flaumenhaft, R. The life cycle of platelet granules. F1000Research 2018, 7, 236. [Google Scholar] [CrossRef]
- Gawaz, M.; Vogel, S. Platelets in tissue repair: Control of apoptosis and interactions with regenerative cells. Blood 2013, 122, 2550–2554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jennings, L.K. Mechanisms of platelet activation: Need for new strategies to protect against platelet-mediated atherothrombosis. Thromb. Haemost. 2009, 102, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Becker, R.C.; Sexton, T.; Smyth, S.S. Translational implications of platelets as vascular first responders. Cir. Res. 2018, 122, 506–522. [Google Scholar] [CrossRef] [PubMed]
- Uchimido, R.; Schmidt, E.P.; Shapiro, N.I. The glycocalyx: A novel diagnostic and therapeutic target in sepsis. Crit. Care 2019, 23, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Periayah, M.H.; Halim, A.S.; Saad, A.Z.M. Mechanism action of platelets and crucial blood coagulation pathways in hemostasis. Int. J. Hematol. Oncol. Stem Cell Res. 2017, 11, 319. [Google Scholar] [PubMed]
- Nieswandt, B.; Brakebusch, C.; Bergmeier, W.; Schulte, V.; Bouvard, D.; Mokhtari-Nejad, R.; Lindhout, T.; Heemskerk, J.W.; Zirngibl, H.; Fässler, R. Glycoprotein VI but not α2β1 integrin is essential for platelet interaction with collagen. EMBO J. 2001, 20, 2120–2130. [Google Scholar] [CrossRef] [Green Version]
- Offermanns, S. Activation of platelet function through G protein–coupled receptors. Cir. Res. 2006, 99, 1293–1304. [Google Scholar] [CrossRef] [Green Version]
- Palta, S.; Saroa, R.; Palta, A. Overview of the coagulation system. Indian J. Anaesth. 2014, 58, 515. [Google Scholar] [CrossRef]
- Alberio, L.; Safa, O.; Clemetson, K.J.; Esmon, C.; Dale, G. Surface expression and functional characterization of α-granule factor V in human platelets: Effects of ionophore A23187, thrombin, collagen, and convulxin. Blood 2000, 95, 1694–1702. [Google Scholar] [CrossRef]
- Hayashi, S.-i.; Watanabe, N.; Nakazawa, K.; Suzuki, J.; Tsushima, K.; Tamatani, T.; Sakamoto, S.; Isobe, M. Roles of P-selectin in inflammation, neointimal formation, and vascular remodeling in balloon-injured rat carotid arteries. Circulation 2000, 102, 1710–1717. [Google Scholar] [CrossRef]
- Sørensen, B.; Tang, M.; Larsen, O.H.; Laursen, P.N.; Fenger-Eriksen, C.; Rea, C.J. The role of fibrinogen: A new paradigm in the treatment of coagulopathic bleeding. Thromb. Res. 2011, 128, S13–S16. [Google Scholar] [CrossRef]
- Peyvandi, F.; Garagiola, I.; Baronciani, L. Role of von Willebrand factor in the haemostasis. Blood Transfus. 2011, 9 (Suppl. S2), s3. [Google Scholar]
- Huveneers, S.; Truong, H.; Fässler, R.; Sonnenberg, A.; Danen, E.H. Binding of soluble fibronectin to integrin α5β1–link to focal adhesion redistribution and contractile shape. J. Cell Sci. 2008, 121, 2452–2462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.; Li, M.; Yin, L.; Fu, G.; Liu, Z. Role of thrombospondin-1 and thrombospondin-2 in cardiovascular diseases. Int. J. Mol. Med. 2020, 45, 1275–1293. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, H.; Kikkawa, Y.; Sanzen, N.; Sekiguchi, K. Purification and Characterization of Human Laminin-8: Laminin-8 stimulates cell adhesion and migration through α3β1 AND α6β1integrins. J. Bio. Chem. 2001, 276, 17550–17558. [Google Scholar] [CrossRef] [Green Version]
- Madsen, C.D.; Ferraris, G.M.S.; Andolfo, A.; Cunningham, O.; Sidenius, N. uPAR-induced cell adhesion and migration: Vitronectin provides the key. J. Cell Biol. 2007, 177, 927–939. [Google Scholar] [CrossRef] [Green Version]
- Wong, R.W.C.; Guillaud, L. The role of epidermal growth factor and its receptors in mammalian CNS. Cytokine Growth Factor Rev. 2004, 15, 147–156. [Google Scholar] [CrossRef]
- Clemmons, D.R.; Snyder, P.; Martin, K. Physiology of Insulin-Like Growth Factor 1. 2014. Available online: https://www.uptodate.com/contents/physiology-of-insulin-like-growth-factor-1 (accessed on 20 April 2022).
- Oliveira, A.G.; Araújo, T.G.; Carvalho, B.d.M.; Rocha, G.Z.; Santos, A.; Saad, M.J. The role of hepatocyte growth factor (HGF) in insulin resistance and diabetes. Front. Endocrinol. 2018, 9, 503. [Google Scholar] [CrossRef]
- Nagaraj, N.S.; Datta, P.K. Targeting the transforming growth factor-β signaling pathway in human cancer. Expert Opin. Investig. Drugs 2010, 19, 77–91. [Google Scholar] [CrossRef] [Green Version]
- Kardas, G.; Daszyńska-Kardas, A.; Marynowski, M.; Brząkalska, O.; Kuna, P.; Panek, M. Role of platelet-derived growth factor (PDGF) in asthma as an immunoregulatory factor mediating airway remodeling and possible pharmacological target. Front. Pharmacol. 2020, 11, 47. [Google Scholar] [CrossRef]
- Niu, G.; Chen, X. Vascular endothelial growth factor as an anti-angiogenic target for cancer therapy. Curr. Drug Targets 2010, 11, 1000–1017. [Google Scholar] [CrossRef] [PubMed]
- Raica, M.; Cimpean, A.M. Platelet-derived growth factor (PDGF)/PDGF receptors (PDGFR) axis as target for antitumor and antiangiogenic therapy. Pharmaceuticals 2010, 3, 572–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, T.; Jacquet, T.; Dalonneau, F.; Coudert, P.; Vaganay, E.; Exbrayat-Héritier, C.; Vollaire, J.; Josserand, V.; Ruggiero, F.; Coll, J.-L. FGF-2 promotes angiogenesis through a SRSF1/SRSF3/SRPK1-dependent axis that controls VEGFR1 splicing in endothelial cells. BMC Biol. 2021, 19, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Bernhard, S.; Hug, S.; Stratmann, A.E.P.; Erber, M.; Vidoni, L.; Knapp, C.L.; Thomaß, B.D.; Fauler, M.; Nilsson, B.; Ekdahl, K.N. Interleukin 8 elicits rapid physiological changes in neutrophils that are altered by inflammatory conditions. J. Innate Immun. 2021, 13, 225–241. [Google Scholar] [CrossRef]
- Brown, A.J.; Sepuru, K.M.; Sawant, K.V.; Rajarathnam, K. Platelet-derived chemokine CXCL7 dimer preferentially exists in the glycosaminoglycan-bound form: Implications for neutrophil–platelet crosstalk. Front. Immunol. 2017, 8, 1248. [Google Scholar] [CrossRef] [Green Version]
- Sawant, K.V.; Poluri, K.M.; Dutta, A.K.; Sepuru, K.M.; Troshkina, A.; Garofalo, R.P.; Rajarathnam, K. Chemokine CXCL1 mediated neutrophil recruitment: Role of glycosaminoglycan interactions. Sci. Rep. 2016, 6, 33123. [Google Scholar] [CrossRef] [Green Version]
- Disteldorf, E.M.; Krebs, C.F.; Paust, H.-J.; Turner, J.-E.; Nouailles, G.; Tittel, A.; Meyer-Schwesinger, C.; Stege, G.; Brix, S.; Velden, J. CXCL5 drives neutrophil recruitment in TH17-mediated GN. J. Am Soc. Nephrol. 2015, 26, 55–66. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Huang, W.; Wang, S.; Wang, J.; Cui, W.; Zhang, W.; Lou, A.; Geng, S.; Li, X. Macrophagic Extracellular Vesicle CXCL2 Recruits and Activates the Neutrophil CXCR2/PKC/NOX4 Axis in Sepsis. J. Immunol. 2021, 207, 2118–2128. [Google Scholar] [CrossRef]
- Jovic, S.; Linge, H.; Shikhagaie, M.; Olin, A.; Lannefors, L.; Erjefält, J.; Mörgelin, M.; Egesten, A. The neutrophil-recruiting chemokine GCP-2/CXCL6 is expressed in cystic fibrosis airways and retains its functional properties after binding to extracellular DNA. Mucosal Immunol. 2016, 9, 112–123. [Google Scholar] [CrossRef]
- Isles, H.M.; Herman, K.D.; Robertson, A.L.; Loynes, C.A.; Prince, L.R.; Elks, P.M.; Renshaw, S.A. The CXCL12/CXCR4 signaling axis retains neutrophils at inflammatory sites in zebrafish. Front. Immunol. 2019, 10, 1784. [Google Scholar] [CrossRef] [Green Version]
- Aldinucci, D.; Colombatti, A. The inflammatory chemokine CCL5 and cancer progression. Mediat. Inflamm. 2014, 2014, 292376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, M.L.; Thakur, A.; Cole, N.; Lloyd, A.; Stapleton, F.; Wakefield, D.; Willcox, M.D. A critical role for CCL2 and CCL3 chemokines in the regulation of polymorphonuclear neutrophils recruitment during corneal infection in mice. Immunol. Cell Biol. 2007, 85, 525–531. [Google Scholar] [PubMed]
- Ford, J.; Hughson, A.; Lim, K.; Bardina, S.V.; Lu, W.; Charo, I.F.; Lim, J.K.; Fowell, D.J. CCL7 is a negative regulator of cutaneous inflammation following Leishmania major infection. Front. Immunol. 2019, 9, 3063. [Google Scholar] [CrossRef]
- Kaneko, N.; Kurata, M.; Yamamoto, T.; Morikawa, S.; Masumoto, J. The role of interleukin-1 in general pathology. Inflamm. Regen. 2019, 39, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, W.; Moosavi, L. Physiology, Factor V; StatPearls Publishing: Tampa, FL, USA, 2019. [Google Scholar]
- Pilli, V.; Plautz, W.; Majumder, R. The journey of protein S from an anticoagulant to a signaling molecule. JSM Biochem. Mol. Biol. 2016, 3, 1014. [Google Scholar]
- Emsley, J.; McEwan, P.A.; Gailani, D. Structure and function of factor XI. Blood Am. J. Hematol. 2010, 115, 2569–2577. [Google Scholar]
- Shaz, B.H.; Hillyer, C.D. Transfusion Medicine and Hemostasis: Clinical and Laboratory Aspects; Elsevier: Amsterdam, The Netherlands, 2013. [Google Scholar]
- Wu, Y. Contact pathway of coagulation and inflammation. Thromb. J. 2015, 13, 17. [Google Scholar] [CrossRef] [Green Version]
- Baker, S.K.; Strickland, S. A critical role for plasminogen in inflammation. J. Exp. Med. 2020, 217, e20191865. [Google Scholar] [CrossRef]
- Albert, F.; Christopher, N.F. The platelet fibrinogen receptor: From megakaryocyte to the mortuary. JRSM Cardiovasc. Dis. 2012, 1, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Andrews, R.K.; Gardiner, E.E.; Shen, Y.; Whisstock, J.C.; Berndt, M.C. Glycoprotein Ib–IX–V. Int. J. Biochem. Cell Biol. 2003, 35, 1170–1174. [Google Scholar] [CrossRef]
- Moroi, M.; Jung, S.M. Platelet glycoprotein VI: Its structure and function. Thromb. Res. 2004, 114, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Schmoker, A.M.; Pearson, L.M.P.; Cruz, C.; Flores, L.G.C.; Branfeild, S.; Torres, F.D.P.; Fonseca, K.; Cantres, Y.M.; Ramirez, C.A.S.; Melendez, L.M. Defining the TLT-1 interactome from resting and activated human platelets. J. Proteom. 2020, 215, 103638. [Google Scholar] [CrossRef] [PubMed]
- Girardi, G.; Lingo, J.J.; Fleming, S.D.; Regal, J.F. Essential role of complement in pregnancy: From implantation to parturition and beyond. Front. Immunol. 2020, 11, 1681. [Google Scholar] [CrossRef] [PubMed]
- Biesma, D.H.; Hannema, A.J.; van Velzen-Blad, H.; Mulder, L.; van Zwieten, R.; Kluijt, I.; Roos, D. A family with complement factor D deficiency. J. Clin. Investig. 2001, 108, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, V.P.; Pangburn, M.K.; Cortés, C. Complement control protein factor H: The good, the bad, and the inadequate. Mol. Immunol. 2010, 47, 2187–2197. [Google Scholar] [CrossRef] [Green Version]
- Schmaier, A.H.; Smith, P.M.; Colman, R.W. Platelet C1-inhibitor. A secreted alpha-granule protein. J. Clin. Investig. 1985, 75, 242–250. [Google Scholar] [CrossRef]
- Schroeder, H.W., Jr.; Cavacini, L. Structure and function of immunoglobulins. J. Allergy Clin. Immunol. 2010, 125, S41–S52. [Google Scholar] [CrossRef] [Green Version]
- Maron, B.A.; Loscalzo, J. The role of platelets in fibrinolysis. In Platelets; Elsevier: Amsterdam, The Netherlands, 2007; pp. 415–430. [Google Scholar]
- Damare, J.; Brandal, S.; Fortenberry, Y.M. Inhibition of PAI-1 antiproteolytic activity against tPA by RNA aptamers. Nucleic Acid Ther. 2014, 24, 239–249. [Google Scholar] [CrossRef] [Green Version]
- Bergin, D.A.; Hurley, K.; McElvaney, N.G.; Reeves, E.P. Alpha-1 antitrypsin: A potent anti-inflammatory and potential novel therapeutic agent. Arch. Immunol. Ther. Exp. 2012, 60, 81–97. [Google Scholar] [CrossRef]
- Borth, W. α2 Macroglobulin, a multifunctional binding protein with targeting characteristics. FASEB J. 1992, 6, 3345–3353. [Google Scholar] [CrossRef]
- Kato, H. Tissue factor pathway inhibitor; its structure, function and clinical significance. Pol. J. Pharmacol. 1996, 48, 67–72. [Google Scholar] [PubMed]
