
Neu3 Sialidase Activates the RISK Cardioprotective Signaling Pathway during Ischemia and Reperfusion Injury (IRI)

 ,
, 
 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Results
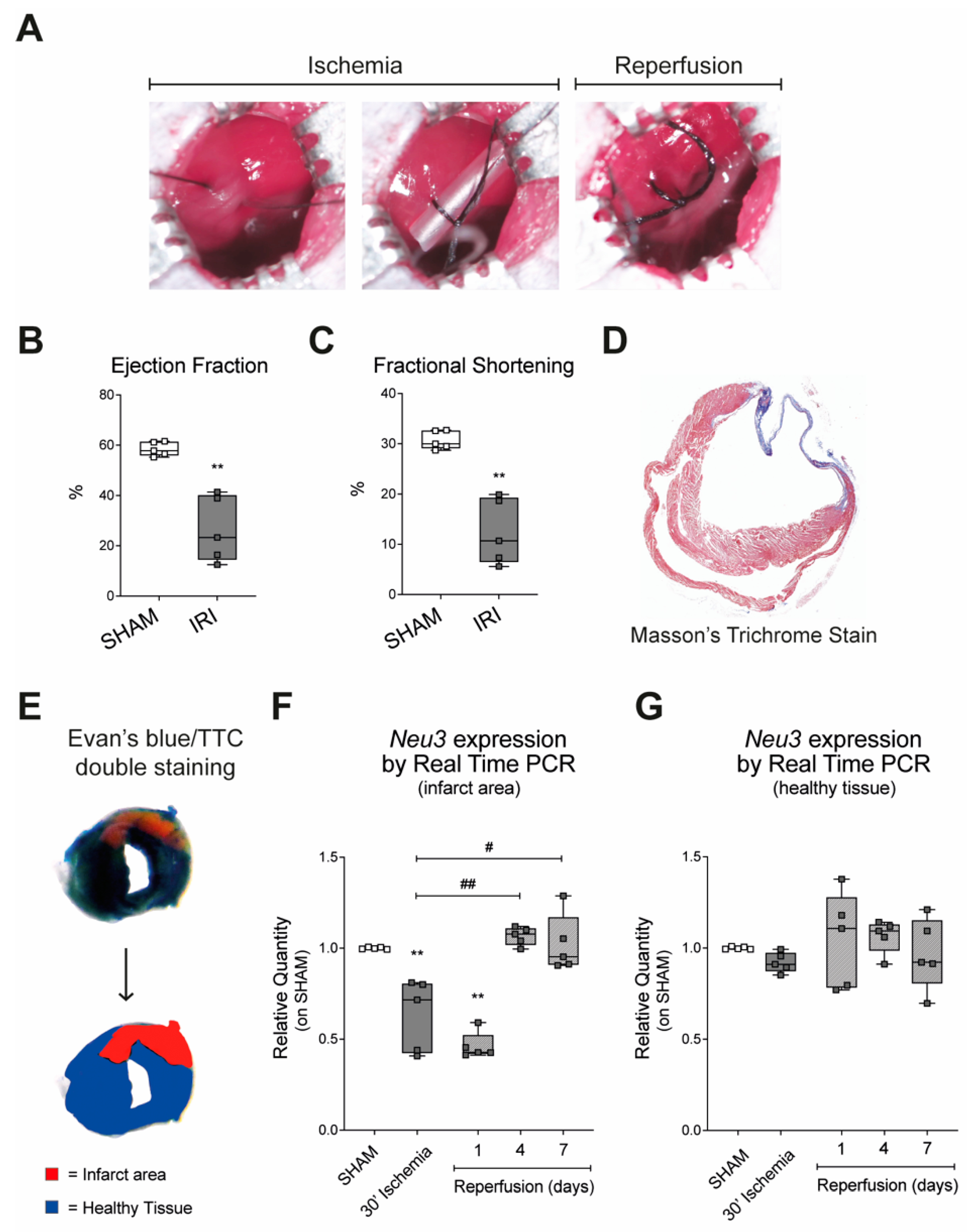
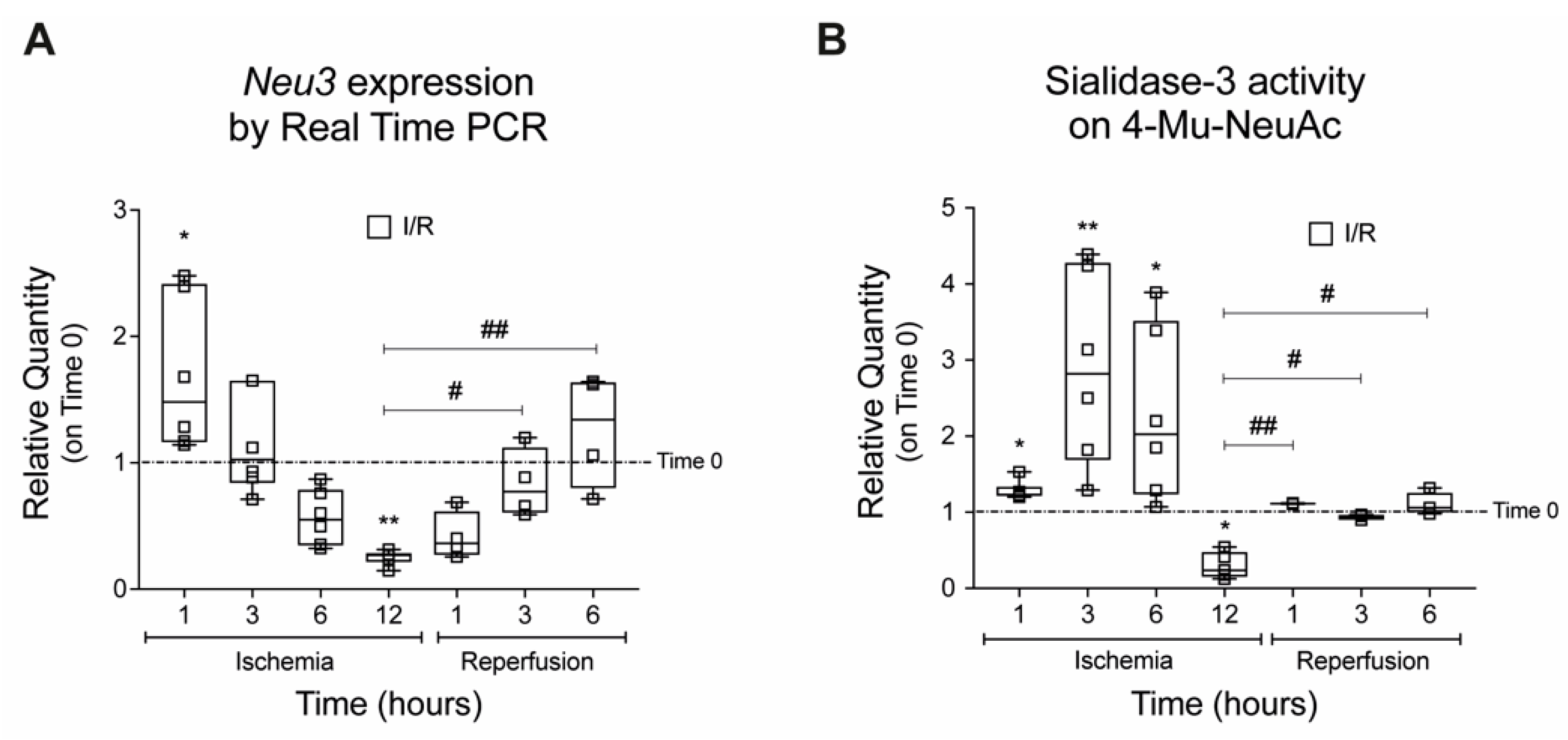
2.1. Sialidase-3 Modulation in a Mouse Model of IRI
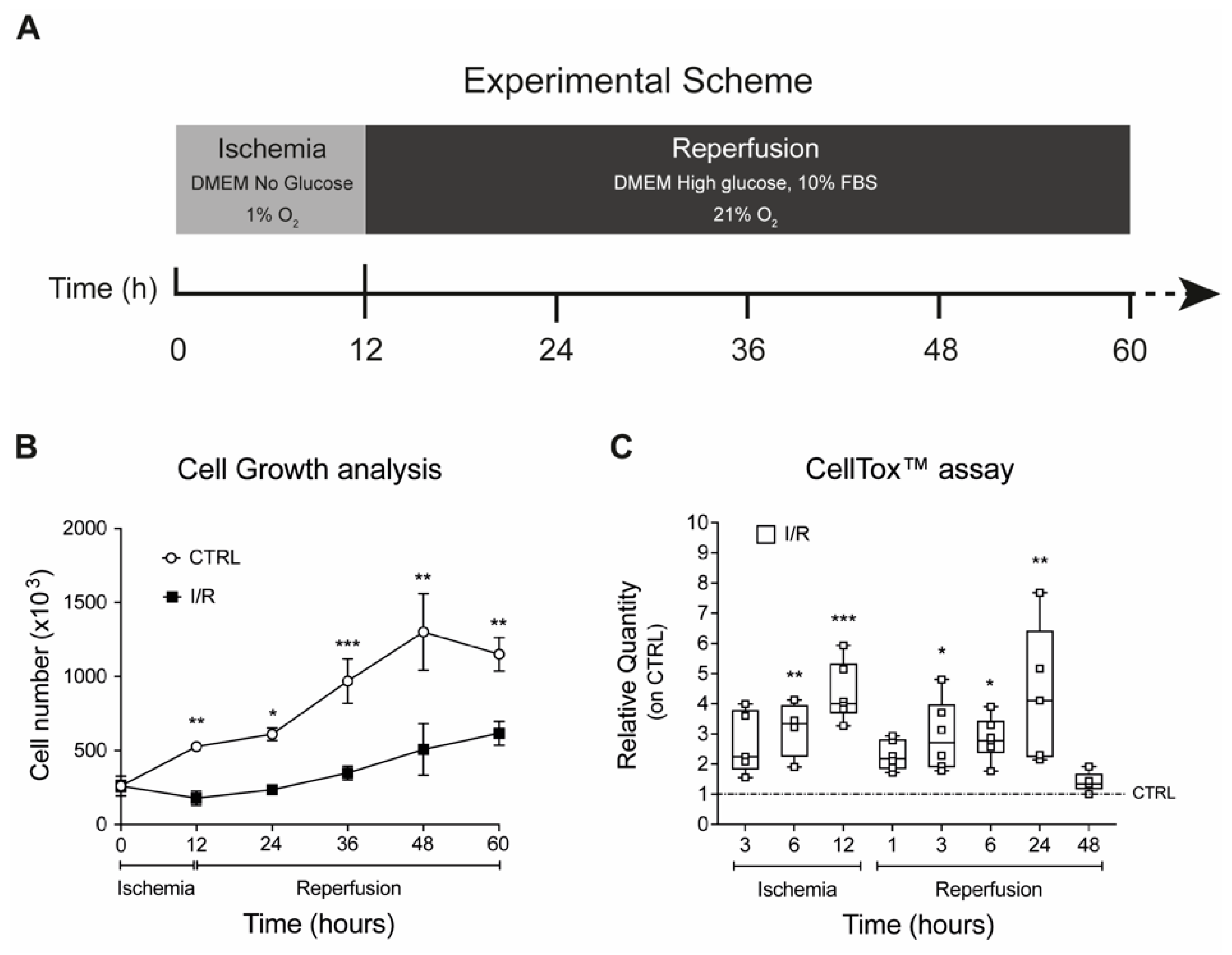
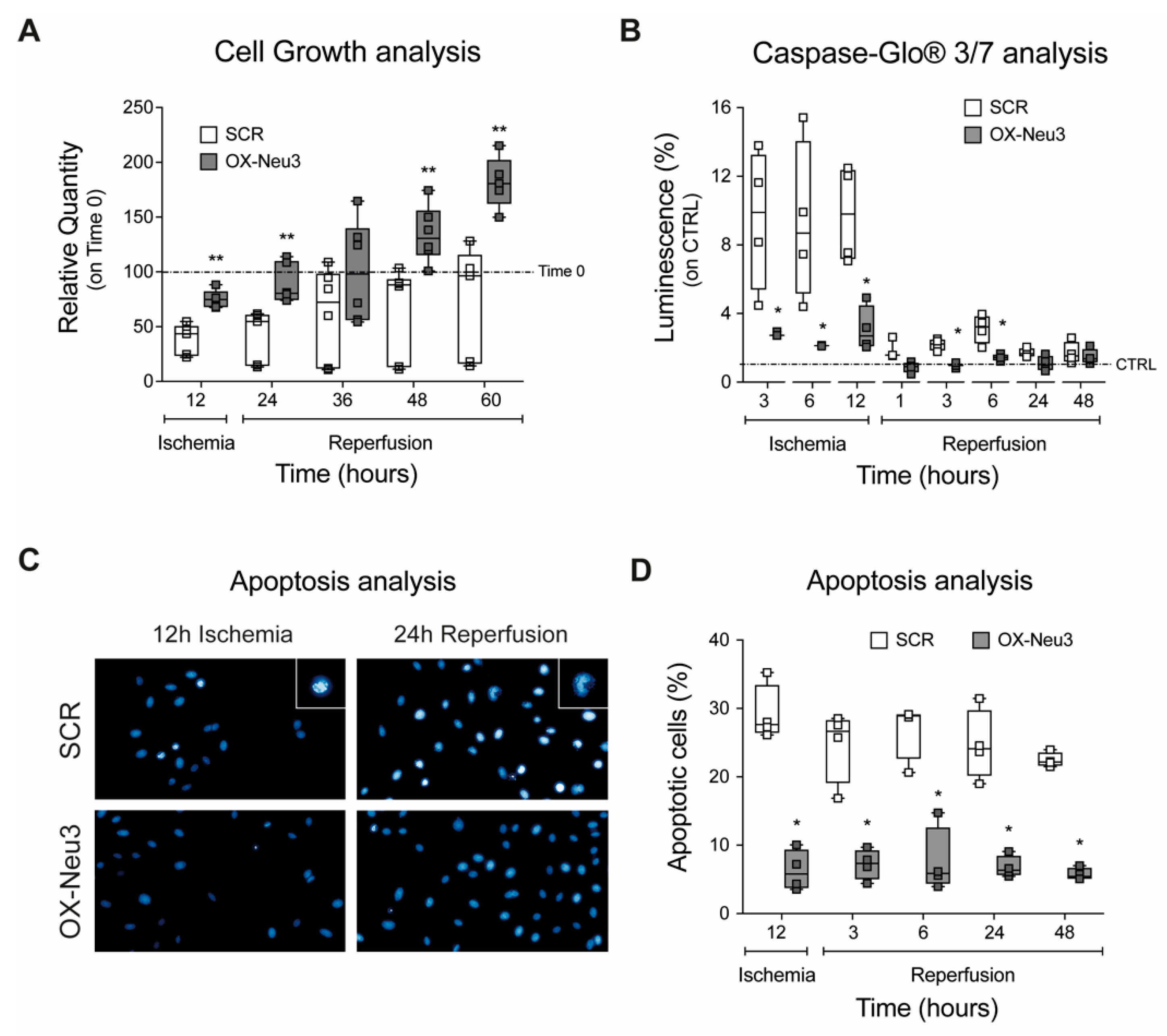
2.2. Effects on Cell Proliferation, Toxicity and Neu3 Expression of an In Vitro Model of IRI
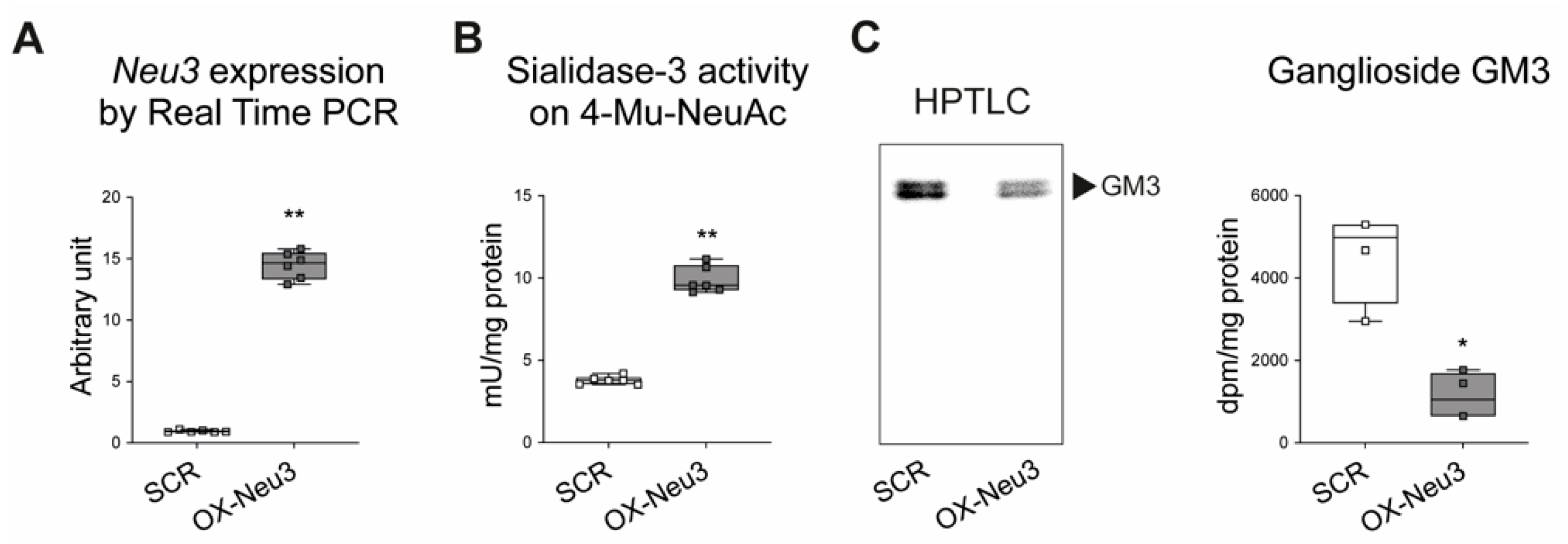
2.3. Effects of Sialidase Neu3 Overexpression in H9c2 Cardiomyoblasts
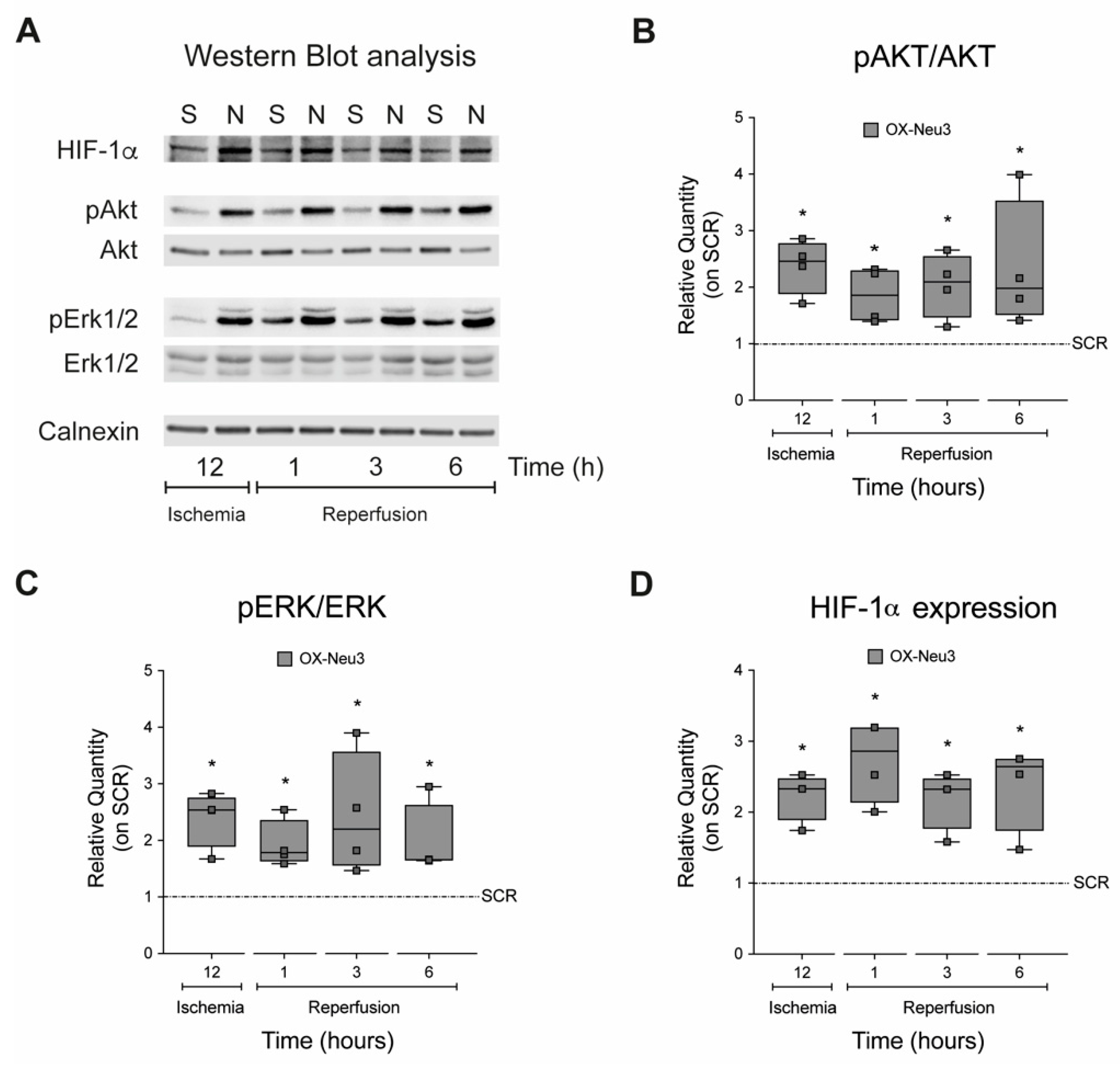
2.4. Sialidase Neu3 Up-Regulation Effects on the RISK Pathway and HIF-1α
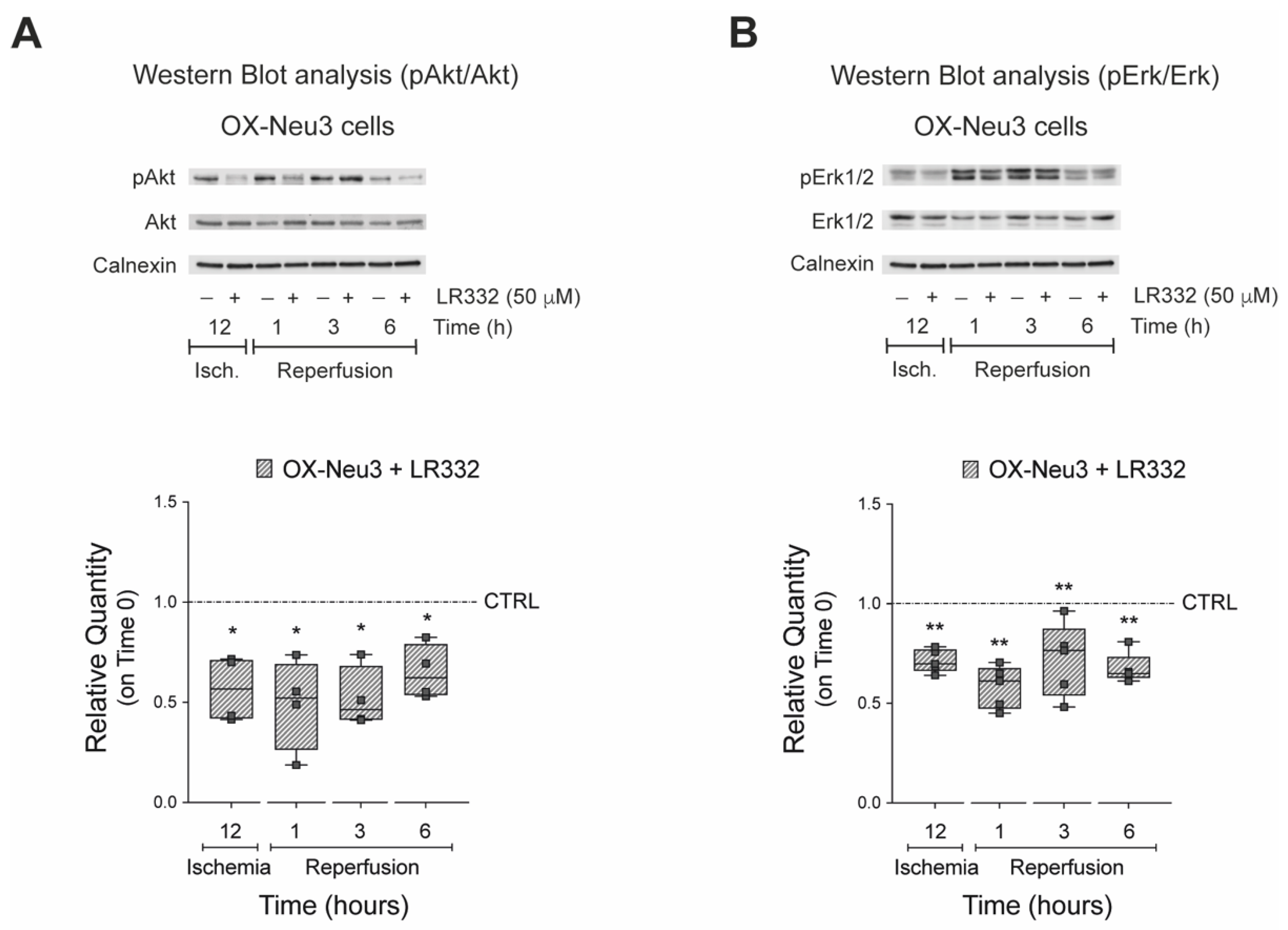
2.5. Effects of Sialidase-3 Inhibition on the RISK Pathway Activation
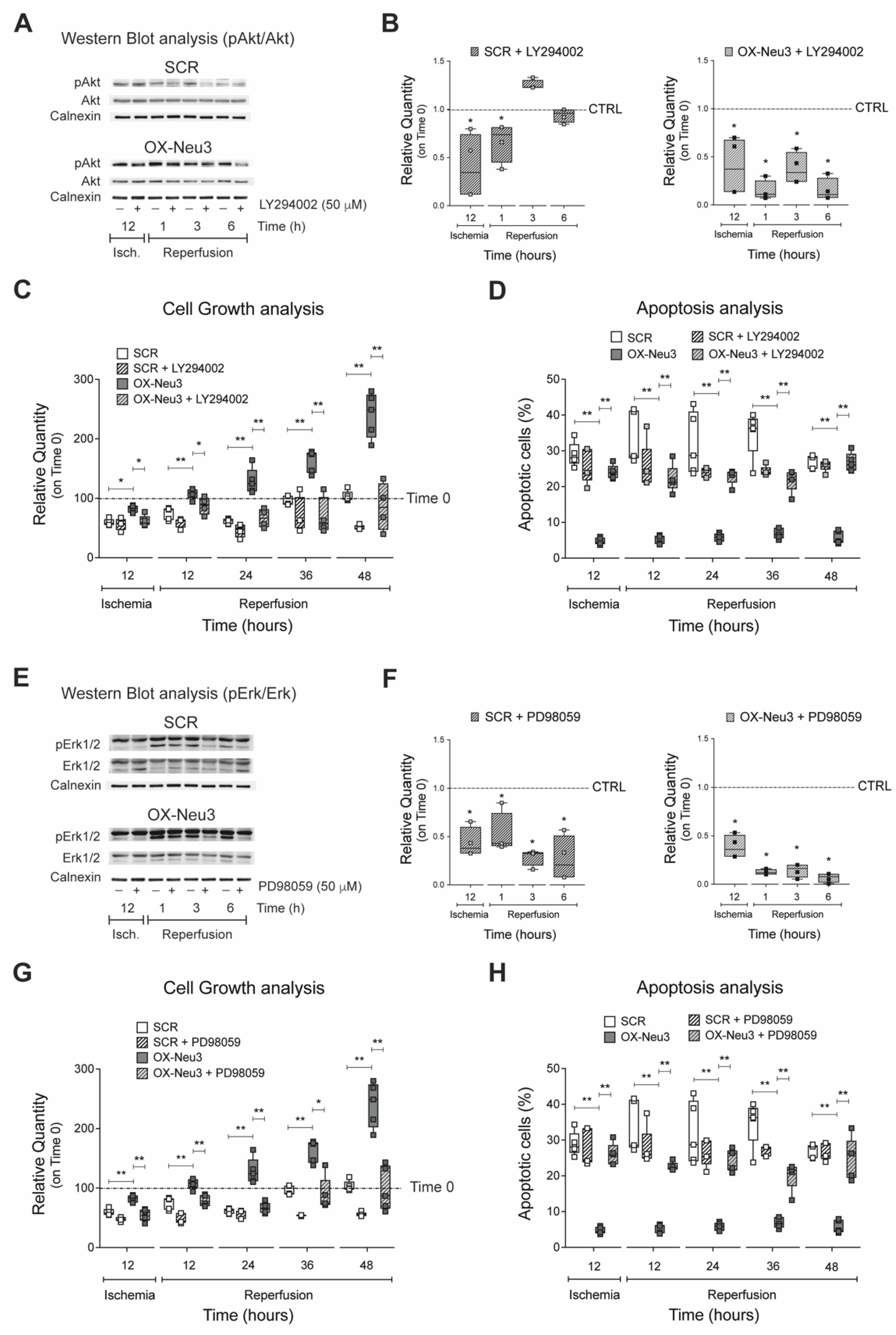
2.6. RISK Pathway Inhibition Reverts Sialidase-3 Cardioprotection
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatments
4.2. Sialidase Neu3 Stable Overexpression
4.3. RNA Extraction and Quantitative PCR (qPCR)
4.4. Sialidase Activity Assay
4.5. Cell Growth Analysis
4.6. Cytotoxicity Detection Test
4.7. Apoptosis Assay by Hoechst 33342 DNA Staining
4.8. Caspase 3/7 Activation Assay
4.9. Protein Extraction and Western Blot Analysis
4.10. Ganglioside GM3 Content Analysis
4.11. Sialidase NEU3 Activity Inhibition
4.12. RISK Pathway Inhibition
4.13. In Vivo Experiments
4.14. Left Anterior Descending (LAD) Coronary Artery Ligation
4.15. Evan’s Blue/TTC Double Staining
4.16. Echocardiography
4.17. Statistical Analysis
5. Study Limitations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, Y.; Huo, C.; Pan, T.; Li, L.; Jin, X.; Lin, X.; Chen, J.; Zhang, J.; Guo, Z.; Xu, J.; et al. Systematic review regulatory principles of non-coding RNAs in cardiovascular diseases. Brief Bioinform. 2019, 20, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Virani, S.S.; Alonso, A.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics-2020 Update: A Report From the American Heart Association. Circulation 2020, 141, e139–e596. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, E.J.; Blaha, M.J.; Chiuve, S.E.; Cushman, M.; Das, S.R.; Deo, R.; de Ferranti, S.D.; Floyd, J.; Fornage, M.; Gillespie, C.; et al. Heart Disease and Stroke Statistics-2017 Update: A Report From the American Heart Association. Circulation 2017, 135, e146–e603. [Google Scholar] [CrossRef] [PubMed]
