
The Endothelium and COVID-19: An Increasingly Clear Link Brief Title: Endotheliopathy in COVID-19

 , and
, and
Abstract
:1. Introduction
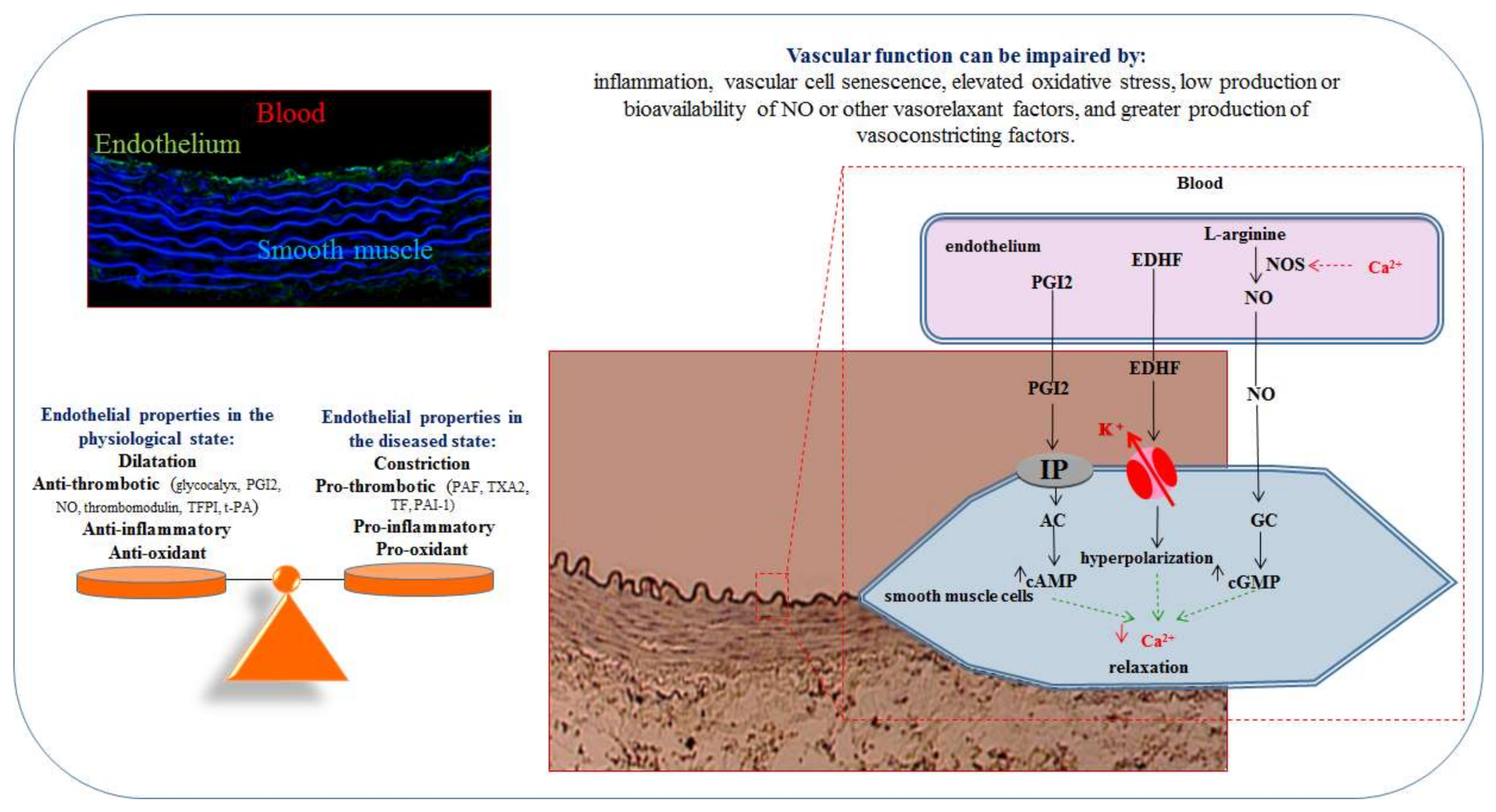
2. The Endothelium
2.1. Endothelial Cells
2.2. Endothelial Dysfunction
2.3. Endothelial Extracellular Vesicles
3. Endothelial Cells and Viruses
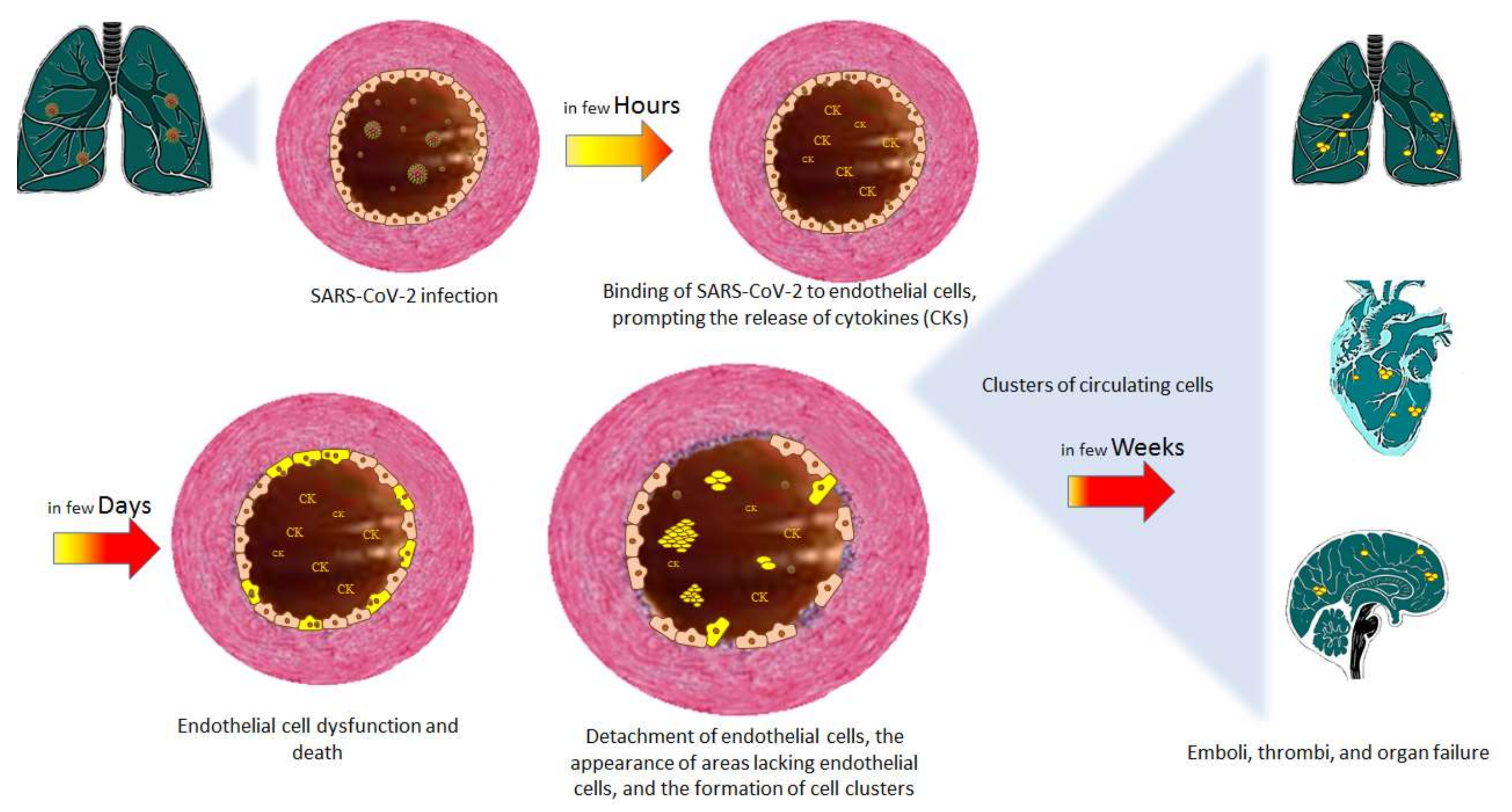
4. The Endothelium and COVID-19
4.1. Endothelial Dysfunction in COVID-19
4.2. Endothelial Extracellular Vesicles in COVID-19
5. Therapeutic Management of Patients with COVID-19
5.1. Antiviral Compounds
5.2. Anti-SARS-CoV-2 Monoclonal Antibodies
6. Treatments That Target Endothelial Dysfunction
7. COVID-19 Treatments That Target the Endothelium or Endothelial Extracellular Vesicles
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| COVID-19 | coronavirus disease 2019 |
| SARS-CoV-2 | severe acute respiratory syndrome coronavirus 2 |
| ACE-2 | angiotensin-converting enzyme 2 |
| NO | nitric oxide |
| NOS | nitric oxide synthase |
| eNOS | endothelial NOS |
| ROS | reactive oxygen species |
| EDHF | endothelium-derived hyperpolarizing factor |
| Ang II | angiotensin II |
| AT1 | angiotensin type 1 |
| EV | extracellular vesicle |
| miRNA | micro RNA |
| SARS-CoV-1 | severe acute respiratory syndrome coronavirus 1 |
| MERS-CoV | Middle East respiratory syndrome coronavirus |
| IL | interleukin |
| TNF-α | tumor necrosis factor alpha |
| TF | tissue factor |
| vWF | von Willebrand factor |
| ICU | intensive care unit |
| PCR | Polymerase chain reaction |
| ACEI | angiotensin-converting enzyme inhibitor |
| ARB | angiotensin II receptor blocker |
References
- Hamming, I.; Timens, W.; Bulthuis, M.L.; Lely, A.T.; Navis, G.; van Goor, H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef] [PubMed]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Chappey, O.; Wautier, M.P.; Boval, B.; Wautier, J.L. Endothelial cells in culture: An experimental model for the study of vascular dysfunctions. Cell. Biol. Toxicol. 1996, 12, 199–205. [Google Scholar] [CrossRef]
- Ryan, U.S.; Ryan, J.W. The ultrastructural basis of endothelial cell surface functions. Biorheology 1984, 21, 155–170. [Google Scholar] [CrossRef]
- Cooke, J.P. Does ADMA cause endothelial dysfunction? Arterioscler. Thromb. Vasc. Biol. 2000, 20, 2032–2037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boerrigter, G.; Burnett, J.C., Jr. Soluble guanylate cyclase: Not a dull enzyme. Circulation 2009, 119, 2752–2754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, M.H.; Cohen, R.; Ullrich, V. Peroxynitrite and vascular endothelial dysfunction in diabetes mellitus. Endothelium 2004, 11, 89–97. [Google Scholar] [CrossRef]
- Münzel, T.; Daiber, A.; Ullrich, V.; Mülsch, A. Vascular consequences of endothelial nitric oxide synthase uncoupling for the activity and expression of the soluble guanylyl cyclase and the cGMP-dependent protein kinase. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1551–1557. [Google Scholar] [CrossRef]
- Zou, M.H.; Shi, C.; Cohen, R.A. Oxidation of the zinc-thiolate complex and uncoupling of endothelial nitric oxide synthase by peroxynitrite. J. Clin. Investig. 2002, 109, 817–826. [Google Scholar] [CrossRef]
- Zweier, J.L.; Chen, C.A.; Druhan, L.J. S-glutathionylation reshapes our understanding of endothelial nitric oxide synthase uncoupling and nitric oxide/reactive oxygen species-mediated signaling. Antioxid. Redox Signal. 2011, 14, 1769–1775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antoniades, C.; Shirodaria, C.; Crabtree, M.; Rinze, R.; Alp, N.; Cunnington, C.; Diesch, J.; Tousoulis, D.; Stefanadis, C.; Leeson, P.; et al. Altered plasma versus vascular biopterins in human atherosclerosis reveal relationships between endothelial nitric oxide synthase coupling, endothelial function, and inflammation. Circulation 2007, 116, 2851–2859. [Google Scholar] [CrossRef] [Green Version]