- Cancemi, P.; Aiello, A.; Accardi, G.; Caldarella, R.; Candore, G.; Caruso, C.; Ciaccio, M.; Cristaldi, L.; Di Gaudio, F.; Siino, V. The role of matrix metalloproteinases (MMP-2 and MMP-9) in ageing and longevity: Focus on sicilian long-living individuals (LLIs). Mediat. Inflamm. 2020, 2020, 8635158. [Google Scholar] [CrossRef] [PubMed]
- Duerschmied, D.; Bode, C. The role of serotonin in haemostasis. Hämostaseologie 2009, 29, 356–359. [Google Scholar] [PubMed] [Green Version]
- Mannaioni, P.; Di Bello, M.; Raspanti, S.; Gambassi, F.; Mugnai, L.; Masini, E. Platelet histamine: Characterization of the proaggregatory effect of histamine in human platelets. Int. Arch. Allergy. Immunol. 1992, 99, 394–396. [Google Scholar] [CrossRef] [PubMed]
- Montenont, E.; Echagarruga, C.; Allen, N.; Araldi, E.; Suarez, Y.; Berger, J.S. Platelet WDR1 suppresses platelet activity and is associated with cardiovascular disease. Blood 2016, 128, 2033–2042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gleissner, C.A. Platelet-derived chemokines in atherogenesis: What’s new? Curr. Vasc. Pharmacol. 2012, 10, 563–569. [Google Scholar] [CrossRef]
- Khodadi, E. Platelet function in cardiovascular disease: Activation of molecules and activation by molecules. Cardiovasc. Toxicol. 2020, 20, 1–10. [Google Scholar] [CrossRef]
- Lv, H.-C.; Wu, H.-Y.; Yin, J.-S.; Ge, J.-B. Thrombin induced platelet-fibrin clot strength in relation to platelet volume indices and inflammatory markers in patients with coronary artery disease. Oncotarget 2017, 8, 64217. [Google Scholar] [CrossRef] [Green Version]
- Smyth, E.M. Thromboxane and the thromboxane receptor in cardiovascular disease. Clin.l Lipidol. 2010, 5, 209–219. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, M.; Rath, D.; Schlotterbeck, J.; Rheinlaender, J.; Walker-Allgaier, B.; Alnaggar, N.; Zdanyte, M.; Müller, I.; Borst, O.; Geisler, T. Regulation of oxidized platelet lipidome: Implications for coronary artery disease. Eur. Heart J. 2017, 38, 1993–2005. [Google Scholar] [CrossRef]
- Akkerman, J.W.N. From low-density lipoprotein to platelet activation. Int. J. Biochem. Cell Biol. 2008, 40, 2374–2378. [Google Scholar] [CrossRef] [PubMed]
- Salomon, R.G. Structural identification and cardiovascular activities of oxidized phospholipids. Cir. Res. 2012, 111, 930–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stellos, K.; Ruf, M.; Sopova, K.; Kilias, A.; Rahmann, A.; Stamatelopoulos, K.; Jorbenadze, R.; Geisler, T.; Gawaz, M.; Bigalke, B. Plasma levels of stromal cell-derived factor-1 in patients with coronary artery disease: Effect of clinical presentation and cardiovascular risk factors. Atherosclerosis 2011, 219, 913–916. [Google Scholar] [CrossRef] [PubMed]
- Kile, B.T.; Panopoulos, A.D.; Stirzaker, R.A.; Hacking, D.F.; Tahtamouni, L.H.; Willson, T.A.; Mielke, L.A.; Henley, K.J.; Zhang, J.-G.; Wicks, I.P. Mutations in the cofilin partner Aip1/Wdr1 cause autoinflammatory disease and macrothrombocytopenia. Blood 2007, 110, 2371–2380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kueh, H.Y.; Charras, G.T.; Mitchison, T.J.; Brieher, W.M. Actin disassembly by cofilin, coronin, and Aip1 occurs in bursts and is inhibited by barbed-end cappers. J. Cell Biol. 2008, 182, 341–353. [Google Scholar] [CrossRef] [Green Version]
- Karpatkin, S. Heterogeneity of human platelets: II. Functional evidence suggestive of young and old platelets. J. Clin. Investig. 1969, 48, 1083–1087. [Google Scholar] [CrossRef]
- Elbatarny, H.S.; Netherton, S.J.; Ovens, J.D.; Ferguson, A.V.; Maurice, D.H. Adiponectin, ghrelin, and leptin differentially influence human platelet and human vascular endothelial cell functions: Implication in obesity-associated cardiovascular diseases. Eur. J. Pharmacol. 2007, 558, 7–13. [Google Scholar] [CrossRef]
- Shoji, T.; Koyama, H.; Fukumoto, S.; Maeno, T.; Yokoyama, H.; Shinohara, K.; Emoto, M.; Shoji, T.; Yamane, T.; Hino, M. Platelet activation is associated with hypoadiponectinemia and carotid atherosclerosis. Atherosclerosis 2006, 188, 190–195. [Google Scholar] [CrossRef]
- Trovati, M.; Anfossi, G. Influence of insulin and of insulin resistance on platelet and vascular smooth muscle cell function. J. Diabetes Complicat. 2002, 16, 35–40. [Google Scholar] [CrossRef]
- Michelson, A.D.; Barnard, M.R.; Krueger, L.A.; Valeri, C.R.; Furman, M.I. Circulating monocyte-platelet aggregates are a more sensitive marker of in vivo platelet activation than platelet surface P-selectin: Studies in baboons, human coronary intervention, and human acute myocardial infarction. Circulation 2001, 104, 1533–1537. [Google Scholar] [CrossRef] [Green Version]
- Elbatarny, H.S.; Maurice, D.H. Leptin-mediated activation of human platelets: Involvement of a leptin receptor and phosphodiesterase 3A-containing cellular signaling complex. Am. J. Physiol. Endocrinol. Metabol. 2005, 289, E695–E702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuentes, E.; Palomo, I. Role of oxidative stress on platelet hyperreactivity during aging. Life Sci. 2016, 148, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Dayal, S.; Wilson, K.M.; Motto, D.G.; Miller, F.J., Jr.; Chauhan, A.K.; Lentz, S.R. Hydrogen peroxide promotes aging-related platelet hyperactivation and thrombosis. Circulation 2013, 127, 1308–1316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastori, D.; Pignatelli, P.; Carnevale, R.; Violi, F. Nox-2 up-regulation and platelet activation: Novel insights. Prostagland. Other Lipid Mediat. 2015, 120, 50–55. [Google Scholar] [CrossRef]
- Pereira, J.; Soto, M.; Palomo, I.; Ocqueteau, M.; Coetzee, L.-M.; Astudillo, S.; Aranda, E.; Mezzano, D. Platelet aging in vivo is associated with activation of apoptotic pathways: Studies in a model of suppressed thrombopoiesis in dogs. Thromb. Haemost. 2002, 87, 905–909. [Google Scholar]
- Koyama, H.; Maeno, T.; Fukumoto, S.; Shoji, T.; Yamane, T.; Yokoyama, H.; Emoto, M.; Shoji, T.; Tahara, H.; Inaba, M. Platelet P-selectin expression is associated with atherosclerotic wall thickness in carotid artery in humans. Circulation 2003, 108, 524–529. [Google Scholar] [CrossRef]
- Flierl, U.; Bauersachs, J.; Schäfer, A. Modulation of platelet and monocyte function by the chemokine fractalkine (CX 3 CL 1) in cardiovascular disease. Eur. J. Clin. Investig. 2015, 45, 624–633. [Google Scholar] [CrossRef]
- Parodi, G.; Marcucci, R.; Valenti, R.; Gori, A.M.; Migliorini, A.; Giusti, B.; Buonamici, P.; Gensini, G.F.; Abbate, R.; Antoniucci, D. High residual platelet reactivity after clopidogrel loading and long-term cardiovascular events among patients with acute coronary syndromes undergoing PCI. JAMA 2011, 306, 1215–1223. [Google Scholar] [CrossRef] [Green Version]
- Mangiacapra, F.; De Bruyne, B.; Muller, O.; Trana, C.; Ntalianis, A.; Bartunek, J.; Heyndrickx, G.; Di Sciascio, G.; Wijns, W.; Barbato, E. High residual platelet reactivity after clopidogrel: Extent of coronary atherosclerosis and periprocedural myocardial infarction in patients with stable angina undergoing percutaneous coronary intervention. JACC: Cardiovas. Interv. 2010, 3, 35–40. [Google Scholar]
- Lievens, D.; von Hundelshausen, P. Platelets in atherosclerosis. Thromb. Haemost. 2011, 106, 827–838. [Google Scholar]
- Semple, J.W.; Italiano, J.E.; Freedman, J. Platelets and the immune continuum. Nat. Rev. Immunol. 2011, 11, 264–274. [Google Scholar] [CrossRef] [PubMed]
- Schulz, C.; Schäfer, A.; Stolla, M.; Kerstan, S.; Lorenz, M.; von Brühl, M.-L.; Schiemann, M.; Bauersachs, J.; Gloe, T.; Busch, D.H. Chemokine fractalkine mediates leukocyte recruitment to inflammatory endothelial cells in flowing whole blood: A critical role for P-selectin expressed on activated platelets. Circulation 2007, 116, 764–773. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, A.; Schulz, C.; Fraccarollo, D.; Tas, P.; Leutke, M.; Eigenthaler, M.; Seidl, S.; Heider, P.; Ertl, G.; Massberg, S. The CX3C chemokine fractalkine induces vascular dysfunction by generation of superoxide anions. Arteroscler. Thromb. Vasc. Biol. 2007, 27, 55–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flierl, U.; Fraccarollo, D.; Lausenmeyer, E.; Rosenstock, T.; Schulz, C.; Massberg, S.; Bauersachs, J.; Schäfer, A. Fractalkine activates a signal transduction pathway similar to P2Y12 and is associated with impaired clopidogrel responsiveness. Arteroscler. Thromb. Vasc. Biol. 2012, 32, 1832–1840. [Google Scholar] [CrossRef] [Green Version]
- Postea, O.; Vasina, E.M.; Cauwenberghs, S.; Projahn, D.; Liehn, E.A.; Lievens, D.; Theelen, W.; Kramp, B.K.; Butoi, E.D.; Soehnlein, O. Contribution of platelet CX3CR1 to platelet–monocyte complex formation and vascular recruitment during hyperlipidemia. Arteroscler. Thromb. Vasc. Biol. 2012, 32, 1186–1193. [Google Scholar] [CrossRef] [Green Version]
- Schäfer, A.; Schulz, C.; Eigenthaler, M.; Fraccarollo, D.; Kobsar, A.; Gawaz, M.; Ertl, G.; Walter, U.; Bauersachs, J. Novel role of the membrane-bound chemokine fractalkine in platelet activation and adhesion. Blood 2004, 103, 407–412. [Google Scholar] [CrossRef] [Green Version]
- Honda, S.; Shirotani-Ikejima, H.; Tadokoro, S.; Maeda, Y.; Kinoshita, T.; Tomiyama, Y.; Miyata, T. Integrin-linked kinase associated with integrin activation. Blood 2009, 113, 5304–5313. [Google Scholar] [CrossRef] [Green Version]
- Tadokoro, S.; Shattil, S.J.; Eto, K.; Tai, V.; Liddington, R.C.; de Pereda, J.M.; Ginsberg, M.H.; Calderwood, D.A. Talin binding to integrin ß tails: A final common step in integrin activation. Science 2003, 302, 103–106. [Google Scholar] [CrossRef]
- Senis, Y.; Antrobus, R.; Severin, S.; Parguina, A.; Rosa, I.; Zitzmann, N.; Watson, S.; García, A. Proteomic analysis of integrin αIIbβ3 outside-in signaling reveals Src-kinase-independent phosphorylation of Dok-1 and Dok-3 leading to SHIP-1 interactions. J. Thromb. Haemost. 2009, 7, 1718–1726. [Google Scholar] [CrossRef]
- Gurbel, P.A.; Fox, K.A.; Tantry, U.S.; Ten Cate, H.; Weitz, J.I. Combination antiplatelet and oral anticoagulant therapy in patients with coronary and peripheral artery disease: Focus on the COMPASS trial. Circulation 2019, 139, 2170–2185. [Google Scholar] [CrossRef]
- Pétillot, P.; Lahorte, C.; Bonanno, E.; Signore, A.; Lancel, S.; Marchetti, P.; Vallet, B.; Slegers, G.; Neviere, R. Annexin V detection of lipopolysaccharide-induced cardiac apoptosis. Shock 2007, 27, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Sun, G.; Wang, G.; Ning, W.; Zhao, K. Soluble P-selectin promotes acute myocardial infarction onset but not severity. Mol. Med. Rep. 2015, 11, 2027–2033. [Google Scholar] [CrossRef] [PubMed]
- Daub, S.; Lutgens, E.; Münzel, T.; Daiber, A. CD40/CD40L and related signaling pathways in cardiovascular health and disease—the pros and cons for cardioprotection. Int. J. Mol. Sci. 2020, 21, 8533. [Google Scholar] [CrossRef] [PubMed]
- Pontén, A.; Bergsten Folestad, E.; Pietras, K.; Eriksson, U. Platelet-derived growth factor D induces cardiac fibrosis and proliferation of vascular smooth muscle cells in heart-specific transgenic mice. Cir. Res. 2005, 97, 1036–1045. [Google Scholar] [CrossRef] [Green Version]
- Kalra, K.; Eberhard, J.; Farbehi, N.; Chong, J.J.; Xaymardan, M. Role of PDGF-A/B Ligands in Cardiac Repair After Myocardial Infarction. Front. Cell Dev. Biol. 2021, 9, 669188. [Google Scholar] [CrossRef]
- Steffel, J.; Lüscher, T.F.; Tanner, F.C. Tissue factor in cardiovascular diseases: Molecular mechanisms and clinical implications. Circulation 2006, 113, 722–731. [Google Scholar] [CrossRef] [Green Version]
- Harry, B.L.; Sanders, J.M.; Feaver, R.E.; Lansey, M.; Deem, T.L.; Zarbock, A.; Bruce, A.C.; Pryor, A.W.; Gelfand, B.D.; Blackman, B.R. Endothelial cell PECAM-1 promotes atherosclerotic lesions in areas of disturbed flow in ApoE-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 2003–2008. [Google Scholar] [CrossRef]