- Reddy, K.; Khaliq, A.; Henning, R.J. Recent advances in the diagnosis and treatment of acute myocardial infarction. World J. Cardiol. 2015, 7, 243–276. [Google Scholar] [CrossRef]
- Roule, V.; Ardouin, P.; Blanchart, K.; Lemaitre, A.; Wain-Hobson, J.; Legallois, D.; Alexandre, J.; Sabatier, R.; Milliez, P.; Beygui, F. Prehospital fibrinolysis versus primary percutaneous coronary intervention in ST-elevation myocardial infarction: A systematic review and meta-analysis of randomized controlled trials. Crit. Care 2016, 20, 359. [Google Scholar] [CrossRef] [Green Version]
- Heusch, G. Myocardial ischaemia-reperfusion injury and cardioprotection in perspective. Nat. Rev. Cardiol. 2020, 17, 773–789. [Google Scholar] [CrossRef]
- Ferdinandy, P.; Schulz, R.; Baxter, G.F. Interaction of cardiovascular risk factors with myocardial ischemia/reperfusion injury, preconditioning, and postconditioning. Pharmacol. Rev. 2007, 59, 418–458. [Google Scholar] [CrossRef]
- Wu, M.Y.; Yiang, G.T.; Liao, W.T.; Tsai, A.P.; Cheng, Y.L.; Cheng, P.W.; Li, C.Y.; Li, C.J. Current Mechanistic Concepts in Ischemia and Reperfusion Injury. Cell. Physiol. Biochem. 2018, 46, 1650–1667. [Google Scholar] [CrossRef]
- Murry, C.E.; Jennings, R.B.; Reimer, K.A. Preconditioning with ischemia: A delay of lethal cell injury in ischemic myocardium. Circulation 1986, 74, 1124–1136. [Google Scholar] [CrossRef] [Green Version]
- Na, H.S.; Kim, Y.I.; Yoon, Y.W.; Han, H.C.; Nahm, S.H.; Hong, S.K. Ventricular premature beat-driven intermittent restoration of coronary blood flow reduces the incidence of reperfusion-induced ventricular fibrillation in a cat model of regional ischemia. Am. Heart J. 1996, 132, 78–83. [Google Scholar] [CrossRef]
- Zhao, Z.Q.; Corvera, J.S.; Halkos, M.E.; Kerendi, F.; Wang, N.P.; Guyton, R.A.; Vinten-Johansen, J. Inhibition of myocardial injury by ischemic postconditioning during reperfusion: Comparison with ischemic preconditioning. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H579–H588. [Google Scholar] [CrossRef] [PubMed]