- Kusama, N.; Kajikuri, J.; Yamamoto, T.; Watanabe, Y.; Suzuki, Y.; Katsuya, H.; Itoh, T. Reduced hyperpolarization in endothelial cells of rabbit aortic valve following chronic nitroglycerine administration. Br. J. Pharmacol. 2005, 146, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Griffith, T.M.; Chaytor, A.T.; Bakker, L.M.; Edwards, D.H. 5-Methyltetrahydrofolate and tetrahydrobiopterin can modulate electrotonically mediated endothelium-dependent vascular relaxation. Proc. Natl. Acad. Sci. USA 2005, 102, 12644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanhoutte, P.M.; Feletou, M.; Taddei, S. Endothelium-dependent contractions in hypertension. Br. J. Pharmacol. 2005, 144, 449–458. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Li, W.; Liu, W.; Altura, B.T.; Altura, B.M. Mechanisms of hydroxyl radical-induced contraction of rat aorta. Eur. J. Pharmacol. 2004, 499, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Morrow, J.D.; Awad, J.A.; Boss, H.J.; Blair, I.A.; Roberts, L.J., 2nd. Non-cyclooxygenase-derived prostanoids (F2-isoprostanes) are formed in situ on phospholipids. Proc. Natl. Acad. Sci. USA 1992, 89, 10721–10725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffman, S.W.; Moore, S.; Ellis, E.F. Isoprostanes: Free radical-generated prostaglandins with constrictor effects on cerebral arterioles. Stroke 1997, 28, 844–849. [Google Scholar] [CrossRef]
- Janssen, L.J.; Premji, M.; Netherton, S.; Catalli, A.; Cox, G.; Keshavjee, S.; Crankshaw, D.J. Excitatory and inhibitory actions of isoprostanes in human and canine airway smooth muscle. J. Pharmacol. Exp. Ther. 2000, 295, 506–511. [Google Scholar]
- Six, I.; Maizel, J.; Barreto, F.C.; Rangrez, A.Y.; Dupont, S.; Slama, M.; Tribouilloy, C.; Choukroun, G.; Mazière, J.C.; Bode-Boeger, S.; et al. Effects of phosphate on vascular function under normal conditions and influence of the uraemic state. Cardiovasc. Res. 2012, 96, 130–139. [Google Scholar] [CrossRef] [Green Version]
- Waqas, M.Y.; Javid, M.A.; Nazir, M.M.; Niaz, N.; Nisar, M.F.; Manzoor, Z.; Bhatti, S.A.; Hameed, S.; Khaliq, M.H. Extracellular vesicles and exosome: Insight from physiological regulatory perspectives. J. Physiol. Biochem. 2022, 2022, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Zhang, Y.; Li, Y.; Luo, L.; Zhao, Y.; Yao, Y. Extracellular vesicles in cardiovascular diseases. Cell Death Discov. 2020, 6, 68. [Google Scholar] [CrossRef] [PubMed]
- Dignat-George, F.; Boulanger, C.M. The many faces of endothelial microparticles. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 27–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenkins, N.T.; Padilla, J.; Boyle, L.J.; Credeur, D.P.; Laughlin, M.H.; Fadel, P.J. Disturbed blood flow acutely induces activation and apoptosis of the human vascular endothelium. Hypertension 2013, 61, 615–621. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.S.; Kim, B.; Lee, H.; Thakkar, S.; Babbitt, D.M.; Eguchi, S.; Brown, M.D.; Park, J.Y. Shear stress-induced mitochondrial biogenesis decreases the release of microparticles from endothelial cells. Am. J. Physiol. Heart. Circ. Physiol. 2015, 309, H425–H433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pichler Hefti, J.; Leichtle, A.; Stutz, M.; Hefti, U.; Geiser, T.; Huber, A.R.; Merz, T.M. Increased endothelial microparticles and oxidative stress at extreme altitude. Eur. J. Appl. Physiol. 2016, 116, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Hosseinkhani, B.; Kuypers, S.; van den Akker, N.M.S.; Molin, D.G.M.; Michiels, L. Extracellular Vesicles Work as a Functional Inflammatory Mediator Between Vascular Endothelial Cells and Immune Cells. Front. Immunol. 2018, 9, 1789. [Google Scholar] [CrossRef] [Green Version]