- Chandra, M.; Miriyala, S.; Panchatcharam, M. PPARγ and its role in cardiovascular diseases. PPAR Res. 2017, 2017, 6404638. [Google Scholar] [CrossRef] [Green Version]
- Khodadi, E.; Asnafi, A.A.; Mohammadi-Asl, J.; Hosseini, S.A.; Malehi, A.S.; Saki, N. Evaluation of miR-21 and miR-150 expression in immune thrombocytopenic purpura pathogenesis: A case-control study. Front. Biol. 2017, 12, 361–369. [Google Scholar] [CrossRef]
- Yao, R.; Ma, Y.; Du, Y.; Liao, M.; Li, H.; Liang, W.; Yuan, J.; Yu, X.; Xiao, H.; Liao, Y. The altered expression of inflammation-related microRNAs with microRNA-155 expression correlates with Th17 differentiation in patients with acute coronary syndrome. Cel. Mol. Immunol. 2011, 8, 486–495. [Google Scholar] [CrossRef] [Green Version]
- Gatsiou, A.; Boeckel, J.-N.; Randriamboavonjy, V.; Stellos, K. MicroRNAs in platelet biogenesis and function: Implications in vascular homeostasis and inflammation. Curr. Vasc. Pharmacol. 2012, 10, 524–531. [Google Scholar] [CrossRef] [PubMed]
- Widera, C.; Gupta, S.K.; Lorenzen, J.M.; Bang, C.; Bauersachs, J.; Bethmann, K.; Kempf, T.; Wollert, K.C.; Thum, T. Diagnostic and prognostic impact of six circulating microRNAs in acute coronary syndrome. J. Mol. Cel. Cardiol. 2011, 51, 872–875. [Google Scholar] [CrossRef] [PubMed]
- Siasos, G.; Kollia, C.; Tsigkou, V.; Basdra, E.K.; Lymperi, M.; Oikonomou, E.; Kokkou, E.; Korompelis, P.; Papavassiliou, A.G. MicroRNAs: Novel diagnostic and prognostic biomarkers in atherosclerosis. Curr. Top. Med. Chem. 2013, 13, 1503–1517. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [Green Version]
- Onselaer, M.B.; Oury, C.; Hunter, R.; Eeckhoudt, S.; Barile, N.; Lecut, C.; Morel, N.; Viollet, B.; Jacquet, L.M.; Bertrand, L. The C a2+/calmodulin-dependent kinase kinase β-AMP-activated protein kinase-α1 pathway regulates phosphorylation of cytoskeletal targets in thrombin-stimulated human platelets. J. Thromb. Haemost. 2014, 12, 973–986. [Google Scholar] [CrossRef]
- Pula, G.; Schuh, K.; Nakayama, K.; Nakayama, K.I.; Walter, U.; Poole, A.W. PKCδ regulates collagen-induced platelet aggregation through inhibition of VASP-mediated filopodia formation. Blood 2006, 108, 4035–4044. [Google Scholar] [CrossRef]
- Pitsilos, S.; Hunt, J.; Mohler, E.R.; Prabhakar, A.M.; Poncz, M.; Dawicki, J.; Khalapyan, T.Z.; Wolfe, M.L.; Fairman, R.; Mitchell, M. Platelet factor 4 localization in carotid atherosclerotic plaques: Correlation with clinical parameters. Thromb. Haemost. 2003, 90, 1112–1120. [Google Scholar] [CrossRef]
- Von Hundelshausen, P.; Koenen, R.R.; Sack, M.; Mause, S.F.; Adriaens, W.; Proudfoot, A.E.; Hackeng, T.M.; Weber, C. Heterophilic interactions of platelet factor 4 and RANTES promote monocyte arrest on endothelium. Blood 2005, 105, 924–930. [Google Scholar] [CrossRef] [Green Version]
- Dann, R.; Hadi, T.; Montenont, E.; Boytard, L.; Alebrahim, D.; Feinstein, J.; Allen, N.; Simon, R.; Barone, K.; Uryu, K.; et al. Platelet-Derived MRP-14 Induces Monocyte Activation in Patients with Symptomatic Peripheral Artery Disease. J. Am. Col. Cardiol. 2018, 71, 53–65. [Google Scholar] [CrossRef]
- Gleissner, C.A.; von Hundelshausen, P.; Ley, K. Platelet chemokines in vascular disease. Arter. Thromb. Vasc. Biol. 2008, 28, 1920–1927. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, M.; Gawaz, M. Platelet-derived CXCL 12 (SDF-1α): Basic mechanisms and clinical implications. J. Thromb. Haemost. 2013, 11, 1954–1967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, C. Platelets and chemokines in atherosclerosis: Partners in crime. Circ. Res. 2005, 96, 612–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Kakutani, M.; Naruko, T.; Ueda, M.; Narumiya, S.; Masaki, T.; Sawamura, T. Activation-dependent surface expression of LOX-1 in human platelets. Biochem. Biophys. Res. Commun. 2001, 282, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Podrez, E.A.; Byzova, T.V.; Febbraio, M.; Salomon, R.G.; Ma, Y.; Valiyaveettil, M.; Poliakov, E.; Sun, M.; Finton, P.J.; Curtis, B.R. Platelet CD36 links hyperlipidemia, oxidant stress and a prothrombotic phenotype. Nat. Med. 2007, 13, 1086–1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, M.; von Ungern-Sternberg, S.N.; Seizer, P.; Schlegel, F.; Büttcher, M.; Sindhu, N.; Müller, S.; Mack, A.; Gawaz, M. Platelet-derived CXCL12 regulates monocyte function, survival, differentiation into macrophages and foam cells through differential involvement of CXCR4–CXCR7. Cell Death Dis. 2015, 6, e1989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallat, Z.; Benamer, H.; Hugel, B.; Benessiano, J.; Steg, P.G.; Freyssinet, J.M.; Tedgui, A. Elevated levels of shed membrane microparticles with procoagulant potential in the peripheral circulating blood of patients with acute coronary syndromes. Circulation 2000, 101, 841–843. [Google Scholar] [CrossRef] [Green Version]
- Chu, S.; Becker, R.; Berger, P.; Bhatt, D.; Eikelboom, J.; Konkle, B.; Mohler, E.; Reilly, M.; Berger, J. Mean platelet volume as a predictor of cardiovascular risk: A systematic review and meta-analysis. J. Thromb. Haemost. 2010, 8, 148–156. [Google Scholar] [CrossRef]
- Powe, C.E.; Levine, R.J.; Karumanchi, S.A. Preeclampsia, a disease of the maternal endothelium: The role of antiangiogenic factors and implications for later cardiovascular disease. Circulation 2011, 123, 2856–2869. [Google Scholar] [CrossRef]
- Walsh, S.W. Preeclampsia: An imbalance in placental prostacyclin and thromboxane production. Am. J. Obstetric. Gynecol. 1985, 152, 335–340. [Google Scholar] [CrossRef]
- Mangos, G.J. Cardiovascular disease following pre-eclampsia: Understanding the mechanisms. J. Hypertens. 2006, 24, 639–641. [Google Scholar] [CrossRef]
- Gurbel, P.A.; Tantry, U.S. Combination antithrombotic therapies. Circulation 2010, 121, 569–583. [Google Scholar] [CrossRef]
- Bonaca, M.P.; Bhatt, D.L.; Cohen, M.; Steg, P.G.; Storey, R.F.; Jensen, E.C.; Magnani, G.; Bansilal, S.; Fish, M.P.; Im, K.; et al. Long-term use of ticagrelor in patients with prior myocardial infarction. N. Eng. J. Med. 2015, 372, 1791–1800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swieringa, F.; Spronk, H.M.H.; Heemskerk, J.W.M.; van der Meijden, P.E.J. Integrating platelet and coagulation activation in fibrin clot formation. Res. Pract. Thromb. Haemost. 2018, 2, 450–460. [Google Scholar] [CrossRef] [PubMed]
- Santilli, F.; Simeone, P.; Liani, R. The role of platelets in diabetes mellitus. In Platelets; Elsevier: Amsterdam, The Netherlands, 2019; pp. 469–503. [Google Scholar]
- Santilli, F.; Simeone, P.; Liani, R.; Davì, G. Platelets and Diabetes. In Platelets Thrombotic Non-Thrombotic Disorders; Springer: Cham, Switzerland, 2017; pp. 1225–1238. [Google Scholar]
- Alexandru, N.; Popov, D.; Sbarcea, A.; Amuzescu, M. Platelet free cytosolic calcium concentration during ageing of type 2 diabetic patients. Platelets 2007, 18, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Nomura, S. Dynamic role of microparticles in type 2 diabetes mellitus. Curr. Diabetes Rev. 2009, 5, 245–251. [Google Scholar] [CrossRef]
- Kakouros, N.; Rade, J.J.; Kourliouros, A.; Resar, J.R. Platelet function in patients with diabetes mellitus: From a theoretical to a practical perspective. Int. J. Endocrinol. 2011, 2011, 742719. [Google Scholar] [CrossRef]
- Domingueti, C.P.; Dusse, L.M.S.A.; das Graças Carvalho, M.; de Sousa, L.P.; Gomes, K.B.; Fernandes, A.P. Diabetes mellitus: The linkage between oxidative stress, inflammation, hypercoagulability and vascular complications. J. Diabetes Complicat. 2016, 30, 738–745. [Google Scholar] [CrossRef]
- Emara, E.; Abdel-Sater, K. Beneficial effects of calcium channel blocker “Nifedipine” on abnormalities of platelets and lipid metabolism in patients with type II diabetes mellitus. J. Diabetes Metab. 2011, 2, 131. [Google Scholar] [CrossRef] [Green Version]
- Guthikonda, S.; Alviar, C.L.; Vaduganathan, M.; Arikan, M.; Tellez, A.; DeLao, T.; Granada, J.F.; Dong, J.-F.; Kleiman, N.S.; Lev, E.I. Role of reticulated platelets and platelet size heterogeneity on platelet activity after dual antiplatelet therapy with aspirin and clopidogrel in patients with stable coronary artery disease. J. Am. Coll. Cardiol. 2008, 52, 743–749. [Google Scholar] [CrossRef] [Green Version]
- Xin, G.; Wei, Z.; Ji, C.; Zheng, H.; Gu, J.; Ma, L.; Huang, W.; Morris-Natschke, S.L.; Yeh, J.-L.; Zhang, R. Metformin uniquely prevents thrombosis by inhibiting platelet activation and mtDNA release. Sci. Rep. 2016, 6, 37841. [Google Scholar] [CrossRef] [Green Version]
- Vinik, A.I.; Erbas, T.; Park, T.S.; Nolan, R.; Pittenger, G.L. Platelet dysfunction in type 2 diabetes. Diabetes Care 2001, 24, 1476–1485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkinson, H.N.; Hardman, M.J. Wound healing: Cellular mechanisms and pathological outcomes. Open Biol. 2020, 10, 200223. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, M.; Kosaric, N.; Bonham, C.A.; Gurtner, G.C. Wound healing: A cellular perspective. Physiol. Rev. 2019, 99, 665–706. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, R.; Toyoda, E.; Maehara, M.; Wasai, S.; Omura, H.; Watanabe, M.; Sato, M. Effect of platelet-rich plasma on M1/M2 macrophage polarization. Int. J. Mol. Sci. 2021, 22, 2336. [Google Scholar] [CrossRef] [PubMed]
- Ho-Tin-Noé, B.; Boulaftali, Y.; Camerer, E. Platelets and vascular integrity: How platelets prevent bleeding in inflammation. Blood 2018, 131, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Eisinger, F.; Patzelt, J.; Langer, H.F. The platelet response to tissue injury. Front. Med. 2018, 5, 317. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Gao, H.; Shi, C.; Erhardt, P.W.; Pavlovsky, A.; A Soloviev, D.; Bledzka, K.; Ustinov, V.; Zhu, L.; Qin, J. Leukocyte integrin Mac-1 regulates thrombosis via interaction with platelet GPIbα. Nat. Commun. 2017, 8, 15559. [Google Scholar] [CrossRef] [Green Version]
- Werner, S.; Grose, R. Regulation of wound healing by growth factors and cytokines. Physiol. Rev. 2003, 83, 835–870. [Google Scholar] [CrossRef]
- Hardwicke, J.; Schmaljohann, D.; Boyce, D.; Thomas, D. Epidermal growth factor therapy and wound healing—Past, present and future perspectives. Surgeon 2008, 6, 172–177. [Google Scholar] [CrossRef]
- Mijiritsky, E.; Assaf, H.D.; Peleg, O.; Shacham, M.; Cerroni, L.; Mangani, L. Use of PRP, PRF and CGF in periodontal regeneration and facial rejuvenation—A narrative review. Biology 2021, 10, 317. [Google Scholar] [CrossRef]
- Arora, G.; Arora, S. Platelet-rich plasma—Where do we stand today? A critical narrative review and analysis. Dermatol. Ther. 2021, 34, e14343. [Google Scholar] [CrossRef] [PubMed]
- Andia, I.; Maffulli, N. Platelet-rich plasma for managing pain and inflammation in osteoarthritis. Nat. Rev. Rheumatol. 2013, 9, 721–730. [Google Scholar] [CrossRef] [PubMed]
- Testa, G.; Giardina, S.M.C.; Culmone, A.; Vescio, A.; Turchetta, M.; Cannavò, S.; Pavone, V. Intra-articular injections in knee osteoarthritis: A review of literature. J. Funct. Morphol. Kinesiol. 2021, 6, 15. [Google Scholar] [CrossRef] [PubMed]
- Jenne, C.; Urrutia, R.; Kubes, P. Platelets: Bridging hemostasis, inflammation, and immunity. Int. J. Lab. Hematol. 2013, 35, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Cognasse, F.; Laradi, S.; Berthelot, P.; Bourlet, T.; Marotte, H.; Mismetti, P.; Garraud, O.; Hamzeh-Cognasse, H. Platelet inflammatory response to stress. Front. Immunol. 2019, 10, 1478. [Google Scholar] [CrossRef]
- Thomas, M.R.; Storey, R.F. The role of platelets in inflammation. Thromb. Haemost. 2015, 114, 449–458. [Google Scholar]