- Przyklenk, K.; Bauer, B.; Ovize, M.; Kloner, R.A.; Whittaker, P. Regional ischemic ‘preconditioning’ protects remote virgin myocardium from subsequent sustained coronary occlusion. Circulation 1993, 87, 893–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yellon, D.M.; Hausenloy, D.J. Realizing the clinical potential of ischemic preconditioning and postconditioning. Nat. Clin. Pract. Cardiovasc. Med. 2005, 2, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Bonezzi, F.; Piccoli, M.; Dei Cas, M.; Paroni, R.; Mingione, A.; Monasky, M.M.; Caretti, A.; Riganti, C.; Ghidoni, R.; Pappone, C.; et al. Sphingolipid Synthesis Inhibition by Myriocin Administration Enhances Lipid Consumption and Ameliorates Lipid Response to Myocardial Ischemia Reperfusion Injury. Front. Physiol. 2019, 10, 986. [Google Scholar] [CrossRef] [Green Version]
- Jayawardena, E.; Medzikovic, L.; Ruffenach, G.; Eghbali, M. Role of miRNA-1 and miRNA-21 in Acute Myocardial Ischemia-Reperfusion Injury and Their Potential as Therapeutic Strategy. Int. J. Mol. Sci. 2022, 23, 1512. [Google Scholar] [CrossRef]
- Yu, Y.W.; Que, J.Q.; Liu, S.; Huang, K.Y.; Qian, L.; Weng, Y.B.; Rong, F.N.; Wang, L.; Zhou, Y.Y.; Xue, Y.J.; et al. Sodium-Glucose Co-transporter-2 Inhibitor of Dapagliflozin Attenuates Myocardial Ischemia/Reperfusion Injury by Limiting NLRP3 Inflammasome Activation and Modulating Autophagy. Front. Cardiovasc. Med. 2021, 8, 768214. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Yellon, D.M. Reperfusion injury salvage kinase signalling: Taking a RISK for cardioprotection. Heart Fail. Rev. 2007, 12, 217–234. [Google Scholar] [CrossRef]
- Heusch, G. Molecular basis of cardioprotection: Signal transduction in ischemic pre-, post-, and remote conditioning. Circ. Res. 2015, 116, 674–699. [Google Scholar] [CrossRef]
- Lecour, S. Activation of the protective Survivor Activating Factor Enhancement (SAFE) pathway against reperfusion injury: Does it go beyond the RISK pathway? J. Mol. Cell. Cardiol. 2009, 47, 32–40. [Google Scholar] [CrossRef]
- Schulman, D.; Latchman, D.S.; Yellon, D.M. Urocortin protects the heart from reperfusion injury via upregulation of p42/p44 MAPK signaling pathway. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H1481–H1488. [Google Scholar] [CrossRef] [Green Version]
- Caccioppo, A.; Franchin, L.; Grosso, A.; Angelini, F.; D’Ascenzo, F.; Brizzi, M.F. Ischemia Reperfusion Injury: Mechanisms of Damage/Protection and Novel Strategies for Cardiac Recovery/Regeneration. Int. J. Mol. Sci. 2019, 20, 5024. [Google Scholar] [CrossRef] [Green Version]