- Jansen, F.; Yang, X.; Franklin, B.S.; Hoelscher, M.; Schmitz, T.; Bedorf, J.; Nickenig, G.; Werner, N. High glucose condition increases NADPH oxidase activity in endothelial microparticles that promote vascular inflammation. Cardiovasc. Res. 2013, 98, 94–106. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, S.; Niida, S.; Azuma, E.; Yanagibashi, T.; Muramatsu, M.; Huang, T.T.; Sagara, H.; Higaki, S.; Ikutani, M.; Nagai, Y.; et al. Inflammation-induced endothelial cell-derived extracellular vesicles modulate the cellular status of pericytes. Sci. Rep. 2015, 5, 8505. [Google Scholar] [CrossRef]
- Bodega, G.; Alique, M.; Bohórquez, L.; Ciordia, S.; Mena, M.C.; Ramírez, M.R. The Antioxidant Machinery of Young and Senescent Human Umbilical Vein Endothelial Cells and Their Microvesicles. Oxid. Med. Cell. Longev. 2017, 2017, 7094781. [Google Scholar] [CrossRef]
- Saeed-Zidane, M.; Linden, L.; Salilew-Wondim, D.; Held, E.; Neuhoff, C.; Tholen, E.; Hoelker, M.; Schellander, K.; Tesfaye, D. Cellular and exosome mediated molecular defense mechanism in bovine granulosa cells exposed to oxidative stress. PLoS ONE 2017, 12, e0187569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janiszewski, M.; Do Carmo, A.O.; Pedro, M.A.; Silva, E.; Knobel, E.; Laurindo, F.R. Platelet-derived exosomes of septic individuals possess proapoptotic NAD(P)H oxidase activity: A novel vascular redox pathway. Crit. Care Med. 2004, 32, 818–825. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.; Hui, D.; Wu, A.; Chan, P.; Cameron, P.; Joynt, G.M.; Ahuja, A.; Yung, M.Y.; Leung, C.B.; To, K.F.; et al. A major outbreak of severe acute respiratory syndrome in Hong Kong. N. Engl. J. Med. 2003, 348, 1986–1994. [Google Scholar] [CrossRef] [PubMed]
- To, K.F.; Tong, J.H.; Chan, P.K.; Au, F.W.; Chim, S.S.; Chan, K.C.; Cheung, J.L.; Liu, E.Y.; Tse, G.M.; Lo, A.W.; et al. Tissue and cellular tropism of the coronavirus associated with severe acute respiratory syndrome: An in-situ hybridization study of fatal cases. J. Pathol. 2004, 202, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Hui, D.S.C.; Zumla, A. Severe Acute Respiratory Syndrome: Historical, Epidemiologic, and Clinical Features. Infect. Dis. Clin. N. Am. 2019, 33, 869–889. [Google Scholar] [CrossRef]
- Azhar, E.I.; Hui, D.S.C.; Memish, Z.A.; Drosten, C.; Zumla, A. The Middle East Respiratory Syndrome (MERS). Infect. Dis. Clin. N. Am. 2019, 33, 891–905. [Google Scholar] [CrossRef]
- Capaldo, C.T.; Nusrat, A. Cytokine regulation of tight junctions. Biochim. Biophys. Acta. 2009, 1788, 864–871. [Google Scholar] [CrossRef] [Green Version]
- Desai, T.R.; Leeper, N.J.; Hynes, K.L.; Gewertz, B.L. Interleukin-6 causes endothelial barrier dysfunction via the protein kinase C pathway. J. Surg. Res. 2002, 104, 118–123. [Google Scholar] [CrossRef]
- Cozzolino, F.; Torcia, M.; Aldinucci, D.; Ziche, M.; Almerigogna, F.; Bani, D.; Stern, D.M. Interleukin 1 is an autocrine regulator of human endothelial cell growth. Proc. Natl. Acad. Sci. USA 1990, 87, 6487–6491. [Google Scholar] [CrossRef] [Green Version]
- Aoki, N.; Siegfried, M.; Lefer, A.M. Anti-EDRF effect of tumor necrosis factor in isolated, perfused cat carotid arteries. Am. J. Physiol. 1989, 256, H1509–H1512. [Google Scholar] [CrossRef] [PubMed]
- Yoshizumi, M.; Perrella, M.A.; Burnett, J.C., Jr.; Lee, M.E. Tumor necrosis factor downregulates an endothelial nitric oxide synthase mRNA by shortening its half-life. Circ. Res. 1993, 73, 205–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scalera, F.; Borlak, J.; Beckmann, B.; Martens-Lobenhoffer, J.; Thum, T.; Täger, M.; Bode-Böger, S.M. Endogenous nitric oxide synthesis inhibitor asymmetric dimethyl L-arginine accelerates endothelial cell senescence. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1816–1822. [Google Scholar] [CrossRef] [Green Version]