- Evangelista, V.; Manarini, S.; Sideri, R.; Rotondo, S.; Martelli, N.; Piccoli, A.; Totani, L.; Piccardoni, P.; Vestweber, D.; De Gaetano, G. Platelet/polymorphonuclear leukocyte interaction: P-selectin triggers protein-tyrosine phosphorylation–dependent CD11b/CD18 adhesion: Role of PSGL-1 as a signaling molecule. Blood 1999, 93, 876–885. [Google Scholar] [CrossRef]
- Mine, S.; Fujisaki, T.; Suematsu, M.; Tanaka, Y. Activated platelets and endothelial cell interaction with neutrophils under flow conditions. Int. Med. 2001, 40, 1085–1092. [Google Scholar] [CrossRef] [Green Version]
- Elzey, B.D.; Schmidt, N.W.; Crist, S.A.; Kresowik, T.P.; Harty, J.T.; Nieswandt, B.; Ratliff, T.L. Platelet-derived CD154 enables T-cell priming and protection against Listeria monocytogenes challenge. Blood 2008, 111, 3684–3691. [Google Scholar] [CrossRef] [Green Version]
- Henn, V.; Slupsky, J.R.; Gräfe, M.; Anagnostopoulos, I.; Förster, R.; Müller-Berghaus, G.; Kroczek, R.A. CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature 1998, 391, 591–594. [Google Scholar] [CrossRef]
- Scheuerer, B.; Ernst, M.; Dürrbaum-Landmann, I.; Fleischer, J.; Grage-Griebenow, E.; Brandt, E.; Flad, H.-D.; Petersen, F. The CXC-chemokine platelet factor 4 promotes monocyte survival and induces monocyte differentiation into macrophages. Blood 2000, 95, 1158–1166. [Google Scholar] [CrossRef] [PubMed]
- Kasper, B.; Brandt, E.; Brandau, S.; Petersen, F. Platelet factor 4 (CXC chemokine ligand 4) differentially regulates respiratory burst, survival, and cytokine expression of human monocytes by using distinct signaling pathways. J. Immunol. 2007, 179, 2584–2591. [Google Scholar] [CrossRef]
- Pitchford, S.; Pan, D.; Welch, H.C. Platelets in neutrophil recruitment to sites of inflammation. Curr. Opin. Hematol. 2017, 24, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maurer, M.; Von Stebut, E. Macrophage inflammatory protein-1. Int. J. Biochem. Cell Biol. 2004, 36, 1882–1886. [Google Scholar] [CrossRef] [PubMed]
- Mause, S.F.; von Hundelshausen, P.; Zernecke, A.; Koenen, R.R.; Weber, C. Platelet microparticles: A transcellular delivery system for RANTES promoting monocyte recruitment on endothelium. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1512–1518. [Google Scholar] [CrossRef] [Green Version]
- Danese, S.; De La Motte, C.; Reyes, B.M.R.; Sans, M.; Levine, A.D.; Fiocchi, C. Cutting edge: T cells trigger CD40-dependent platelet activation and granular RANTES release: A novel pathway for immune response amplification. J. Immunol. 2004, 172, 2011–2015. [Google Scholar] [CrossRef] [Green Version]
- Krensky, A.M.; Ahn, Y.-T. Mechanisms of disease: Regulation of RANTES (CCL5) in renal disease. Nat. Clin. Pract. Nephrol. 2007, 3, 164–170. [Google Scholar] [CrossRef] [Green Version]
- Lindemann, S.; Tolley, N.D.; Dixon, D.A.; McIntyre, T.M.; Prescott, S.M.; Zimmerman, G.A.; Weyrich, A.S. Activated platelets mediate inflammatory signaling by regulated interleukin 1β synthesis. J. Cell Biol. 2001, 154, 485–490. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.G.; Heur, M. Interleukin-1β enhances cell migration through AP-1 and NF-κB pathway-dependent FGF2 expression in human corneal endothelial cells. Biol. Cell 2013, 105, 175–189. [Google Scholar] [CrossRef] [Green Version]
- Ben-Sasson, S.Z.; Hu-Li, J.; Quiel, J.; Cauchetaux, S.; Ratner, M.; Shapira, I.; Dinarello, C.A.; Paul, W.E. IL-1 acts directly on CD4 T cells to enhance their antigen-driven expansion and differentiation. Proc. Nat. Acad. Sci. USA 2009, 106, 7119–7124. [Google Scholar] [CrossRef] [Green Version]
- Yeaman, M.R.; Bayer, A.S.; Koo, S.-P.; Foss, W.; Sullam, P.M. Platelet microbicidal proteins and neutrophil defensin disrupt the Staphylococcus aureus cytoplasmic membrane by distinct mechanisms of action. J. Clin. Investig. 1998, 101, 178–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghasemzadeh, M.; Kaplan, Z.S.; Alwis, I.; Schoenwaelder, S.M.; Ashworth, K.J.; Westein, E.; Hosseini, E.; Salem, H.H.; Slattery, R.; McColl, S.R. The CXCR1/2 ligand NAP-2 promotes directed intravascular leukocyte migration through platelet thrombi. Blood 2013, 121, 4555–4566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herr, N.; Bode, C.; Duerschmied, D. The effects of serotonin in immune cells. Front. Cardiovasc. Med. 2017, 4, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashina, K.; Tsubosaka, Y.; Nakamura, T.; Omori, K.; Kobayashi, K.; Hori, M.; Ozaki, H.; Murata, T. Histamine induces vascular hyperpermeability by increasing blood flow and endothelial barrier disruption in vivo. PLoS ONE 2015, 10, e0132367. [Google Scholar] [CrossRef]
- Vischer, U.M.; Jornot, L.; Wollheim, C.B.; Theler, J.-M. Reactive oxygen intermediates induce regulated secretion of von Willebrand factor from cultured human vascular endothelial cells. Blood 1995, 85, 3164–3172. [Google Scholar] [CrossRef]
- Marcus, A.J.; Broekman, M.J.; Drosopoulos, J.H.; Olson, K.E.; Islam, N.; Pinsky, D.J.; Levi, R. In Role of CD39 (NTPDase-1) in thromboregulation, cerebroprotection, and cardioprotection. Semin. Thromb. Hemost. 2005, 31, 234–246. [Google Scholar] [CrossRef]
- Wagner, D.D.; Marder, V.J. Biosynthesis of von Willebrand protein by human endothelial cells: Processing steps and their intracellular localization. J. Cell Biol. 1984, 99, 2123–2130. [Google Scholar] [CrossRef]
- Sutton, N.R.; Baek, A.; Pinsky, D.J. Endothelial Cells and Inflammation. In Encyclopedia of Medical Immunology: Autoimmune Diseases; Mackay, I.R., Rose, N.R., Diamond, B., Davidson, A., Eds.; Springer New York: New York, NY, USA, 2014; pp. 367–381. [Google Scholar]
- Elalamy, I.; Chakroun, T.; Gerotziafas, G.; Petropoulou, A.; Robert, F.; Karroum, A.; Elgrably, F.; Samama, M.-M.; Hatmi, M. Circulating platelet–leukocyte aggregates: A marker of microvascular injury in diabetic patients. Thromb. Res. 2008, 121, 843–848. [Google Scholar] [CrossRef]
- Turgut, B.; Turgut, N.; Çelik, Y.; Tekgündüz, E.; Pamuk, G.E.; Demir, M. Differences in platelet–leukocyte aggregates among subtypes of acute cerebral ischemia. J. Neurol. Sci. 2011, 305, 126–130. [Google Scholar] [CrossRef]
- Evangelista, V.; Manarini, S.; Rotondo, S.; Martelli, N.; Polischuk, R.; McGregor, J.L.; De Gaetano, G.; Cerletti, C. Platelet/polymorphonuclear leukocyte interaction in dynamic conditions: Evidence of adhesion cascade and cross talk between P-selectin and the beta 2 integrin CD11b/CD18. Blood 1996, 88, 4183–4194. [Google Scholar] [CrossRef]
- Wang, H.-B.; Wang, J.-T.; Zhang, L.; Geng, Z.H.; Xu, W.-L.; Xu, T.; Huo, Y.; Zhu, X.; Plow, E.F.; Chen, M. P-selectin primes leukocyte integrin activation during inflammation. Nat. Immunol. 2007, 8, 882–892. [Google Scholar] [CrossRef] [PubMed]
- Weber, C.; Springer, T.A. Neutrophil accumulation on activated, surface-adherent platelets in flow is mediated by interaction of Mac-1 with fibrinogen bound to alphaIIbbeta3 and stimulated by platelet-activating factor. J. Clin. Investig. 1997, 100, 2085–2093. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.I.; Chen, Z.; Xu, H.; Li, C.Q.; Dong, J.-F.; McIntire, L.V.; Ballantyne, C.M.; Zhang, L.; Furman, M.I.; Berndt, M.C. Platelet glycoprotein Ibα is a counterreceptor for the leukocyte integrin Mac-1 (CD11b/CD18). J. Exp. Med. 2000, 192, 193–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghasemzadeh, M.; Hosseini, E. Platelet-leukocyte crosstalk: Linking proinflammatory responses to procoagulant state. Thromb. Res. 2013, 131, 191–197. [Google Scholar] [CrossRef]
- Plescia, J.; Altieri, D.C. Activation of Mac-1 (CD11b/CD18)-bound factor X by released cathepsin G defines an alternative pathway of leucocyte initiation of coagulation. Biochem. J. 1996, 319, 873–879. [Google Scholar] [CrossRef] [Green Version]
- Müller, F.; Mutch, N.J.; Schenk, W.A.; Smith, S.A.; Esterl, L.; Spronk, H.M.; Schmidbauer, S.; Gahl, W.A.; Morrissey, J.H.; Renné, T. Platelet polyphosphates are proinflammatory and procoagulant mediators in vivo. Cell 2009, 139, 1143–1156. [Google Scholar] [CrossRef] [Green Version]
- Amor, S.; Peferoen, L.A.; Vogel, D.Y.; Breur, M.; van der Valk, P.; Baker, D.; van Noort, J.M. Inflammation in neurodegenerative diseases—An update. Immunology 2014, 142, 151–166. [Google Scholar] [CrossRef]
- Flad, H.-D.; Brandt, E. Platelet-derived chemokines: Pathophysiology and therapeutic aspects. Cell. Mol. Life Sci. 2010, 67, 2363–2386. [Google Scholar] [CrossRef]
- Massberg, S.; Enders, G.; Matos, F.C.d.M.; Tomic, L.I.D.; Leiderer, R.; Eisenmenger, S.; Messmer, K.; Krombach, F. Fibrinogen deposition at the postischemic vessel wall promotes platelet adhesion during ischemia-reperfusion in vivo. Blood 1999, 94, 3829–3838. [Google Scholar] [CrossRef]
- Katz, J.N.; Kolappa, K.P.; Becker, R.C. Beyond thrombosis: The versatile platelet in critical illness. Chest 2011, 139, 658–668. [Google Scholar] [CrossRef]
- Pigozzi, L.; Aron, J.P.; Ball, J.; Cecconi, M. Understanding platelet dysfunction in sepsis. Intensive Care Med. 2016, 42, 583–586. [Google Scholar] [CrossRef] [PubMed]
- Nieswandt, B.; Kleinschnitz, C.; Stoll, G. Ischaemic stroke: A thrombo-inflammatory disease? J. Physiol. 2011, 589, 4115–4123. [Google Scholar] [CrossRef] [PubMed]
- Czapiga, M.; Kirk, A.D.; Lekstrom-Himes, J. Platelets deliver costimulatory signals to antigen-presenting cells: A potential bridge between injury and immune activation. Exp. Hematol. 2004, 32, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Silva-Cardoso, S.C.; Affandi, A.J.; Spel, L.; Cossu, M.; Van Roon, J.A.; Boes, M.; Radstake, T.R. CXCL4 exposure potentiates TLR-driven polarization of human monocyte-derived dendritic cells and increases stimulation of T cells. J. Immunol. 2017, 199, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Klockenbusch, C.; Walsh, G.M.; Brown, L.M.; Hoffman, M.D.; Ignatchenko, V.; Kislinger, T.; Kast, J. Global proteome analysis identifies active immunoproteasome subunits in human platelets. Mol. Cell. Proteom. 2014, 13, 3308–3319. [Google Scholar] [CrossRef] [Green Version]
- Semple, J.W.; Speck, E.R.; Milev, Y.P.; Blanchette, V.; Freedman, J. Indirect allorecognition of platelets by T helper cells during platelet transfusions correlates with anti-major histocompatibility complex antibody and cytotoxic T lymphocyte formation. Blood 1995, 86, 805–812, Erratum in Blood 1995, 86, 4710. [Google Scholar]
- Boegel, S.; Löwer, M.; Bukur, T.; Sorn, P.; Castle, J.C.; Sahin, U. HLA and proteasome expression body map. BMC Med. Gen. 2018, 11, 36. [Google Scholar] [CrossRef]
- Saunders, R.N.; Metcalfe, M.S.; Nicholson, M.L. Rapamycin in transplantation: A review of the evidence. Kidney Int. 2001, 59, 3–16. [Google Scholar] [CrossRef] [Green Version]
- Śledź, K.M.; Moore, S.F.; Durrant, T.N.; Blair, T.A.; Hunter, R.W.; Hers, I. Rapamycin restrains platelet procoagulant responses via FKBP-mediated protection of mitochondrial integrity. Biochem. Pharmacol. 2020, 177, 113975. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Lu, W.-J.; Lin, K.-C.; Huang, S.-Y.; Thomas, P.A.; Wu, Y.-H.; Wu, H.-C.; Lin, K.-H.; Sheu, J.-R. Role of a Janus kinase 2-dependent signaling pathway in platelet activation. Thromb. Res. 2014, 133, 1088–1096. [Google Scholar] [CrossRef] [PubMed]
- Yellaturu, C.R.; Rao, G.N. Cytosolic phospholipase A2 is an effector of Jak/STAT signaling and is involved in platelet-derived growth factor BB-induced growth in vascular smooth muscle cells. J. Biol. Chem. 2003, 278, 9986–9992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suades, R.; Padró, T.; Alonso, R.; Mata, P.; Badimon, L. Lipid-lowering therapy with statins reduces microparticle shedding from endothelium, platelets and inflammatory cells. Thromb. Haemost. 2013, 110, 366–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Gamal, H.; Parray, A.S.; Mir, F.A.; Shuaib, A.; Agouni, A. Circulating microparticles as biomarkers of stroke: A focus on the value of endothelial-and platelet-derived microparticles. J. Cell. Physiol. 2019, 234, 16739–16754. [Google Scholar] [CrossRef]