- Halestrap, A.P.; Clarke, S.J.; Javadov, S.A. Mitochondrial permeability transition pore opening during myocardial reperfusion—A target for cardioprotection. Cardiovasc. Res. 2004, 61, 372–385. [Google Scholar] [CrossRef] [Green Version]
- Tahrir, F.G.; Langford, D.; Amini, S.; Mohseni Ahooyi, T.; Khalili, K. Mitochondrial quality control in cardiac cells: Mechanisms and role in cardiac cell injury and disease. J. Cell. Physiol. 2019, 234, 8122–8133. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.G.; Lee, W.H.; Theodorou, L.; Kodo, K.; Lim, S.Y.; Shukla, D.H.; Briston, T.; Kiriakidis, S.; Ashcroft, M.; Davidson, S.M.; et al. HIF-1 reduces ischaemia-reperfusion injury in the heart by targeting the mitochondrial permeability transition pore. Cardiovasc. Res. 2014, 104, 24–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, J.; Chen, P.; Zhong, J.; Cheng, Y.; Chen, H.; He, Y.; Chen, C. HIF1alpha in myocardial ischemiareperfusion injury (Review). Mol. Med. Rep. 2021, 23, 352. [Google Scholar] [CrossRef] [PubMed]
- Scaringi, R.; Piccoli, M.; Papini, N.; Cirillo, F.; Conforti, E.; Bergante, S.; Tringali, C.; Garatti, A.; Gelfi, C.; Venerando, B.; et al. NEU3 sialidase is activated under hypoxia and protects skeletal muscle cells from apoptosis through the activation of the epidermal growth factor receptor signaling pathway and the hypoxia-inducible factor (HIF)-1alpha. J. Biol. Chem. 2013, 288, 3153–3162. [Google Scholar] [CrossRef] [Green Version]
- Ghiroldi, A.; Piccoli, M.; Creo, P.; Cirillo, F.; Rota, P.; D’Imperio, S.; Ciconte, G.; Monasky, M.M.; Micaglio, E.; Garatti, A.; et al. Role of sialidase Neu3 and ganglioside GM3 in cardiac fibroblasts activation. Biochem. J. 2020, 477, 3401–3415. [Google Scholar] [CrossRef]
- Guo, T.; Datwyler, P.; Demina, E.; Richards, M.R.; Ge, P.; Zou, C.; Zheng, R.; Fougerat, A.; Pshezhetsky, A.V.; Ernst, B.; et al. Selective Inhibitors of Human Neuraminidase 3. J. Biol. Chem. 2008, 61, 1990–2008. [Google Scholar] [CrossRef]
- Rossello, X.; Yellon, D.M. The RISK pathway and beyond. Basic Res. Cardiol. 2018, 113, 2. [Google Scholar] [CrossRef] [Green Version]