- Shiraki, A.; Oyama, J.; Komoda, H.; Asaka, M.; Komatsu, A.; Sakuma, M.; Kodama, K.; Sakamoto, Y.; Kotooka, N.; Hirase, T.; et al. The glucagon-like peptide 1 analog liraglutide reduces TNF-α-induced oxidative stress and inflammation in endothelial cells. Atherosclerosis 2012, 221, 375–382. [Google Scholar] [CrossRef]
- Wassmann, S.; Stumpf, M.; Strehlow, K.; Schmid, A.; Schieffer, B.; Böhm, M.; Nickenig, G. Interleukin-6 induces oxidative stress and endothelial dysfunction by overexpression of the angiotensin II type 1 receptor. Circ. Res. 2004, 94, 534–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bombeli, T.; Karsan, A.; Tait, J.F.; Harlan, J.M. Apoptotic vascular endothelial cells become procoagulant. Blood 1997, 89, 2429–2442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bikdeli, B.; Madhavan, M.V.; Jimenez, D.; Chuich, T.; Dreyfus, I.; Driggin, E.; Nigoghossian, C.; Ageno, W.; Madjid, M.; Guo, Y.; et al. COVID-19 and Thrombotic or Thromboembolic Disease: Implications for Prevention, Antithrombotic Therapy, and Follow-Up. J. Am. Coll. Cardiol. 2020, 75, 2950–2973. [Google Scholar] [CrossRef] [PubMed]
- Guan, W.J.; Ni, Z.Y.; Hu, Y.; Liang, W.H.; Ou, C.Q.; He, J.X.; Liu, L.; Shan, H.; Lei, C.L.; Hui, D.S.C.; et al. China Medical Treatment Expert Group for Covid-19. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in COVID-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef]
- Goshua, G.; Pine, A.B.; Meizlish, M.L.; Chang, C.H.; Zhang, H.; Bahel, P.; Baluha, A.; Bar, N.; Bona, R.D.; Burns, A.J.; et al. Endotheliopathy in COVID-19-associated coagulopathy: Evidence from a single-centre, cross-sectional study. Lancet Haematol. 2020, 7, e575–e582. [Google Scholar] [CrossRef]
- Pine, A.B.; Meizlish, M.L.; Goshua, G.; Chang, C.H.; Zhang, H.; Bishai, J.; Bahel, P.; Patel, A.; Gbyli, R.; Kwan, J.M.; et al. Circulating markers of angiogenesis and endotheliopathy in COVID-19. Pulm. Circ. 2020, 10, 1–4. [Google Scholar] [CrossRef]
- Cimino, G.; Vizzardi, E.; Calvi, E.; Pancaldi, E.; Pascariello, G.; Bernardi, N.; Cersosimo, A.; Amore, L.; Inciardi, R.M.; Raddino, R.; et al. Endothelial dysfunction in COVID-19 patients assessed with Endo-PAT2000. Monaldi Arch. Chest. Dis. 2022. [Google Scholar] [CrossRef]
- Fraser, D.D.; Patterson, E.K.; Slessarev, M.; Gill, S.E.; Martin, C.; Daley, M.; Miller, M.R.; Patel, M.A.; Dos Santos, C.C.; Bosma, K.J.; et al. Endothelial Injury and Glycocalyx Degradation in Critically Ill Coronavirus Disease 2019 Patients: Implications for Microvascular Platelet Aggregation. Crit. Care Explor. 2020, 24, e0194. [Google Scholar] [CrossRef] [PubMed]
- Krishnamachary, B.; Cook, C.; Spikes, L.; Chalise, P.; Dhillon, N.K. The Potential Role of Extracellular Vesicles in COVID-19 Associated Endothelial injury and Pro-inflammation. medRxiv 2020. [Google Scholar] [CrossRef]
- Rosell, A.; Havervall, S.; von Meijenfeldt, F.; Hisada, Y.; Aguilera, K.; Grover, S.P.; Lisman, T.; Mackman, N.; Thålin, C. Patients with COVID-19 Have Elevated Levels of Circulating Extracellular Vesicle Tissue Factor Activity That Is Associated with Severity and Mortality-Brief Report. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 878–882. [Google Scholar] [CrossRef] [PubMed]
- Jing, H.; Zuo, N.; Novakovic, V.A.; Shi, J. The Central Role of Extracellular Vesicles in the Mechanisms of Thrombosis in COVID-19 Patients With Cancer and Therapeutic Strategies. Front. Cell. Dev. Biol. 2022, 9, 792335. [Google Scholar] [CrossRef] [PubMed]
- Vince, R.V.; Chrismas, B.; Midgley, A.W.; McNaughton, L.R.; Madden, L.A. Hypoxia mediated release of endothelial microparticles and increased association of S100A12 with circulating neutrophils. Oxid. Med. Cell. Longev. 2009, 2, 2–6. [Google Scholar] [CrossRef] [PubMed]