- Fitzgerald, D.J. Vascular biology of thrombosis: The role of platelet-vessel wall adhesion. Neurology 2001, 57 (Suppl. S2), S1–S4. [Google Scholar] [CrossRef]
- Coller, B.S. A Brief and Highly Selective History of Ideas. In Platelets; Elsevier Science: San Diego, CA, USA, 2002. [Google Scholar]
- Semeraro, N.; Ammollo, C.T.; Semeraro, F.; Colucci, M. Sepsis, thrombosis and organ dysfunction. Thromb. Res. 2012, 129, 290–295. [Google Scholar] [CrossRef]
- Rittirsch, D.; Flierl, M.A.; Ward, P.A. Harmful molecular mechanisms in sepsis. Nat. Rev. Immunol. 2008, 8, 776–787. [Google Scholar] [CrossRef] [Green Version]
- Beristain-Covarrubias, N.; Perez-Toledo, M.; Flores-Langarica, A.; Zuidscherwoude, M.; Hitchcock, J.R.; Channell, W.M.; King, L.D.; Thomas, M.R.; Henderson, I.R.; Rayes, J. Salmonella-induced thrombi in mice develop asynchronously in the spleen and liver and are not effective bacterial traps. Blood 2019, 133, 600–604. [Google Scholar] [CrossRef] [Green Version]
- Engelmann, B.; Massberg, S. Thrombosis as an intravascular effector of innate immunity. Nat. Rev. Immunol. 2013, 13, 34–45. [Google Scholar] [CrossRef]
- Levi, M.; Löwenberg, E.C. In thrombocytopenia in critically ill patients. Semin. Thromb. Hemost. 2008, 34, 417–424. [Google Scholar] [CrossRef]
- Shibazaki, M.; Nakamura, M.; Endo, Y. Biphasic, organ-specific, and strain-specific accumulation of platelets induced in mice by a lipopolysaccharide from Escherichia coli and its possible involvement in shock. Infect. Immun. 1996, 64, 5290–5294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andonegui, G.; Kerfoot, S.M.; McNagny, K.; Ebbert, K.V.; Patel, K.D.; Kubes, P. Platelets express functional Toll-like receptor-4. Blood 2005, 106, 2417–2423. [Google Scholar] [CrossRef] [PubMed]
- Claushuis, T.A.; van Vught, L.A.; Scicluna, B.P.; Wiewel, M.A.; Klein Klouwenberg, P.M.; Hoogendijk, A.J.; Ong, D.S.; Cremer, O.L.; Horn, J.; Franitza, M. Thrombocytopenia is associated with a dysregulated host response in critically ill sepsis patients. Blood 2016, 127, 3062–3072. [Google Scholar] [CrossRef] [Green Version]
- Tsirigotis, P.; Chondropoulos, S.; Frantzeskaki, F.; Stamouli, M.; Gkirkas, K.; Bartzeliotou, A.; Papanikolaou, N.; Atta, M.; Papassotiriou, I.; Dimitriadis, G. Thrombocytopenia in critically ill patients with severe sepsis/septic shock: Prognostic value and association with a distinct serum cytokine profile. J. Crit. Care 2016, 32, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Aslam, R.; Speck, E.R.; Kim, M.; Crow, A.R.; Bang, K.A.; Nestel, F.P.; Ni, H.; Lazarus, A.H.; Freedman, J.; Semple, J.W. Platelet Toll-like receptor expression modulates lipopolysaccharide-induced thrombocytopenia and tumor necrosis factor-α production in vivo. Blood 2006, 107, 637–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Handtke, S.; Steil, L.; Greinacher, A.; Thiele, T. Toward the relevance of platelet subpopulations for transfusion medicine. Front. Med. 2018, 5, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russwurm, S.; Vickers, J.; Meier-Hellmann, A.; Spangenberg, P.; Bredle, D.; Reinhart, K.; Lösche, W. Platelet and leukocyte activation correlate with the severity of septic organ dysfunction. Shock 2002, 17, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Salat, A.; Bodingbauer, G.; Boehm, D.; Murabito, M.; Tochkow, E.; Sautner, T.; Mueller, M.R.; Fuegger, R. Changes of platelet surface antigens in patients suffering from abdominal septic shock. Thromb Res. 1999, 95, 289–294. [Google Scholar] [CrossRef]
- Yaguchi, A.; Lobo, F.L.; Vincent, J.L.; Pradier, O. Platelet function in sepsis. J. Thromb. Haemost. 2004, 2, 2096–2102. [Google Scholar] [CrossRef]
- Inwald, D.P.; Faust, S.N.; Lister, P.; Peters, M.J.; Levin, M.; Heyderman, R.; Klein, N.J. Platelet and soluble CD40L in meningococcal sepsis. Intensive Care Med. 2006, 32, 1432–1437. [Google Scholar] [CrossRef]
- Mavrommatis, A.C.; Theodoridis, T.; Orfanidou, A.; Roussos, C.; Christopoulou-Kokkinou, V.; Zakynthinos, S. Coagulation system and platelets are fully activated in uncomplicated sepsis. Crit. Care Med. 2000, 28, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Washington, A.V.; Gibot, S.; Acevedo, I.; Gattis, J.; Quigley, L.; Feltz, R.; De La Mota, A.; Schubert, R.L.; Gomez-Rodriguez, J.; Cheng, J.; et al. TREM-like transcript-1 protects against inflammation-associated hemorrhage by facilitating platelet aggregation in mice and humans. J. Clin. Investig. 2009, 119, 1489–1501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esponda, O.; Morales, J.; Aguilar, A.; Gomez, M.; Washington, A.V. Clinical studies support a role for trem-like transcript-1 during the progression of sepsis. Boletin Asociacion Medica Puerto Rico 2010, 102, 59–61. [Google Scholar]
- Morales, J.; Villa, K.; Gattis, J.; Castro, W.; Colon, K.; Lubkowski, J.; Sanabria, P.; Hunter, R.; Washington, A.V. Soluble TLT-1 modulates platelet-endothelial cell interactions and actin polymerization. Blood Coagul. Fibrinol. Int. J. Haemost. Thromb. 2010, 21, 229–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derive, M.; Bouazza, Y.; Sennoun, N.; Marchionni, S.; Quigley, L.; Washington, V.; Massin, F.; Max, J.P.; Ford, J.; Alauzet, C.; et al. Soluble TREM-like transcript-1 regulates leukocyte activation and controls microbial sepsis. J. Immunol. 2012, 188, 5585–5592. [Google Scholar] [CrossRef]
- Gawaz, M.; Dickfeld, T.; Bogner, C.; Fateh-Moghadam, S.; Neumann, F.J. Platelet function in septic multiple organ dysfunction syndrome. Intensive Care Med. 1997, 23, 379–385. [Google Scholar] [CrossRef]
- Gawaz, M.; Fateh-Moghadam, S.; Pilz, G.; Gurland, H.J.; Werdan, K. Platelet activation and interaction with leucocytes in patients with sepsis or multiple organ failure. Eur. J. Clin. Investig. 1995, 25, 843–851. [Google Scholar] [CrossRef]
- Cox, D.; Kerrigan, S.W.; Watson, S.P. Platelets and the innate immune system: Mechanisms of bacterial-induced platelet activation. J. Thromb. Haemost. 2011, 9, 1097–1107. [Google Scholar] [CrossRef]
- Koupenova, M.; Vitseva, O.; MacKay, C.R.; Beaulieu, L.M.; Benjamin, E.J.; Mick, E.; Kurt-Jones, E.A.; Ravid, K.; Freedman, J.E. Platelet-TLR7 mediates host survival and platelet count during viral infection in the absence of platelet-dependent thrombosis. Blood 2014, 124, 791–802. [Google Scholar] [CrossRef] [Green Version]
- Thon, J.N.; Peters, C.G.; Machlus, K.R.; Aslam, R.; Rowley, J.; Macleod, H.; Devine, M.T.; Fuchs, T.A.; Weyrich, A.S.; Semple, J.W. T granules in human platelets function in TLR9 organization and signaling. J. Cell Biol. 2012, 198, 561–574. [Google Scholar] [CrossRef]
- Clark, S.R.; Ma, A.C.; Tavener, S.A.; McDonald, B.; Goodarzi, Z.; Kelly, M.M.; Patel, K.D.; Chakrabarti, S.; McAvoy, E.; Sinclair, G.D. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 2007, 13, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Klouche, M.; Klinger, M.H.; Kühnel, W.; Wilhelm, D. Endocytosis, storage, and release of IgE by human platelets: Differences in patients with type I allergy and nonatopic subjects. J. Allergy Clin. Immunol. 1997, 100, 235–241. [Google Scholar] [CrossRef]
- Yang, W.H.; Heithoff, D.M.; Aziz, P.V.; Haslund-Gourley, B.; Westman, J.S.; Narisawa, S.; Pinkerton, A.B.; Millán, J.L.; Nizet, V.; Mahan, M.J.; et al. Accelerated Aging and Clearance of Host Anti-inflammatory Enzymes by Discrete Pathogens Fuels Sepsis. Cell Host Microb. 2018, 24, 500–513.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grewal, P.K.; Aziz, P.V.; Uchiyama, S.; Rubio, G.R.; Lardone, R.D.; Le, D.; Varki, N.M.; Nizet, V.; Marth, J.D. Inducing host protection in pneumococcal sepsis by preactivation of the Ashwell-Morell receptor. Proc. Nat. Acad. Sci. USA 2013, 110, 20218–20223. [Google Scholar] [CrossRef] [Green Version]
- Plummer, C.; Wu, H.; Kerrigan, S.W.; Meade, G.; Cox, D.; Ian Douglas, C. A serine-rich glycoprotein of Streptococcus sanguis mediates adhesion to platelets via GPIb. Br. J. Haematol. 2005, 129, 101–109. [Google Scholar] [CrossRef]
- Hartleib, J.; Köhler, N.; Dickinson, R.B.; Chhatwal, G.S.; Sixma, J.J.; Hartford, O.M.; Foster, T.J.; Peters, G.; Kehrel, B.E.; Herrmann, M. Protein A is the von Willebrand factor binding protein on Staphylococcus aureus. Blood 2000, 96, 2149–2156. [Google Scholar]
- Brennan, M.P.; Loughman, A.; Devocelle, M.; Arasu, S.; Chubb, A.J.; Foster, T.; Cox, D. Elucidating the role of Staphylococcus epidermidis serine–aspartate repeat protein G in platelet activation. J. Thromb. Haemost. 2009, 7, 1364–1372. [Google Scholar] [CrossRef]
- Coburn, J.; Leong, J.M.; Erban, J.K. Integrin alpha IIb beta 3 mediates binding of the Lyme disease agent Borrelia burgdorferi to human platelets. Proc. Nat. Acad. Sci. USA 1993, 90, 7059–7063. [Google Scholar] [CrossRef] [Green Version]
- Siboo, I.R.; Cheung, A.L.; Bayer, A.S.; Sullam, P.M. Clumping factor A mediates binding of Staphylococcus aureus to human platelets. Infect. Immun. 2001, 69, 3120–3127. [Google Scholar] [CrossRef] [Green Version]
- Arman, M.; Krauel, K. Human platelet IgG Fc receptor Fcγ RIIA in immunity and thrombosis. J. Thromb. Haemost. 2015, 13, 893–908. [Google Scholar] [CrossRef]
- Riaz, A.H.; Tasma, B.E.; Woodman, M.E.; Wooten, R.M.; Worth, R.G. Human platelets efficiently kill IgG-opsonized E. coli. FEMS Immunol. Med. Microbiol. 2012, 65, 78–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montague, S.J.; Delierneux, C.; Lecut, C.; Layios, N.; Dinsdale, R.J.; Lee, C.S.-M.; Poulter, N.S.; Andrews, R.K.; Hampson, P.; Wearn, C.M. Soluble GPVI is elevated in injured patients: Shedding is mediated by fibrin activation of GPVI. Blood Adv. 2018, 2, 240–251. [Google Scholar] [CrossRef] [PubMed]
- Laursen, M.A.; Larsen, J.B.; Hvas, A.-M. Platelet function in disseminated intravascular coagulation: A systematic review. Platelets 2018, 29, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Woth, G.; Varga, A.; Ghosh, S.; Krupp, M.; Kiss, T.; Bogár, L.; Mühl, D. Platelet aggregation in severe sepsis. J. Thromb. Thrombolysis 2011, 31, 6–12. [Google Scholar] [CrossRef]
- Dewitte, A.; Lepreux, S.; Villeneuve, J.; Rigothier, C.; Combe, C.; Ouattara, A.; Ripoche, J. Blood platelets and sepsis pathophysiology: A new therapeutic prospect in critical ill patients? Ann. Intensive Care 2017, 7, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Lösche, W.; Boettel, J.; Kabisch, B.; Winning, J.; Claus, R.A.; Bauer, M. Do aspirin and other antiplatelet drugs reduce the mortality in critically ill patients? Thrombosis 2012, 2012, 720254. [Google Scholar] [CrossRef]
- Winning, J.; Neumann, J.; Kohl, M.; Claus, R.A.; Reinhart, K.; Bauer, M.; Lösche, W. Antiplatelet drugs and outcome in mixed admissions to an intensive care unit. Crit. Care Med. 2010, 38, 32–37. [Google Scholar] [CrossRef]
- Thomas, M.R.; Outteridge, S.N.; Ajjan, R.A.; Phoenix, F.; Sangha, G.K.; Faulkner, R.E.; Ecob, R.; Judge, H.M.; Khan, H.; West, L.E. Platelet P2Y12 inhibitors reduce systemic inflammation and its prothrombotic effects in an experimental human model. Atertio. Thromb. Vasc. Biol. 2015, 35, 2562–2570. [Google Scholar] [CrossRef] [Green Version]
- Sossdorf, M.; Otto, G.P.; Boettel, J.; Winning, J.; Lösche, W. Benefit of low-dose aspirin and non-steroidal anti-inflammatory drugs in septic patients. Crit. Care 2013, 17, 402. [Google Scholar] [CrossRef] [Green Version]
- Eisen, D.P.; Reid, D.; McBryde, E.S. Acetyl salicylic acid usage and mortality in critically ill patients with the systemic inflammatory response syndrome and sepsis. Crit. Care Med. 2012, 40, 1761–1767. [Google Scholar] [CrossRef]