- Haq, S.; Choukroun, G.; Lim, H.; Tymitz, K.M.; del Monte, F.; Gwathmey, J.; Grazette, L.; Michael, A.; Hajjar, R.; Force, T.; et al. Differential activation of signal transduction pathways in human hearts with hypertrophy versus advanced heart failure. Circulation 2001, 103, 670–677. [Google Scholar] [CrossRef]
- Nagoshi, T.; Matsui, T.; Aoyama, T.; Leri, A.; Anversa, P.; Li, L.; Ogawa, W.; del Monte, F.; Gwathmey, J.K.; Grazette, L.; et al. PI3K rescues the detrimental effects of chronic Akt activation in the heart during ischemia/reperfusion injury. J. Clin. Investig. 2005, 115, 2128–2138. [Google Scholar] [CrossRef] [PubMed]
- Hata, K.; Tochigi, T.; Sato, I.; Kawamura, S.; Shiozaki, K.; Wada, T.; Takahashi, K.; Moriya, S.; Yamaguchi, K.; Hosono, M.; et al. Increased sialidase activity in serum of cancer patients: Identification of sialidase and inhibitor activities in human serum. Cancer Sci. 2015, 106, 383–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Proshin, S.; Yamaguchi, K.; Yamashita, Y.; Katakura, R.; Yamamoto, K.; Shima, H.; Hosono, M.; Miyagi, T. Sialidase NEU3 defines invasive potential of human glioblastoma cells by regulating calpain-mediated proteolysis of focal adhesion proteins. BBA-Gen. Subj. 2017, 1861, 2778–2788. [Google Scholar] [CrossRef] [PubMed]
- Tringali, C.; Silvestri, I.; Testa, F.; Baldassari, P.; Anastasia, L.; Mortarini, R.; Anichini, A.; Lopez-Requena, A.; Tettamanti, G.; Venerando, B. Molecular subtyping of metastatic melanoma based on cell ganglioside metabolism profiles. BMC Cancer 2014, 14, 560. [Google Scholar] [CrossRef] [Green Version]
- Anastasia, L.; Papini, N.; Colazzo, F.; Palazzolo, G.; Tringali, C.; Dileo, L.; Piccoli, M.; Conforti, E.; Sitzia, C.; Monti, E.; et al. NEU3 sialidase strictly modulates GM3 levels in skeletal myoblasts C2C12 thus favoring their differentiation and protecting them from apoptosis. J. Biol. Chem. 2008, 283, 36265–36271. [Google Scholar] [CrossRef] [Green Version]
- Piccoli, M.; Conforti, E.; Varrica, A.; Ghiroldi, A.; Cirillo, F.; Resmini, G.; Pluchinotta, F.; Tettamanti, G.; Giamberti, A.; Frigiola, A.; et al. NEU3 sialidase role in activating HIF-1alpha in response to chronic hypoxia in cyanotic congenital heart patients. Int. J. Cardiol. 2017, 230, 6–13. [Google Scholar] [CrossRef]
- Kierans, S.J.; Taylor, C.T. Regulation of glycolysis by the hypoxia-inducible factor (HIF): Implications for cellular physiology. J. Physiol. 2021, 599, 23–37. [Google Scholar] [CrossRef]