- Szotowski, B.; Antoniak, S.; Goldin-Lang, P.; Tran, Q.V.; Pels, K.; Rosenthal, P.; Bogdanov, V.Y.; Borchert, H.H.; Schultheiss, H.P.; Rauch, U. Antioxidative treatment inhibits the release of thrombogenic tissue factor from irradiation- and cytokine-induced endothelial cells. Cardiovasc. Res. 2007, 73, 806–812. [Google Scholar] [CrossRef]
- Brodsky, S.V.; Zhang, F.; Nasjletti, A.; Goligorsky, M.S. Endothelium-derived microparticles impair endothelial function in vitro. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H1910–H1915. [Google Scholar] [CrossRef] [Green Version]
- Ju, R.; Zhuang, Z.W.; Zhang, J.; Lanahan, A.A.; Kyriakides, T.; Sessa, W.C.; Simons, M. Angiopoietin-2 secretion by endothelial cell exosomes: Regulation by the phosphatidylinositol 3-kinase (PI3K)/Akt/endothelial nitric oxide synthase (eNOS) and syndecan-4/syntenin pathways. J. Biol. Chem. 2014, 289, 510–519. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.M.; Wang, Y.; Huang, J.Y.; Yang, Z.; Chen, L.; Wang, L.C.; Tang, A.L.; Lou, Z.F.; Tao, J. C-Reactive protein-induced endothelial microparticle generation in HUVECs is related to BH4-dependent NO formation. J. Vasc. Res. 2007, 44, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Amabile, N.; Guérin, A.P.; Leroyer, A.; Mallat, Z.; Nguyen, C.; Boddaert, J.; London, G.M.; Tedgui, A.; Boulanger, C.M. Circulating endothelial microparticles are associated with vascular dysfunction in patients with end-stage renal failure. J. Am. Soc. Nephrol. 2005, 16, 3381–3388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rautou, P.E.; Vion, A.C.; Amabile, N.; Chironi, G.; Simon, A.; Tedgui, A.; Boulanger, C.M. Microparticles, vascular function, and atherothrombosis. Circ. Res. 2011, 109, 593–606. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, A.M.; Wilkinson, F.L.; McCarthy, E.M.; Moreno-Martinez, D.; Langford-Smith, A.; Romero, M.; Duarte, J.; Alexander, M.Y. Endothelial microparticles prevent lipid-induced endothelial damage via Akt/eNOS signaling and reduced oxidative stress. FASEB J. 2017, 31, 4636–4648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Densmore, J.C.; Signorino, P.R.; Ou, J.; Hatoum, O.A.; Rowe, J.J.; Shi, Y.; Kaul, S.; Jones, D.W.; Sabina, R.E.; Pritchard, K.A., Jr.; et al. Endothelium-derived microparticles induce endothelial dysfunction and acute lung injury. Shock 2006, 26, 464–471. [Google Scholar] [CrossRef]
- Burger, D.; Turner, M.; Munkonda, M.N.; Touyz, R.M. Endothelial Microparticle-Derived Reactive Oxygen Species: Role in Endothelial Signaling and Vascular Function. Oxid. Med. Cell. Longev. 2016, 2016, 5047954. [Google Scholar] [CrossRef] [Green Version]
- Leroyer, A.S.; Ebrahimian, T.G.; Cochain, C.; Récalde, A.; Blanc-Brude, O.; Mees, B.; Vilar, J.; Tedgui, A.; Levy, B.I.; Chimini, G.; et al. Microparticles from ischemic muscle promotes postnatal vasculogenesis. Circulation 2009, 119, 2808–2817. [Google Scholar] [CrossRef] [Green Version]
- Bodega, G.; Alique, M.; Bohórquez, L.; Morán, M.; Magro, L.; Puebla, L.; Ciordia, S.; Mena, M.C.; Arza, E.; Ramírez, M.R. Young and Especially Senescent Endothelial Microvesicles Produce NADPH: The Fuel for Their Antioxidant Machinery. Oxid. Med. Cell. Longev. 2018, 2018, 3183794. [Google Scholar] [CrossRef] [Green Version]
- Benedikter, B.J.; Weseler, A.R.; Wouters, E.F.M.; Savelkoul, P.H.M.; Rohde, G.G.U.; Stassen, F.R.M. Redox-dependent thiol modifications: Implications for the release of extracellular vesicles. Cell. Mol. Life Sci. 2018, 75, 2321–2337. [Google Scholar] [CrossRef] [Green Version]
- Haute Autorité de Santé. Available online: https://www.has-sante.fr (accessed on 8 April 2022).