- Kaur, C.; Ling, E.A. Blood brain barrier in hypoxic-ischemic conditions. Curr. Neurovasc. Res. 2008, 5, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 2008, 57, 178–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langer, H.F.; Chavakis, T. Platelets and neurovascular inflammation. Thromb. Haemost. 2013, 110, 888–893. [Google Scholar] [PubMed]
- Behari, M.; Shrivastava, M. Role of platelets in neurodegenerative diseases: A universal pathophysiology. Int. J. Neurosci. 2013, 123, 287–299. [Google Scholar] [CrossRef]
- Sotnikov, I.; Veremeyko, T.; Starossom, S.C.; Barteneva, N.; Weiner, H.L.; Ponomarev, E.D. Platelets recognize brain-specific glycolipid structures, respond to neurovascular damage and promote neuroinflammation. PLoS ONE 2013, 8, e58979. [Google Scholar] [CrossRef] [Green Version]
- Danese, E.; Montagnana, M.; Lippi, G. Platelets and migraine. Thromb. Res. 2014, 134, 17–22. [Google Scholar] [CrossRef]
- D’Andrea, G.; Cananzi, A.R.; Toldo, M.; Ferro-Milone, F. Platelet activation and migraine: A study with flunarizine. Headache 1986, 26, 339–342. [Google Scholar] [CrossRef]
- Govitrapong, P.; Limthavon, C.; Srikiatkhachorn, A. 5-HT2 serotonin receptor on blood platelet of migraine patients. Headache 1992, 32, 480–484. [Google Scholar] [CrossRef]
- Zeller, J.A.; Frahm, K.; Baron, R.; Stingele, R.; Deuschl, G. Platelet-leukocyte interaction and platelet activation in migraine: A link to ischemic stroke? J. Neurol. Neurosurg. Psychiatry 2004, 75, 984–987. [Google Scholar] [CrossRef]
- Wilmshurst, P.T.; Nightingale, S.; Walsh, K.P.; Morrison, W.L. Clopidogrel reduces migraine with aura after transcatheter closure of persistent foramen ovale and atrial septal defects. Heart 2005, 91, 1173–1175. [Google Scholar] [CrossRef] [Green Version]
- Langer, H.F.; Choi, E.Y.; Zhou, H.; Schleicher, R.; Chung, K.J.; Tang, Z.; Göbel, K.; Bdeir, K.; Chatzigeorgiou, A.; Wong, C.; et al. Platelets contribute to the pathogenesis of experimental autoimmune encephalomyelitis. Circ. Res. 2012, 110, 1202–1210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kihara, Y.; Ishii, S.; Kita, Y.; Toda, A.; Shimada, A.; Shimizu, T. Dual phase regulation of experimental allergic encephalomyelitis by platelet-activating factor. J. Exp. Med. 2005, 202, 853–863. [Google Scholar] [CrossRef] [PubMed]
- Callea, L.; Arese, M.; Orlandini, A.; Bargnani, C.; Priori, A.; Bussolino, F. Platelet activating factor is elevated in cerebral spinal fluid and plasma of patients with relapsing-remitting multiple sclerosis. J. Neuroimmunol. 1999, 94, 212–221. [Google Scholar] [CrossRef]
- Giles, J.A.; Greenhalgh, A.D.; Denes, A.; Nieswandt, B.; Coutts, G.; McColl, B.W.; Allan, S.M. Neutrophil infiltration to the brain is platelet-dependent, and is reversed by blockade of platelet GPIbα. Immunology 2018, 154, 322–328. [Google Scholar] [CrossRef] [Green Version]
- Gupta, P.; Bigley, A.B.; Markofski, M.; Laughlin, M.; LaVoy, E.C. Autologous serum collected 1 h post-exercise enhances natural killer cell cytotoxicity. Brain Behav. Immun. 2018, 71, 81–92. [Google Scholar] [CrossRef]
- Sardi, F.; Fassina, L.; Venturini, L.; Inguscio, M.; Guerriero, F.; Rolfo, E.; Ricevuti, G. Alzheimer’s disease, autoimmunity and inflammation. The good, the bad and the ugly. Autoimmun. Rev. 2011, 11, 149–153. [Google Scholar] [CrossRef] [PubMed]
- De Silva, H.A.; Aronson, J.K.; Grahame-Smith, D.G.; Jobst, K.A.; Smith, A.D. Abnormal function of potassium channels in platelets of patients with Alzheimer’s disease. Lancet 1998, 352, 1590–1593. [Google Scholar] [CrossRef]
- Kniewallner, K.M.; Ehrlich, D.; Kiefer, A.; Marksteiner, J.; Humpel, C. Platelets in the Alzheimer’s disease brain: Do they play a role in cerebral amyloid angiopathy? Curr. Neurovasc. Res. 2015, 12, 4–14. [Google Scholar] [CrossRef]
- Kleinschnitz, C.; Pozgajova, M.; Pham, M.; Bendszus, M.; Nieswandt, B.; Stoll, G. Targeting platelets in acute experimental stroke: Impact of glycoprotein Ib, VI, and IIb/IIIa blockade on infarct size, functional outcome, and intracranial bleeding. Circulation 2007, 115, 2323–2330. [Google Scholar] [CrossRef] [Green Version]
- Kleinschnitz, C.; De Meyer, S.F.; Schwarz, T.; Austinat, M.; Vanhoorelbeke, K.; Nieswandt, B.; Deckmyn, H.; Stoll, G. Deficiency of von Willebrand factor protects mice from ischemic stroke. Blood 2009, 113, 3600–3603. [Google Scholar] [CrossRef] [Green Version]
- Thornton, P.; McColl, B.W.; Greenhalgh, A.; Denes, A.; Allan, S.M.; Rothwell, N.J. Platelet interleukin-1α drives cerebrovascular inflammation. Blood 2010, 115, 3632–3639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasperska-Zajac, A.; Rogala, B. Platelet activation during allergic inflammation. Inflammation 2007, 30, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Katoh, N. Platelets as versatile regulators of cutaneous inflammation. J. Dermatol. Sci. 2009, 53, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Tamagawa-Mineoka, R.; Katoh, N.; Ueda, E.; Masuda, K.; Kishimoto, S. Elevated platelet activation in patients with atopic dermatitis and psoriasis: Increased plasma levels of beta-thromboglobulin and platelet factor 4. Allergol. Int. Off. J. Jpn. Soc. Allergol. 2008, 57, 391–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benton, A.S.; Kumar, N.; Lerner, J.; Wiles, A.A.; Foerster, M.; Teach, S.J.; Freishtat, R.J. Airway platelet activation is associated with airway eosinophilic inflammation in asthma. J. Investig. Med. Off. Publ. Am. Fed. Clin. Res. 2010, 58, 987–990. [Google Scholar] [CrossRef]
- Pitchford, S.C.; Yano, H.; Lever, R.; Riffo-Vasquez, Y.; Ciferri, S.; Rose, M.J.; Giannini, S.; Momi, S.; Spina, D.; O’Connor, B.; et al. Platelets are essential for leukocyte recruitment in allergic inflammation. J. Allergy Clin. Immunol. 2003, 112, 109–118. [Google Scholar] [CrossRef]
- Pitchford, S.C.; Riffo-Vasquez, Y.; Sousa, A.; Momi, S.; Gresele, P.; Spina, D.; Page, C.P. Platelets are necessary for airway wall remodeling in a murine model of chronic allergic inflammation. Blood 2004, 103, 639–647. [Google Scholar] [CrossRef] [Green Version]
- Kameyoshi, Y.; Schröder, J.M.; Christophers, E.; Yamamoto, S. Identification of the cytokine RANTES released from platelets as an eosinophil chemotactic factor. Int. Arch. Allergy Immunol. 1994, 104 (Suppl. S1), 49–51. [Google Scholar] [CrossRef]
- Benveniste, J.; Henson, P.M.; Cochrane, C.G. Leukocyte-dependent histamine release from rabbit platelets: The role of IgE, basophils, and a platelet-activating factor. J. Exp. Med. 1972, 136, 1356–1377. [Google Scholar] [CrossRef]
- Johansson, M.W.; Han, S.-T.; Gunderson, K.A.; Busse, W.W.; Jarjour, N.N.; Mosher, D.F. Platelet activation, P-selectin, and eosinophil β1-integrin activation in asthma. Am. J. Respir. Crit. Care Med. 2012, 185, 498–507. [Google Scholar] [CrossRef] [Green Version]
- Gallagher, J.; Bernstein, I.; Maccia, C.; Splansky, G.; Glueck, H. Cyclic platelet dysfunction in IgE-mediated allergy. J. Allergy Clin. Immunol. 1978, 62, 229–235. [Google Scholar] [CrossRef]
- Palma-Carlos, A.; Palma-Carlos, L.; Santos, C.B.; de Sousa, C. Platelet aggregation in allergic reactions. Int. Arch. Allergy Immunol. 1991, 94, 251–253. [Google Scholar] [CrossRef] [PubMed]
- Szczeklik, A.; Milner, P.; Birch, J.; Watkins, J.; Martin, J. Prolonged bleeding time, reduced platelet aggregation, altered PAF-acether sensitivity and increased platelet mass are a trait of asthma and hay fever. Thromb. Haemost. 1986, 56, 283–287. [Google Scholar] [CrossRef]
- Kowal, K.; Pampuch, A.; Kowal-Bielecka, O.; DuBuske, L.; Bodzenta-Łukaszyk, A. Platelet activation in allergic asthma patients during allergen challenge with Dermatophagoides pteronyssinus. Clin. Exp. Allergy 2006, 36, 426–432. [Google Scholar] [CrossRef] [PubMed]
- Taytard, A.; Guenard, H.; Vuillemin, L.; Bouvot, J.; Vergeret, J.; Ducassou, D.; Piquet, Y.; Freour, P. Platelet kinetics in stable atopic asthmatic patients. Am. Rev. Res. Dis. 1986, 134, 983–985. [Google Scholar] [CrossRef] [PubMed]
- Taytard, A.; Vuillemin, L. Platelet kinetics in stable asthmatic patients. Agents Actions Suppl. 1987, 21, 161–167. [Google Scholar] [PubMed]
- Ind, P.; Peters, A.; Malik, F.; Lavender, J.; Dollery, C. Pulmonary platelet kinetics in asthma. Thorax 1985, 40, 412. [Google Scholar] [CrossRef] [Green Version]
- Hemmendinger, S.; Pauli, G.; Tenabene, A.; Pujol, J.L.; Bessot, J.C.; Eber, M.; Cazenave, J.-P. Platelet function: Aggregation by PAF or sequestration in lung is not modified during immediate or late allergen-induced bronchospasm in man. J. Allergy Clin. Immunol. 1989, 83, 990–996. [Google Scholar] [CrossRef]
- Tunon-de-Lara, J.; Rio, P.; Marthan, R.; Vuillemin, L.; Ducassou, D.; Taytard, A. The effect of sodium cromoglycate on platelets: An in vivo and in vitro approach. J. Allergy Clin. Immunol. 1992, 89, 994–1000. [Google Scholar] [CrossRef]
- Shah, S.A.; Page, C.P.; Pitchford, S.C. Platelet–eosinophil interactions as a potential therapeutic target in allergic inflammation and asthma. Front. Med. 2017, 4, 129. [Google Scholar] [CrossRef] [Green Version]
- Page, C. The involvement of platelets in non-thrombotic processes. Trend. Pharmacol. Sci. 1988, 9, 66–71. [Google Scholar] [CrossRef]
- Haemmerle, M.; Stone, R.L.; Menter, D.G.; Afshar-Kharghan, V.; Sood, A.K. The platelet lifeline to cancer: Challenges and opportunities. Cancer Cell 2018, 33, 965–983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franco, A.T.; Corken, A.; Ware, J. Platelets at the interface of thrombosis, inflammation, and cancer. Blood 2015, 126, 582–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, A.K.; Singhi, E.K.; Arroyo, J.P.; Ikizler, T.A.; Gould, E.R.; Brown, J.; Beckman, J.A.; Harrison, D.G.; Moslehi, J. Mechanisms of VEGF (vascular endothelial growth factor) inhibitor-associated hypertension and vascular disease. Hypertension 2018, 71, e1–e8. [Google Scholar] [CrossRef] [PubMed]
- Ucuzian, A.A.; Gassman, A.A.; East, A.T.; Greisler, H.P. Molecular mediators of angiogenesis. J. Burn Care Res. 2010, 31, 158–175. [Google Scholar] [CrossRef]
- Rouwkema, J.; Khademhosseini, A. Vascularization and angiogenesis in tissue engineering: Beyond creating static networks. Trend. Biotechnol. 2016, 34, 733–745. [Google Scholar] [CrossRef]
- Teven, C.M.; Farina, E.M.; Rivas, J.; Reid, R.R. Fibroblast growth factor (FGF) signaling in development and skeletal diseases. Genes Dis. 2014, 1, 199–213. [Google Scholar] [CrossRef] [Green Version]
- Shen, Z.; Yao, C.; Wang, Z.; Yue, L.; Fang, Z.; Yao, H.; Lin, F.; Zhao, H.; Sun, Y.-J.; Bian, X.-W. Vastatin, an endogenous antiangiogenesis polypeptide that is lost in hepatocellular carcinoma, effectively inhibits tumor metastasis. Mol. Ther. 2016, 24, 1358–1368. [Google Scholar] [CrossRef] [Green Version]
- O’Reilly, M.S.; Boehm, T.; Shing, Y.; Fukai, N.; Vasios, G.; Lane, W.S.; Flynn, E.; Birkhead, J.R.; Olsen, B.R.; Folkman, J. Endostatin: An endogenous inhibitor of angiogenesis and tumor growth. Cell 1997, 88, 277–285. [Google Scholar] [CrossRef] [Green Version]
- Mancuso, M.E.; Santagostino, E. Platelets: Much more than bricks in a breached wall. Br. J. Haematol. 2017, 178, 209–219. [Google Scholar] [CrossRef] [Green Version]
- Raman, D.; Baugher, P.J.; Thu, Y.M.; Richmond, A. Role of chemokines in tumor growth. Cancer Lett. 2007, 256, 137–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gay, L.J.; Felding-Habermann, B. Contribution of platelets to tumour metastasis. Nat. Rev. Cancer 2011, 11, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Wiesner, T.; Bugl, S.; Mayer, F.; Hartmann, J.T.; Kopp, H.-G. Differential changes in platelet VEGF, Tsp, CXCL12, and CXCL4 in patients with metastatic cancer. Clin. Exp. Metastasis 2010, 27, 141–149. [Google Scholar] [CrossRef]