- Hargreaves, M.; Spriet, L.L. Author Correction: Skeletal muscle energy metabolism during exercise. Nat. Metab. 2020, 2, 990. [Google Scholar] [CrossRef]
- Cai, Z.Q.; Manalo, D.J.; Wei, G.; Rodriguez, E.R.; Fox-Talbot, K.; Lu, H.S.; Zweier, J.L.; Semenza, G.L. Hearts from rodents exposed to intermittent hypoxia or erythropoietin are protected against ischemia-reperfusion injury. Circulation 2003, 108, 79–85. [Google Scholar] [CrossRef] [Green Version]
- Ockaili, R.; Natarajan, R.; Salloum, F.; Fisher, B.J.; Jones, D.; Fowler, A.A.; Kukreja, R.C. HIF-1 activation attenuates postischemic myocardial injury: Role for heme oxygenase-1 in modulating microvascular chemokine generation. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H542–H548. [Google Scholar] [CrossRef] [Green Version]
- Natarajan, R.; Salloum, F.N.; Fisher, B.J.; Kukreja, R.C.; Fowler, A.A. Hypoxia inducible factor-1 activation by prolyl 4-hydroxylase-2 gene silencing attenuates myocardial ischemia reperfusion injury. Circ. Res. 2006, 98, 133–140. [Google Scholar] [CrossRef] [Green Version]
- Ong, S.G.; Hausenloy, D.J. Hypoxia-inducible factor as a therapeutic target for cardioprotection. Pharmacol. Therapeut. 2012, 136, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, R.D.; Banik, A.; Mandal, B.; Sarkar, S. Cardiac-specific overexpression of HIF-1 alpha during acute myocardial infarction ameliorates cardiomyocyte apoptosis via differential regulation of hypoxia-inducible pro-apoptotic and anti-oxidative genes. Biochem. Biophys. Res. Commun. 2021, 537, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.A.; Liu, D.W.; Hu, H.J.; Zhang, P.Q.; Xie, R.Q.; Cui, W. HIF-1 alpha/BNIP3 signaling pathway-induced-autophagy plays protective role during myocardial ischemia-reperfusion injury. Biomed. Pharmacother. 2019, 120, 109464. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, R.D.; Banerjee, D.; Banik, A.; Sarkar, S. Severity and duration of hypoxic stress differentially regulates HIF-1 alpha-mediated cardiomyocyte apoptotic signaling milieu during myocardial infarction. Arch. Biochem. Biophys. 2020, 690, 108430. [Google Scholar] [CrossRef]