- Oka, R.K.; Szuba, A.; Giacomini, J.C.; Cooke, J.P. A pilot study of L-arginine supplementation on functional capacity in peripheral arterial disease. Vasc. Med. 2005, 10, 265–274. [Google Scholar] [CrossRef] [Green Version]
- Maxwell, A.J.; Anderson, B.E.; Cooke, J.P. Nutritional therapy for peripheral arterial disease: A double-blind, placebo-controlled, randomized trial of HeartBar. Vasc. Med. 2000, 5, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Böger, R.H.; Bode-Böger, S.M.; Thiele, W.; Creutzig, A.; Alexander, K.; Frölich, J.C. Restoring vascular nitric oxide formation by L-arginine improves the symptoms of intermittent claudication in patients with peripheral arterial occlusive disease. J. Am. Coll. Cardiol. 1998, 32, 1336–1344. [Google Scholar] [CrossRef] [Green Version]
- Kashyap, V.S.; Lakin, R.O.; Campos, P.; Allemang, M.; Kim, A.; Sarac, T.P.; Hausladen, A.; Stamler, J.S. The LargPAD Trial: Phase IIA evaluation of l-arginine infusion in patients with peripheral arterial disease. J. Vasc. Surg. 2017, 66, 187–194. [Google Scholar] [CrossRef] [Green Version]
- Regensteiner, J.G.; Ware, J.E., Jr.; McCarthy, W.J.; Zhang, P.; Forbes, W.P.; Heckman, J.; Hiatt, W.R. Effect of cilostazol on treadmill walking, community-based walking ability, and health-related quality of life in patients with intermittent claudication due to peripheral arterial disease: Meta-analysis of six randomized controlled trials. J. Am. Geriatr. Soc. 2002, 50, 1939–1946. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Touyz, R.M.; Park, J.B.; Schiffrin, E.L. Antioxidant effects of vitamins C and E are associated with altered activation of vascular NADPH oxidase and superoxide dismutase in stroke-prone SHR. Hypertension 2001, 38, 606–611. [Google Scholar] [CrossRef] [Green Version]
- Ashor, A.W.; Siervo, M.; Lara, J.; Oggioni, C.; Afshar, S.; Mathers, J.C. Effect of vitamin C and vitamin E supplementation on endothelial function: A systematic review and meta-analysis of randomised controlled trials. Br. J. Nutr. 2015, 113, 1182–1194. [Google Scholar] [CrossRef] [Green Version]
- Hussin, A.M.; Ashor, A.W.; Schoenmakers, I.; Hill, T.; Mathers, J.C.; Siervo, M. Effects of vitamin D supplementation on endothelial function: A systematic review and meta-analysis of randomised clinical trials. Eur. J. Nutr. 2017, 56, 1095–1104. [Google Scholar] [CrossRef]
- Schiffrin, E.L.; Deng, L.Y. Comparison of effects of angiotensin I-converting enzyme inhibition and beta-blockade for 2 years on function of small arteries from hypertensive patients. Hypertension 1995, 25, 699–703. [Google Scholar] [CrossRef]
- Ghiadoni, L.; Magagna, A.; Versari, D.; Kardasz, I.; Huang, Y.; Taddei, S.; Salvetti, A. Different effect of antihypertensive drugs on conduit artery endothelial function. Hypertension 2003, 41, 1281–1286. [Google Scholar] [CrossRef] [Green Version]
- Rizzoni, D.; Muiesan, M.L.; Porteri, E.; Castellano, M.; Zulli, R.; Bettoni, G.; Salvetti, M.; Monteduro, C.; Agabiti-Rosei, E. Effects of long-term antihypertensive treatment with lisinopril on resistance arteries in hypertensive patients with left ventricular hypertrophy. J. Hypertens. 1997, 15, 197–204. [Google Scholar] [CrossRef]
- Anderson, T.J.; Elstein, E.; Haber, H.; Charbonneau, F. Comparative study of ACE-inhibition, angiotensin II antagonism, and calcium channel blockade on flow-mediated vasodilation in patients with coronary disease (BANFF study). J. Am. Coll. Cardiol. 2000, 35, 60–66. [Google Scholar] [CrossRef] [Green Version]
- Pasini, A.F.; Garbin, U.; Nava, M.C.; Stranieri, C.; Pellegrini, M.; Boccioletti, V.; Luchetta, M.L.; Fabrizzi, P.; Lo Cascio, V.; Cominacini, L. Effect of sulfhydryl and non-sulfhydryl angiotensin-converting enzyme inhibitors on endothelial function in essential hypertensive patients. Am. J. Hypertens. 2007, 20, 443–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghiadoni, L.; Versari, D.; Magagna, A.; Kardasz, I.; Plantinga, Y.; Giannarelli, C.; Taddei, S.; Salvetti, A. Ramipril dose-dependently increases nitric oxide availability in the radial artery of essential hypertension patients. J. Hypertens. 2007, 25, 361–366. [Google Scholar] [CrossRef]
- Mancini, G.B.; Henry, G.C.; Macaya, C.; O’Neill, B.J.; Pucillo, A.L.; Carere, R.G.; Wargovich, T.J.; Mudra, H.; Lüscher, T.F.; Klibaner, M.I.; et al. Angiotensin-converting enzyme inhibition with quinapril improves endothelial vasomotor dysfunction in patients with coronary artery disease. The TREND (Trial on Reversing ENdothelial Dysfunction) Study. Circulation 1996, 94, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Schiffrin, E.L.; Park, J.B.; Intengan, H.D.; Touyz, R.M. Correction of arterial structure and endothelial dysfunction in human essential hypertension by the angiotensin receptor antagonist losartan. Circulation 2000, 101, 1653–1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiffrin, E.L.; Park, J.B.; Pu, Q. Effect of crossing over hypertensive patients from a beta-blocker to an angiotensin receptor antagonist on resistance artery structure and on endothelial function. J. Hypertens. 2002, 20, 71–78. [Google Scholar] [CrossRef]
- Tzemos, N.; Lim, P.O.; MacDonald, T.M. Valsartan improves endothelial dysfunction in hypertension: A randomized, double-blind study. Cardiovasc. Ther. 2009, 27, 151–158. [Google Scholar] [CrossRef]
- Hirooka, Y.; Kimura, Y.; Sagara, Y.; Ito, K.; Sunagawa, K. Effects of valsartan or amlodipine on endothelial function and oxidative stress after one year follow-up in patients with essential hypertension. Clin. Exp. Hypertens. 2008, 30, 267–276. [Google Scholar] [CrossRef]