- Suzuki, A.; Takahashi, T.; Nakamura, K.; Tsuyuoka, R.; Okuno, Y.; Enomoto, T.; Fukumoto, M.; Imura, H. Thrombocytosis in patients with tumors producing colony-stimulating factor. Blood 1992, 80, 2052–2059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmes, C.E.; Levis, J.E.; Schneider, D.J.; Bambace, N.M.; Sharma, D.; Lal, I.; Wood, M.E.; Muss, H.B. Platelet phenotype changes associated with breast cancer and its treatment. Platelets 2016, 27, 703–711. [Google Scholar] [CrossRef]
- Chadha, A.S.; Kocak-Uzel, E.; Das, P.; Minsky, B.D.; Delclos, M.E.; Mahmood, U.; Guha, S.; Ahmad, M.; Varadhachary, G.R.; Javle, M. Paraneoplastic thrombocytosis independently predicts poor prognosis in patients with locally advanced pancreatic cancer. Acta Oncol. 2015, 54, 971–978. [Google Scholar] [CrossRef] [PubMed]
- Monreal, M.; Fernandez-Llamazares, J.; Piñol, M.; Julian, J.F.; Broggi, M.; Abad, A. Platelet count and survival in patients with colorectal cancer–a preliminary study. Thromb. Haemost. 1998, 79, 916–918. [Google Scholar] [CrossRef]
- Jefferson, K.; Persad, R. Poor prognosis associated with thrombocytosis in patients with renal cell carcinoma. BJU Int. 2001, 87, 715. [Google Scholar] [CrossRef]
- Buergy, D.; Wenz, F.; Groden, C.; Brockmann, M.A. Tumor–platelet interaction in solid tumors. Int. J. Cancer 2012, 130, 2747–2760. [Google Scholar] [CrossRef]
- Haemmerle, M.; Taylor, M.L.; Gutschner, T.; Pradeep, S.; Cho, M.S.; Sheng, J.; Lyons, Y.M.; Nagaraja, A.S.; Dood, R.L.; Wen, Y. Platelets reduce anoikis and promote metastasis by activating YAP1 signaling. Nat. Commun. 2017, 8, 310. [Google Scholar] [CrossRef]
- Carr, B.I.; Cavallini, A.; D’Alessandro, R.; Refolo, M.G.; Lippolis, C.; Mazzocca, A.; Messa, C. Platelet extracts induce growth, migration and invasion in human hepatocellular carcinoma in vitro. BMC Cancer 2014, 14, 43. [Google Scholar] [CrossRef] [PubMed]
- Labelle, M.; Begum, S.; Hynes, R.O. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell 2011, 20, 576–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clere, N.; Renault, S.; Corre, I. Endothelial-to-mesenchymal transition in cancer. Front. Cell Dev. Biol. 2020, 8, 747. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Lan, X.; Liu, M.; Zhou, B.; Wang, B.; Chen, P. Direct TGF-β1 signaling between activated platelets and pancreatic cancer cells primes cisplatin insensitivity. Cell Biol. Int. 2013, 37, 478–484. [Google Scholar] [CrossRef]
- Köck, K.; Grube, M.; Jedlitschky, G.; Oevermann, L.; Siegmund, W.; Ritter, C.A.; Kroemer, H.K. Expression of adenosine triphosphate-binding cassette (ABC) drug transporters in peripheral blood cells. Clin. Pharmacokin. 2007, 46, 449–470. [Google Scholar]
- Huijbers, E.J.; van Beijnum, J.R.; Thijssen, V.L.; Sabrkhany, S.; Nowak-Sliwinska, P.; Griffioen, A.W. Role of the tumor stroma in resistance to anti-angiogenic therapy. Drug Resist. Updates 2016, 25, 26–37. [Google Scholar] [CrossRef]
- Van Der Zee, A.G.; De Bruijn, H.W.; Krans, M.; De Cuyper, E.M.; Hollema, H.; Limburg, P.C.; Bijzet, J.; De Vries, E.G. Higher levels of interleukin-6 in cystic fluids from patients with malignant versus benign ovarian tumors correlate with decreased hemoglobin levels and increased platelet counts. Cancer 1995, 75, 1004–1009. [Google Scholar] [CrossRef]
- Feng, S.; Kroll, M.H.; Nick, A.M.; Sood, A.K.; Afshar-Kharghan, V. Platelets are not hyperreactive in patients with ovarian cancer. Platelets 2016, 27, 716–718. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Cui, W.; Pei, Y.; Xu, D. Platelets promote invasion and induce epithelial to mesenchymal transition in ovarian cancer cells by TGF-β signaling pathway. Gynecol. Oncol. 2019, 153, 639–650. [Google Scholar] [CrossRef]
- Andrade, S.S.; Sumikawa, J.T.; Castro, E.D.; Batista, F.P.; Paredes-Gamero, E.; Oliveira, L.C.; Guerra, I.M.; Peres, G.B.; Cavalheiro, R.P.; Juliano, L. Interface between breast cancer cells and the tumor microenvironment using platelet-rich plasma to promote tumor angiogenesis-influence of platelets and fibrin bundles on the behavior of breast tumor cells. Oncotarget 2017, 8, 16851. [Google Scholar] [CrossRef]
- Ibele, G.M.; Kay, N.E.; Johnson, G.J.; Jacob, H.S. Human platelets exert cytotoxic effects on tumor cells. Blood 1985, 65, 1252–1255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pacchiarini, L.; Serra, L.; Grignani, G.; Gamba, G.; Gorini, M. In vitro effect of culture fluids from neoplastic tissues on platelet aggregation. I. Human tumors of the gastrointestinal tract. Boll. Della Soc. Ital. Biol. Sper. 1982, 58, 847–853. [Google Scholar]
- Gasic, G.J.; Gasic, T.B.; Galanti, N.; Johnson, T.; Murphy, S. Platelet—tumor-cell interactions in mice. The role of platelets in the spread of malignant disease. Int. J. Cancer 1973, 11, 704–718. [Google Scholar] [CrossRef] [PubMed]
- Mezouar, S.; Darbousset, R.; Dignat-George, F.; Panicot-Dubois, L.; Dubois, C. Inhibition of platelet activation prevents the P-selectin and integrin-dependent accumulation of cancer cell microparticles and reduces tumor growth and metastasis in vivo. Int. J. Cancer 2015, 136, 462–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pucci, F.; Rickelt, S.; Newton, A.P.; Garris, C.; Nunes, E.; Evavold, C.; Pfirschke, C.; Engblom, C.; Mino-Kenudson, M.; Hynes, R.O. PF4 promotes platelet production and lung cancer growth. Cell Rep. 2016, 17, 1764–1772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hicks, B.M.; Murray, L.J.; Hughes, C.; Cardwell, C.R. Clopidogrel use and cancer-specific mortality: A population-based cohort study of colorectal, breast and prostate cancer patients. Pharmacoepidemiol. Drug Saf. 2015, 24, 830–840. [Google Scholar] [CrossRef]
- Gebremeskel, S.; LeVatte, T.; Liwski, R.S.; Johnston, B.; Bezuhly, M. The reversible P2Y12 inhibitor ticagrelor inhibits metastasis and improves survival in mouse models of cancer. Int. J. Cancer 2015, 136, 234–240. [Google Scholar] [CrossRef]
- Sierko, E.; Wojtukiewicz, M.Z. Inhibition of Platelet Function: Does It Offer a Chance of Better Cancer Progression Control? Semin. Thromb. Hemost. 2007, 33, 712–721. [Google Scholar] [CrossRef]
- Connors, J.M.; Levy, J.H. COVID-19 and its implications for thrombosis and anticoagulation. Blood 2020, 135, 2033–2040. [Google Scholar] [CrossRef]
- Bonaventura, A.; Vecchié, A.; Dagna, L.; Martinod, K.; Dixon, D.L.; Van Tassell, B.W.; Dentali, F.; Montecucco, F.; Massberg, S.; Levi, M. Endothelial dysfunction and immunothrombosis as key pathogenic mechanisms in COVID-19. Nat. Rev. Immunol. 2021, 21, 319–329. [Google Scholar] [CrossRef]
- Gawaz, M.; Langer, H.; May, A.E. Platelets in inflammation and atherogenesis. J Clin. Investig. 2005, 115, 3378–3384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolai, L.; Leunig, A.; Brambs, S.; Kaiser, R.; Weinberger, T.; Weigand, M.; Muenchhoff, M.; Hellmuth, J.C.; Ledderose, S.; Schulz, H. Immunothrombotic dysregulation in COVID-19 pneumonia is associated with respiratory failure and coagulopathy. Circulation 2020, 142, 1176–1189. [Google Scholar] [CrossRef] [PubMed]
- Bi, X.; Su, Z.; Yan, H.; Du, J.; Wang, J.; Chen, L.; Peng, M.; Chen, S.; Shen, B.; Li, J. Prediction of severe illness due to COVID-19 based on an analysis of initial Fibrinogen to Albumin Ratio and Platelet count. Platelets 2020, 31, 674–679. [Google Scholar] [CrossRef] [PubMed]
- Grobler, C.; Maphumulo, S.C.; Grobbelaar, L.M.; Bredenkamp, J.C.; Laubscher, G.J.; Lourens, P.J.; Steenkamp, J.; Kell, D.B.; Pretorius, E. COVID-19: The Rollercoaster of Fibrin(Ogen), D-Dimer, Von Willebrand Factor, P-Selectin and Their Interactions with Endothelial Cells, Platelets and Erythrocytes. Int. J. Mol. Sci. 2020, 21, 5168. [Google Scholar] [CrossRef] [PubMed]
- Ruberto, F.; Chistolini, A.; Curreli, M.; Frati, G.; Marullo, A.G.; Biondi-Zoccai, G.; Mancone, M.; Sciarretta, S.; Miraldi, F.; Alessandri, F. Von Willebrand factor with increased binding capacity is associated with reduced platelet aggregation but enhanced agglutination in COVID-19 patients: Another COVID-19 paradox? J. Thromb. Thrombolysis 2021, 52, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Zaid, Y.; Puhm, F.; Allaeys, I.; Naya, A.; Oudghiri, M.; Khalki, L.; Limami, Y.; Zaid, N.; Sadki, K.; Ben El Haj, R. Platelets can associate with SARS-Cov-2 RNA and are hyperactivated in COVID-19. Circ. Res. 2020, 127, 1404–1418. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, Y.; Wang, X.; Yang, L.; Li, H.; Wang, Y.; Liu, M.; Zhao, X.; Xie, Y.; Yang, Y.; et al. SARS-CoV-2 binds platelet ACE2 to enhance thrombosis in COVID-19. J. Hematol. Oncol. 2020, 13, 120. [Google Scholar] [CrossRef]
- Bikdeli, B.; Madhavan, M.V.; Jimenez, D.; Chuich, T.; Dreyfus, I.; Driggin, E.; Nigoghossian, C.; Ageno, W.; Madjid, M.; Guo, Y. Global COVID-19 Thrombosis Collaborative Group, Endorsed by the ISTH, NATF, ESVM, and the IUA, Supported by the ESC Working Group on Pulmonary Circulation and Right Ventricular Function. COVID-19 and thrombotic or thromboembolic disease: Implications for prevention, antithrombotic therapy, and follow-up: JACC state-of-the-art review. J. Am. Coll. Cardiol. 2020, 75, 2950–2973. [Google Scholar]
- Alnor, A.; Sandberg, M.B.; Toftanes, B.E.; Vinholt, P.J. Platelet parameters and leukocyte morphology is altered in COVID-19 patients compared to non-COVID-19 patients with similar symptomatology. Scand. J. Clin. Lab. Investig. 2021, 81, 213–217. [Google Scholar] [CrossRef]
- Boeckh-Behrens, T.; Golkowski, D.; Ikenberg, B.; Schlegel, J.; Protzer, U.; Schulz, C.; Novotny, J.; Kreiser, K.; Zimmer, C.; Hemmer, B.; et al. COVID-19-associated Large Vessel Stroke in a 28-year-old Patient: NETs and Platelets Possible Key Players in Acute Thrombus Formation. Clin. Neuroradiol. 2021, 31, 511–514. [Google Scholar] [CrossRef]
- Althaus, K.; Marini, I.; Zlamal, J.; Pelzl, L.; Singh, A.; Haberle, H.; Mehrlander, M.; Hammer, S.; Schulze, H.; Bitzer, M.; et al. Antibody-induced procoagulant platelets in severe COVID-19 infection. Blood 2021, 137, 1061–1071. [Google Scholar] [CrossRef] [PubMed]
- Saleh, J.; Peyssonnaux, C.; Singh, K.K.; Edeas, M. Mitochondria and microbiota dysfunction in COVID-19 pathogenesis. Mitochondrion 2020, 54, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Poterucha, T.J.; Libby, P.; Goldhaber, S.Z. More than an anticoagulant: Do heparins have direct anti-inflammatory effects? Thromb. Haemost. 2017, 117, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Tang, N.; Bai, H.; Chen, X.; Gong, J.; Li, D.; Sun, Z. Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J. Thromb. Haemost. 2020, 18, 1094–1099. [Google Scholar] [CrossRef]
- Kistangari, G.; McCrae, K.R. Immune thrombocytopenia. Hematol. Oncol. Clin. 2013, 27, 495–520. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharjee, S.; Banerjee, M. Immune Thrombocytopenia Secondary to COVID-19: A Systematic Review. SN Compr. Clin. Med. 2020, 2, 2048–2058. [Google Scholar] [CrossRef]
- Wang, J.; Hajizadeh, N.; Moore, E.E.; McIntyre, R.C.; Moore, P.K.; Veress, L.A.; Yaffe, M.B.; Moore, H.B.; Barrett, C.D. Tissue plasminogen activator (tPA) treatment for COVID-19 associated acute respiratory distress syndrome (ARDS): A case series. J. Thromb. Haemost. 2020, 18, 1752–1755. [Google Scholar] [CrossRef]
- Zhang, X.; Li, M.; Chen, T.; Lv, D.; Xia, P.; Qian, W. Management of COVID-19-related immune thrombocytopenia by rhTPO. Blood Res. 2021, 56, 205–207. [Google Scholar] [CrossRef]


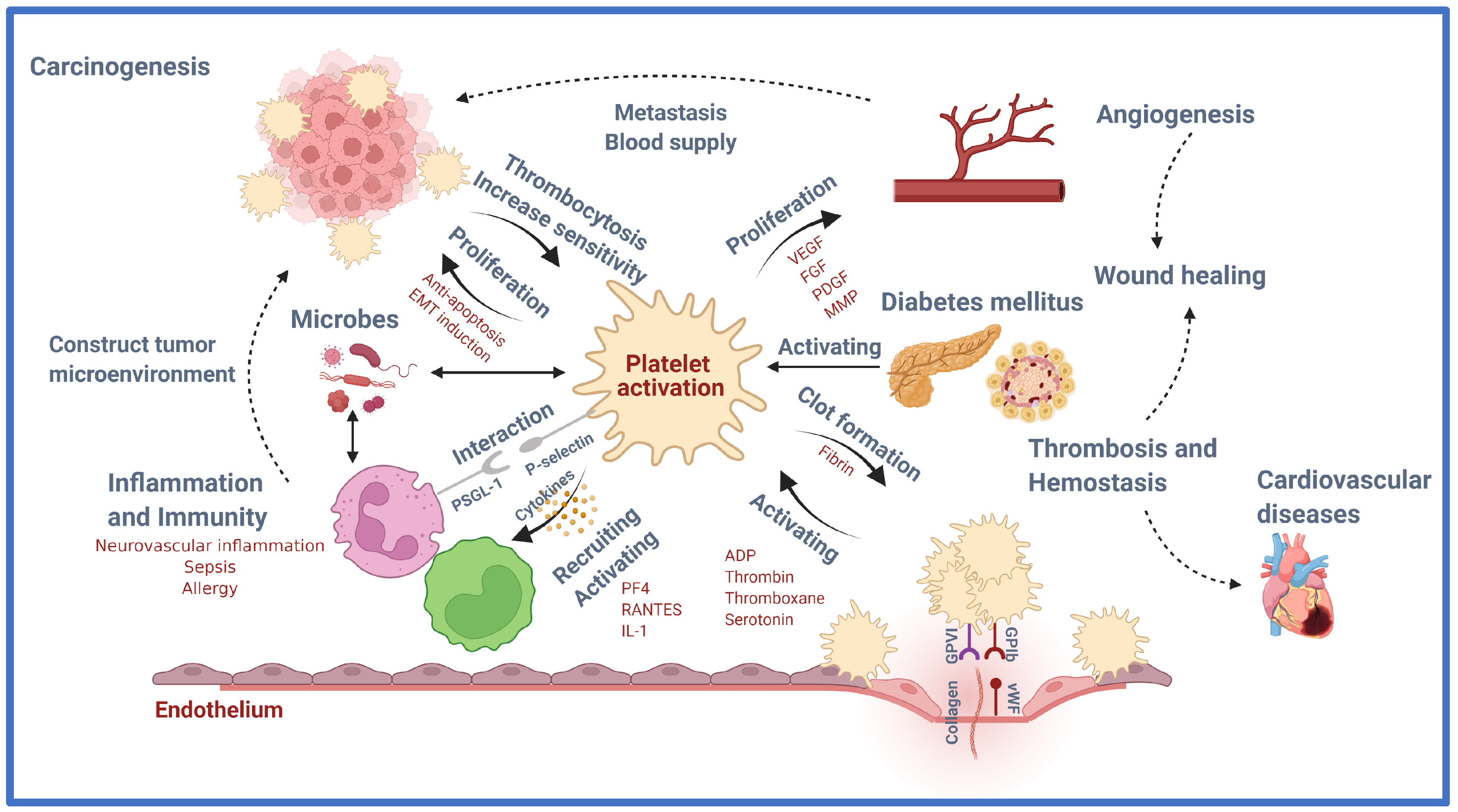{kind=link}
| Granule | Type | Contents | Role | References |
|---|---|---|---|---|
| α-granules | Adhesive proteins | P-selectin | Promoting adherence of leukocytes to activated platelets and endothelium | [20] |
| Fibrinogen | Binding to GpIIb/IIIa receptors on the surface of platelets | [21] | ||
| Von Willebrand factor | Binding to FVIII on the surface of platelets | [22] | ||
| Fibronectin | Binding to integrin α5β1 and αvβ3 on the surface of platelets | [23] | ||