- Cho, S.; Cho, M.; Kim, J.; Kaeberlein, M.; Lee, S.J.; Suh, Y. Syringaresinol protects against hypoxia/reoxygenation-induced cardiomyocytes injury and death by destabilization of HIF-1 alpha in a FOXO3-dependent mechanism. Oncotarget 2015, 6, 43–55. [Google Scholar] [CrossRef]
- Malhotra, R.; Tyson, D.W.; Rosevear, H.M.; Brosius, F.C. Hypoxia-inducible factor-1 alpha is a critical mediator of hypoxia induced apoptosis in cardiac H9c2 and kidney epithelial HK-2 cells. BMC Cardiovasc. Disord. 2008, 8, 9. [Google Scholar] [CrossRef] [Green Version]
- Thu, V.T.; Kim, H.K. Majonoside-R2 Postconditioning Protects Cardiomyocytes Against Hypoxia/Reoxygenation Injury by Attenuating the Expression of HIF1 alpha and Activating RISK Pathway. J. Med. Food 2021, 24, 1222–1229. [Google Scholar] [CrossRef]
- Rota, P.; Cirillo, F.; Piccoli, M.; Gregorio, A.; Tettamanti, G.; Allevi, P.; Anastasia, L. Synthesis and Biological Evaluation of Several Dephosphonated Analogues of CMP-Neu5Ac as Inhibitors of GM3-Synthase. Chem.-Eur. J. 2015, 21, 14614–14629. [Google Scholar] [CrossRef]
- Cirillo, F.; Ghiroldi, A.; Fania, C.; Piccoli, M.; Torretta, E.; Tettamanti, G.; Gelfi, C.; Anastasia, L. NEU3 Sialidase Protein Interactors in the Plasma Membrane and in the Endosomes. J. Biol. Chem. 2016, 291, 10615–10624. [Google Scholar] [CrossRef] [Green Version]
- Reforgiato, M.R.; Milano, G.; Fabrias, G.; Casas, J.; Gasco, P.; Paroni, R.; Samaja, M.; Ghidoni, R.; Caretti, A.; Signorelli, P. Inhibition of ceramide de novo synthesis as a postischemic strategy to reduce myocardial reperfusion injury. Basic Res. Cardiol. 2016, 111, 12. [Google Scholar] [CrossRef] [PubMed]
- Palmisano, A.; Piccoli, M.; Monti, C.B.; Canu, T.; Cirillo, F.; Napolitano, A.; Perani, L.; Signorelli, P.; Vignale, D.; Anastasia, L.; et al. Single-shot morpho-functional and structural characterization of the left-ventricle in a mouse model of acute ischemia-reperfusion injury with an optimized 3D IntraGate cine FLASH sequence at 7T MR. Magn. Reson. Imaging 2020, 68, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Ling, X.; Zhou, H.; Zhang, C. Dexmedetomidine at a dose of 1 microM attenuates H9c2 cardiomyocyte injury under 3 h of hypoxia exposure and 3 h of reoxygenation through the inhibition of endoplasmic reticulum stress. Exp. Ther. Med. 2021, 21, 132. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, A.V.; Javadov, S.; Sickinger, S.; Frotschnig, S.; Grimm, M. H9c2 and HL-1 cells demonstrate distinct features of energy metabolism, mitochondrial function and sensitivity to hypoxia-reoxygenation. Biochim. Biophys. Acta 2015, 1853, 276–284. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Li, X.; Zhang, F.; Zhao, B.; Du, W.; Sun, D.; Li, G. Protective effect of nicorandil against myocardial ischemia/reperfusion injury mediated via IL33/ST2 signaling pathway. Mol. Cell. Biochem. 2022. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| Rat Neu3 | 5′-ATGCCCTCTGATGGACAGAT-3′ | 5′-CATGTCCCTGATGGTGCTC-3′ |
| Rat Rpl13a | 5′-TCTCCGAAAGCGGATGAACAC-3′ | 5′-CAACACCTTGAGGCGTTCCA-3′ |
| Mouse Neu3 | 5′-TGCGTGTTCAGTCAAGCC-3′ | 5′-GCAGTAGAGCACAGGGTTAC-3′ |
| Mouse Rpl13a | 5′-CTCGGCCGTTCCTGTAT-3′ | 5′-GTGGAAGTGGGGCTTCAGTA-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piccoli, M.; Coviello, S.; Canali, M.E.; Rota, P.; La Rocca, P.; Cirillo, F.; Lavota, I.; Tarantino, A.; Ciconte, G.; Pappone, C.; et al. Neu3 Sialidase Activates the RISK Cardioprotective Signaling Pathway during Ischemia and Reperfusion Injury (IRI). Int. J. Mol. Sci. 2022, 23, 6090. https://doi.org/10.3390/ijms23116090
Piccoli M, Coviello S, Canali ME, Rota P, La Rocca P, Cirillo F, Lavota I, Tarantino A, Ciconte G, Pappone C, et al. Neu3 Sialidase Activates the RISK Cardioprotective Signaling Pathway during Ischemia and Reperfusion Injury (IRI). International Journal of Molecular Sciences. 2022; 23(11):6090. https://doi.org/10.3390/ijms23116090
Chicago/Turabian StylePiccoli, Marco, Simona Coviello, Maria Elena Canali, Paola Rota, Paolo La Rocca, Federica Cirillo, Ivana Lavota, Adriana Tarantino, Giuseppe Ciconte, Carlo Pappone, and et al. 2022. "Neu3 Sialidase Activates the RISK Cardioprotective Signaling Pathway during Ischemia and Reperfusion Injury (IRI)" International Journal of Molecular Sciences 23, no. 11: 6090. https://doi.org/10.3390/ijms23116090
APA StylePiccoli, M., Coviello, S., Canali, M. E., Rota, P., La Rocca, P., Cirillo, F., Lavota, I., Tarantino, A., Ciconte, G., Pappone, C., Ghiroldi, A., & Anastasia, L. (2022). Neu3 Sialidase Activates the RISK Cardioprotective Signaling Pathway during Ischemia and Reperfusion Injury (IRI). International Journal of Molecular Sciences, 23(11), 6090. https://doi.org/10.3390/ijms23116090