- Takiguchi, S.; Ayaori, M.; Uto-Kondo, H.; Iizuka, M.; Sasaki, M.; Komatsu, T.; Takase, B.; Adachi, T.; Ohsuzu, F.; Ikewaki, K. Olmesartan improves endothelial function in hypertensive patients: Link with extracellular superoxide dismutase. Hypertens. Res. 2011, 34, 686–692. [Google Scholar] [CrossRef]
- Ozdemir, B.; Yazici, A. Could the decrease in the endothelial nitric oxide (NO) production and NO bioavailability be the crucial cause of COVID-19 related deaths? Med. Hypotheses 2020, 144, 109970. [Google Scholar] [CrossRef]
- Kamenshchikov, N.O.; Berra, L.; Carroll, R.W. Therapeutic Effects of Inhaled Nitric Oxide Therapy in COVID-19 Patients. Biomedicines 2022, 10, 369. [Google Scholar] [CrossRef] [PubMed]
- Akaberi, D.; Krambrich, J.; Ling, J.; Luni, C.; Hedenstierna, G.; Järhult, J.D.; Lennerstrand, J.; Lundkvist, Å. Mitigation of the replication of SARS-CoV-2 by nitric oxide in vitro. Redox Biol. 2020, 37, 101734. [Google Scholar] [CrossRef] [PubMed]
- Abosheasha, M.A.; El-Gowily, A.H.; Elfiky, A.A. Potential antiviral properties of antiplatelet agents against SARS-CoV-2 infection: An in silico perspective. J. Thromb. Thrombolysis 2021, 12, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Abosheasha, M.A.; El-Gowily, A.H. Superiority of cilostazol among antiplatelet FDA-approved drugs against COVID 19 M(pro) and spike protein: Drug repurposing approach. Drug Dev. Res. 2021, 82, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Motta, N.A.V.; Autran, L.J.; Brazão, S.C.; Lopes, R.O.; Scaramello, C.B.V.; Lima, G.F.; Brito, F.C.F. Could cilostazol be beneficial in COVID-19 treatment? Thinking about phosphodiesterase-3 as a therapeutic target. Int. Immunopharmacol. 2021, 92, 107336. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Abbas, A.K.; Fausto, N.; Aster, J.C. Robbins & Cotran Pathologic Basis of Disease, 8th ed.; Saunders: Philadelphia, PA, USA, 2010. [Google Scholar]
- Kawabe, J.; Yuhki, K.; Okada, M.; Kanno, T.; Yamauchi, A.; Tashiro, N.; Sasaki, T.; Okumura, S.; Nakagawa, N.; Aburakawa, Y.; et al. Prostaglandin I2 promotes recruitment of endothelial progenitor cells and limits vascular remodeling. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 464–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birukova, A.A.; Tian, Y.; Dubrovskyi, O.; Zebda, N.; Sarich, N.; Tian, X.; Wang, Y.; Birukov, K.G. VE-cadherin trans-interactions modulate Rac activation and enhancement of lung endothelial barrier by iloprost. J. Cell. Physiol. 2012, 227, 3405–3416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, C.F.; Lian, W.S.; Chen, S.H.; Lai, P.F.; Li, H.F.; Lan, Y.F.; Cheng, W.T.; Lin, H. Protective effects of adiponectin against renal ischemia-reperfusion injury via prostacyclin-PPARα-Heme oxygenase-1 signaling pathway. J. Cell. Physiol. 2012, 227, 239–249. [Google Scholar] [CrossRef]
- Chen, H.H.; Chen, T.W.; Lin, H. Prostacyclin-induced peroxisome proliferator-activated receptor-α translocation attenuates NF-κB and TNF-α activation after renal ischemia-reperfusion injury. Am. J. Physiol. Ren. Physiol. 2009, 297, 1109–1118. [Google Scholar] [CrossRef]
- Czeslick, E.G.; Simm, A.; Grond, S.; Silber, R.E.; Sablotzki, A. Inhibition of intracellular tumour necrosis factor (TNF)-α and interleukin (IL)-6 production in human monocytes by iloprost. Eur. J. Clin. Investig. 2003, 33, 1013–1017. [Google Scholar] [CrossRef]
- Müller, T.; Dürk, T.; Blumenthal, B.; Herouy, Y.; Sorichter, S.; Grimm, M.; Panther, E.; Cicko, S.; Norgauer, J.; Idzko, M. Iloprost has potent anti-inflammatory properties on human monocyte-derived dendritic cells. Clin. Exp. Allergy. 2010, 40, 1214–1221. [Google Scholar] [CrossRef] [PubMed]
- D’Amelio, P.; Cristofaro, M.A.; D’Amico, L.; Veneziano, L.; Roato, I.; Sassi, F.; Bisignano, G.; Saracco, M.; Pellerito, R.; Patanè, S.; et al. Iloprost modulates the immune response in systemic sclerosis. BMC Immunol. 2010, 11, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birukova, A.A.; Wu, T.; Tian, Y.; Meliton, A.; Sarich, N.; Tian, X.; Leff, A.; Birukov, K.G. Iloprost improves endothelial barrier function in lipopolysaccharide-induced lung injury. Eur. Respir. J. 2013, 41, 165–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filippini, A.; Bnà, C.; Bellosta, R.; Bazzani, R.; Luzzani, L.; Pegorer, M.A.; Zandalasini, M.; Baldon, M.; Codazzi, M.; Sabatini, T. COVID-19 acute respiratory distress syndrome: Can iloprost have a role for the treatment? Respir. Med. Case Rep. 2021, 32, 101358. [Google Scholar] [CrossRef] [PubMed]
- Johansson, P.I.; Søe-Jensen, P.; Bestle, M.H.; Clausen, N.E.; Kristiansen, K.T.; Lange, T.; Stensballe, J.; Perner, A. Prostacyclin in Intubated Patients with COVID-19 and Severe Endotheliopathy: A Multicenter, Randomized Clinical Trial. Am. J. Respir. Crit. Care Med. 2022, 205, 324–329. [Google Scholar] [CrossRef]
- Vigstedt, M.; Søe-Jensen, P.; Bestle, M.H.; Clausen, N.E.; Kristiansen, K.T.; Lange, T.; Stensballe, J.; Perner, A.; Johansson, P.I. The effect of prostacyclin infusion on markers of endothelial activation and damage in mechanically ventilated patients with SARS-CoV-2 infection. J. Crit. Care 2022, 69, 154010. [Google Scholar] [CrossRef]
- Gavrielatou, E.; Xourgia, E.; Xixi, N.A.; Mantelou, A.G.; Ischaki, E.; Kanavou, A.; Zervakis, D.; Routsi, C.; Kotanidou, A.; Siempos, I.I. Effect of Vitamin C on Clinical Outcomes of Critically Ill Patients With COVID-19: An Observational Study and Subsequent Meta-Analysis. Front. Med. 2022, 9, 814587. [Google Scholar] [CrossRef]