| Thrombospondin-1 Thrombospondin-2 | Binding to β1, αIIβ3, and αvβ3 on the surface of platelets | [24] | ||
| Laminin-8 | Binding to α3β1, and α6β1 on the surface of platelets | [25] | ||
| Vitronectin | Binding to αvβ3 on the surface of platelets, and uPAR | [26] | ||
| Growth factors | Epidermal growth factor (EGF) | Stimulating the proliferation of fibroblasts and epithelial cells | [27] | |
| Insulin-like growth factor 1 (IGF-1) | The major mediator of growth hormone-stimulated somatic growth and growth hormone-independent anabolic responses | [28] | ||
| Hepatocyte growth factor (HGF) | Metabolic flux of glucose in different insulin-sensitive cell types; plays a key role in β-cell homeostasis | [29] | ||
| Transforming growth factor β (TGF-β) | Inhibiting the proliferation of epithelial cells | [30] | ||
| Platelet-derived growth factor (PDGF) | Growth control of mesenchymal cells such as fibroblasts and smooth muscle cells | [31] | ||
| Angiogenic factors | Vascular endothelium growth factor (VEGF) | Enhanced endothelial cell proliferation and survival, increased migration and invasion of endothelial cells, increased permeability of existing vessels, forming a lattice network for endothelial cell migration, enhanced chemotaxis, and homing of bone marrow-derived vascular precursor cells | [32] | |
| Platelet-derived growth factor (PDGF) | Up-regulating VEGF production, modulating the proliferation and recruitment of perivascular cells | [33] | ||
| Fibroblast growth factor (FGF) | Activating a serine-rich proteins/serine-rich phosphorylating kinases network that regulates VEGFR1 alternative spicing in endothelial cells. | [34] | ||
| Chemokines | CXCL8 (IL 8) CXCL7 (platelet basic protein/NAP-2) CXCL1 (GRO- α) CXCL5 (ENA-78) CXCL2 (MIP-2) CXCL6 (LIX) CXCL-12 (SDF-1α) | Activating and recruiting neutrophils | [35] [36] [37] [38] [39] [40] [41] | |
| CCL5 (RANTES) | Recruiting T cells, macrophages, eosinophils, and basophils | [42] | ||
| CCL3 (MIP-1α) | Recruiting polymorphonuclear leukocyte | [43] | ||
| CCL2 (MCP-1) | Recruiting polymorphonuclear leukocyte | [43] | ||
| CCL7 (MCP-3) | Recruiting monocytes, neutrophils, eosinophils, and basophils | [44] | ||
| IL1β | Recruiting and activating leukocytes | [45] | ||
| CD40L Proteases | - | |||
| Coagulation factors | Factor V | Cleaving prothrombin to thrombin | [46] | |
| Protein S | Anticoagulant by inhibiting FIXa | [47] | ||
| Factor XI | Hemostasis through activation of factor IX | [48] | ||
| Factor XIII | Stabilizing fibrin networks | [49] | ||
| Kininogens | Activating FXI | [50] | ||
| Plasminogen | Fibrinolysis by binding to the fibrin clot | [51] | ||
| Integral membrane proteins | Integrin αIIbβ3 | By binding fibrinogen, facilitates irreversible binding of platelets to the exposed extracellular matrix and enables the cross-linking of adjacent platelets | [52] | |
| GPIba-IX-V | By binding the von Willebrand factor, initiating platelet aggregation and thrombus formation | [53] | ||
| GPVI | By binding collagen, initiating platelet aggregation | [54] | ||
| TLT-1 | Binds fibrinogen and plays a role in bleeding initiated by inflammatory insults | [55] | ||
| P-selectin | Promoting adherence of leukocytes to activated platelets and endothelium | [20] | ||
| Immune mediators | Complement C3 precursor | Cleaved into C3a and C3b by foreign invaders, and triggering inflammation, phagocytosis, cell lysis, and cell activation | [56] | |
| Complement C4 precursor | Activating classical and lectin pathways and the formation of C3 convertase | [56] | ||
| Factor D | Initiating the alternative pathway of complement activation and amplification loop of C3 activation | [57] | ||
| Factor H | Controlling the alternative pathway | [58] | ||
| C1 inhibitor | Inhibiting the activation of the proteins of early blood coagulation and the classical complement pathways | [59] | ||
| Immunoglobulins | Binding and neutralizing antigens | [60] | ||
| Protease inhibitors | α2-antiplasmin | Inhibiting plasminogen binding to fibrin, and cross-linking fibrin | [61] | |
| PAI-1 | Binding and inhibiting the tissue-type and urokinase-type plasminogen activator | [62] | ||
| α2-antitrypsin | Anti-inflammatory properties by the destructive effect of major proteases | [63] | ||
| α2-macroglobulin | Binding foreign peptides, thereby serving as humoral defense barriers against pathogens | [64] | ||
| TFPI | Inhibiting FXa and FVIIa, thereby blocking the initial steps of the extrinsic coagulation pathway | [65] | ||
| C1-inhibitor | Inhibiting the activation of the proteins of early blood coagulation and the classical complement pathways | [59] | ||
| Proteoglycans | MMP2, MMP9 | Degrading collagen, elastin, fibronectin, gelatin, and laminin and remodeling the extracellular matrix | [66] | |
| Dense granules | Amines | Serotonin | Inducing constriction of injured blood vessels and enhancing platelet aggregation to minimize blood loss | [67] |
| Histamine | Aggregatory and immunological stimuli | [68] | ||
| Bivalent cations | Ca2+ Mg2+ | |||
| Nucleotides | ATP ADP GTP GDP | |||
| Polyphosphates | ||||
| Lysosome granules | Acid proteases | Cathepsin D, E Carboxypeptidases (A, B) Prolinecarboxypeptidase Collagenase Acid phosphatase Arylsulphatase | ||
| Glycohydrolases | Heparinase β-N-acetyl-glucosaminidase | |||
| Function | Platelet Molecule | Mechanism | References |
|---|---|---|---|
| Thrombin | Strengthn platelet-fibrin clot | [72] | |
| Platelet activation molecule | Nitric oxide | Enhance thromboxane generation and platelet aggregation | [73] |
| LDL | Increase ROS formation Platelet activation Prothrombinase complex thrombin and thrombosis formation | [74,75,76] | |
| WDR1 | Increase ADF-cofilin Actin disassembling Decrease actin cytoskeleton and thrombin activation | [77,78,79] | |
| Leptin | Increase PDE3A Decrease cAMP | [80,81] | |
| Adiponectin | Insulin resistance Hypoadiponectinemia | [82,83,84,85] | |
| ROS | 8-isoPGF2α increase Platelets aggregation Drugs’ non-responsiveness | [86,87,88,89,90] | |
| ADP | Increase platelet activation Active leukocytes, endothelial cells, and SMCs | [91,92,93] | |
| Fractalkine | Increase platelet activation Inducing P-selectin Platelet adhesion to fibrinogen and collagen | [94,95] | |
| GPVI | Platelet aggregation Initiated signaling cascades with SFKs | [96,97] | |
| Integrin αIIbβ3 | Platelet aggregation Initiated signaling cascades with SFKs | [96,97] | |
| Talin | Bind and regulate αIIbβ3 | [96,97] | |
| ILK | Integrin activation α-granule secretion and platelet activation | [98,99,100] | |
| MPs | Induce angiogenesis and revascularization improvement | [101,102,103] | |
| SPARC | Angiogenesis formation Cardiac integrity Platelet reorganization | [101,102,103] | |
| miRNA-223 | Regulate EPB41L3 gene ACS formation and vascular dysfunction | [104] | |
| miRNA-126 | Regulate VCAM-1 gene ACS formation and vascular dysfunction | [104] | |
| Molecule activation by platelet | Thromboxane | ROS generation | [73] |
| Annexin V | Inducing cardiac myocytes apoptosis | [105] | |
| P-selectin | By linking with PSGL-1, induce several inflammation, such as myocardial infarction, stroke, and peripheral artery diseases. | [106] | |
| CD40L | Induce plaque initiation, thrombus stabilization, platelet activation, and vascular inflammation | [107] | |
| PDGF-D | Stimulates proliferation of cardiac interstitial fibroblasts and arterial smooth muscle cells. | [108] | |
| PDGF-A PDGF-B | Induce cardiac hypertrophy and fibrosis mediate by mesenchymal Fibroblasts | [109] | |
| Tissue factor | Initiating the coagulation cascade Induce migration and proliferation of vascular smooth muscle cells | [110] | |
| PECAM-1 | Regulating NF-kB-mediated gene expression | [111] | |
| Peroxisome proliferator-activated receptor gamma (PPAR-r) | Induce insulin resistance, Increase vasodilation | [112] | |
| ACC | Fatty acid metabolism in platelets | [113,114] | |
| AMPK | Changes in platelet shape and secrete the granules | [113,114] | |
| MLCs | Changes in platelet shape and secrete the granules | [113,114] | |
| Cofilin | Changes in platelet shape and secrete the granules | [113,114] | |
| Thrombin | AMPK activation in platelets and phosphorylation of MLCs and cofilin | [113,114] | |
| CXCL7 | Most expressions in platelet Role in CVD | [70] | |
| CXCL4 | Increased atherosclerosis Heterodimer with CCL5 Bind to monocytes Modulate signaling pathways in the cells | [115,116,117] | |
| CCL5 | Increase the monocytes and T-lymphocytes adhesion by ICAM-1 and VCAM-1 Increase angiogenesis | [118] | |
| CXCL1 | Bind to CXCR2 Induce monocyte activation to atherosclerotic lesions | [119] | |
| CXCL12 | Atherosclerotic lesions formation Activate platelets via CXCR4 Secrete by macrophages and increase the atherosclerosis | [120,121] | |
| CCL2 | Trigger atherosclerotic lesions formation | [70,122] | |
| CCL3 | Express in atherosclerosis Bind to CCR1 and CCR5 | [70,122] |
| Mediator | Main Interactions | Main Role in Inflammation |
|---|---|---|
| P-selectin (CD62P) | Monocytes Neutrophils Endothelium | Formation of PLA [163] Formation of bridges between leukocytes and endothelium [164] |
| CD40L (CD154) | T cells B cells Monocytes DCs Endothelial cells | Important mediator of T cell immune response [165] Link between innate and adaptive immune responses [165] Promotes leukocyte recruitment to the endothelium [166] |
| PF4 (CXCL4) | Monocytes Neutrophils | Induces leukocyte pro-inflammatory cytokine release, phagocytosis, chemotaxis, generation of ROS [122,167,168] Inhibits leukocyte apoptosis [167] Promote neutrophil firm adhesion on the endothelium [169] |
| MIP-1α (CCL3) | Monocytes Macrophages Neutrophil | Promotes monocyte, macrophage, and neutrophil chemotaxis [170] Upregulates monocyte and macrophage release of pro-inflammatory mediators [170] |
| RANTES (CCL5) | Monocytes Macrophages T cells Endothelial cells | Promotes monocyte, macrophage, and T cell chemotaxis and recruitment to the endothelium [171,172] Induce expression of MMP [173] |
| IL-1 | SMC Monocyte Macrophage Endothelium T cells | Central to pro-inflammatory cytokine cascade and vascular inflammation [174] Increase the expression of adhesion factors on endothelial cells to enable migration [175] Vasodilation and hypotension Increase the expansion of naïve and memory CD4 T cells [176] |
| Microbicidal proteins | Bacteria | Disrupt cell membrane [177] |
| NAP-2 (CXCL7) | Neutrophil | Promote neutrophil firm adhesion on the endothelium and transmigration [169] |
| SDF-1α | Monocyte | Regulating leukocyte polarization and motility [178] |
| Serotonin | Neutrophil T-cell | Neutrophil and T-cell recruitment, vasodilation, and increasing vascular permeability [179] |
| Histamine | Endothelium | Vasodilation, increasing vascular permeability, and endothelial activation [180] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chaudhary, P.K.; Kim, S.; Kim, S. An Insight into Recent Advances on Platelet Function in Health and Disease. Int. J. Mol. Sci. 2022, 23, 6022. https://doi.org/10.3390/ijms23116022
Chaudhary PK, Kim S, Kim S. An Insight into Recent Advances on Platelet Function in Health and Disease. International Journal of Molecular Sciences. 2022; 23(11):6022. https://doi.org/10.3390/ijms23116022
Chicago/Turabian StyleChaudhary, Preeti Kumari, Sanggu Kim, and Soochong Kim. 2022. "An Insight into Recent Advances on Platelet Function in Health and Disease" International Journal of Molecular Sciences 23, no. 11: 6022. https://doi.org/10.3390/ijms23116022
APA StyleChaudhary, P. K., Kim, S., & Kim, S. (2022). An Insight into Recent Advances on Platelet Function in Health and Disease. International Journal of Molecular Sciences, 23(11), 6022. https://doi.org/10.3390/ijms23116022