- Murai, I.H.; Fernandes, A.L.; Sales, L.P.; Pinto, A.J.; Goessler, K.F.; Duran, C.S.C.; Silva, C.B.R.; Franco, A.S.; Macedo, M.B.; Dalmolin, H.H.H.; et al. Effect of a Single High Dose of Vitamin D3 on Hospital Length of Stay in Patients With Moderate to Severe COVID-19: A Randomized Clinical Trial. JAMA 2021, 325, 1053–1060. [Google Scholar] [CrossRef]
- Rawat, D.; Roy, A.; Maitra, S.; Shankar, V.; Khanna, P.; Baidya, D.K. Vitamin D supplementation and COVID-19 treatment: A systematic review and meta-analysis. Diabetes Metab. Syndr. 2021, 15, 102189. [Google Scholar] [CrossRef]
- Talebi-Taher, M.; Najafi, M.H.; Behzad, S. COVID-19 and RAAS inhibitors: Is there a final conclusion? Iran. J. Microbiol. 2021, 13, 728–736. [Google Scholar] [CrossRef]
- Labandeira-Garcia, J.L.; Labandeira, C.M.; Valenzuela, R.; Pedrosa, M.A.; Quijano, A.; Rodriguez-Perez, A.I. Drugs Modulating Renin-Angiotensin System in COVID-19 Treatment. Biomedicines 2022, 10, 502. [Google Scholar] [CrossRef] [PubMed]
- Monteil, V.; Kwon, H.; Prado, P.; Hagelkrüys, A.; Wimmer, R.A.; Stahl, M.; Leopoldi, A.; Garreta, E.; Hurtado Del Pozo, C.; Prosper, F.; et al. Inhibition of SARS-CoV-2 Infections in Engineered Human Tissues Using Clinical-Grade Soluble Human ACE2. Cell 2020, 181, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Wysocki, J.; Ye, M.; Hassler, L.; Gupta, A.K.; Wang, Y.; Nicoleascu, V.; Randall, G.; Wertheim, J.A.; Batlle, D. A Novel Soluble ACE2 Variant with Prolonged Duration of Action Neutralizes SARS-CoV-2 Infection in Human Kidney Organoids. J. Am. Soc. Nephrol. 2021, 32, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. First antibody against COVID-19 spike protein enters phase I. Nat. Rev. Drug. Discov. 2020, 19, 435. [Google Scholar] [CrossRef] [PubMed]
- Chi, X.; Yan, R.; Zhang, J.; Zhang, G.; Zhang, Y.; Hao, M.; Zhang, Z.; Fan, P.; Dong, Y.; Yang, Y.; et al. A neutralizing human antibody binds to the N-terminal domain of the Spike protein of SARS-CoV-2. Science 2020, 369, 650–655. [Google Scholar] [CrossRef]
- Shi, R.; Shan, C.; Duan, X.; Chen, Z.; Liu, P.; Song, J.; Song, T.; Bi, X.; Han, C.; Wu, L.; et al. A human neutralizing antibody targets the receptor-binding site of SARS-CoV-2. Nature 2020, 584, 120–124. [Google Scholar] [CrossRef]
- Chen, X.; Li, R.; Pan, Z.; Qian, C.; Yang, Y.; You, R.; Zhao, J.; Liu, P.; Gao, L.; Li, Z.; et al. Human monoclonal antibodies block the binding of SARS-CoV-2 spike protein to angiotensin converting enzyme 2 receptor. Cell. Mol. Immunol. 2020, 17, 647–649. [Google Scholar] [CrossRef]
- El-Shennawy, L.; Hoffmann, A.D.; Dashzeveg, N.K.; McAndrews, K.M.; Mehl, P.J.; Cornish, D.; Yu, Z.; Tokars, V.L.; Nicolaescu, V.; Tomatsidou, A.; et al. Circulating ACE2-expressing extracellular vesicles block broad strains of SARS-CoV-2. Nat. Commun. 2022, 13, 405. [Google Scholar] [CrossRef]
- Lener, T.; Gimona, M.; Aigner, L.; Börger, V.; Buzas, E.; Camussi, G.; Chaput, N.; Chatterjee, D.; Court, F.A.; Del Portillo, H.A.; et al. Applying extracellular vesicles based therapeutics in clinical trials-an ISEV position paper. J. Extracell. Vesicles 2015, 4, 30087. [Google Scholar] [CrossRef]
- Zhang, Q.; Jeppesen, D.K.; Higginbotham, J.N.; Franklin, J.L.; Crowe, J.E., Jr.; Coffey, R.J. Angiotensin-converting Enzyme 2-containing Small Extracellular Vesicles and Exomeres Bind the Severe Acute Respiratory Syndrome Coronavirus 2 Spike Protein. Gastroenterology 2021, 160, 958–961.e3. [Google Scholar] [CrossRef]
- Cocozza, F.; Névo, N.; Piovesana, E.; Lahaye, X.; Buchrieser, J.; Schwartz, O.; Manel, N.; Tkach, M.; Théry, C.; Martin-Jaular, L. Extracellular vesicles containing ACE2 efficiently prevent infection by SARS-CoV-2 Spike protein-containing virus. J. Extracell. Vesicles 2020, 10, e12050. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Su, P.; Pan, T.; Zhou, X.; Li, H.; Huang, H.; Wang, A.; Wang, F.; Huang, J.; Yan, H.; et al. Engineering Extracellular Vesicles Enriched with Palmitoylated ACE2 as COVID-19 Therapy. Adv. Mater. 2021, 19, e2103471. [Google Scholar] [CrossRef] [PubMed]
- Scott, T.A.; Supramaniam, A.; Idris, A.; Cardoso, A.A.; Shrivastava, S.; Kelly, G.; Grepo, N.A.; Soemardy, C.; Ray, R.M.; McMillan, N.A.J.; et al. Engineered extracellular vesicles directed to the spike protein inhibit SARS-CoV-2. Mol. Ther. Methods Clin. Dev. 2022, 24, 355–366. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Pre-Exposure Prophylaxis | Post-Exposure Prophylaxis | Curative Treatment at Home | Curative Treatment in Hospital | |
|---|---|---|---|---|
| Evusheld® (tixagevimab + cilgavimab) | Yes | No | No | No |
| Ronapreve® (casirivimab + indevimab) | No | Yes (delta) | Yes (delta) | Yes (delta) |
| Xevudy® (sotrovimab) | No | No | No | Yes |
| Therapeutic Approaches for Endothelial Injury in COVID-19 | In Vitro Effects | In Vivo Effects |
|---|---|---|
| Inhaled NO | Has antiviral activty against SARS-CoV-2 | Has pulmonary vasodilatation activity |
| Cilostazol | Binds effectively to SARS-CoV-2’s main protease and spike protein | Unstudied |
| Prostacyclin | Protects the endothelium and has anti-inflammatory effects | Improves endothelial damage repairing, has neoangiogenetic and antithrombotic activities |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Six, I.; Guillaume, N.; Jacob, V.; Mentaverri, R.; Kamel, S.; Boullier, A.; Slama, M. The Endothelium and COVID-19: An Increasingly Clear Link Brief Title: Endotheliopathy in COVID-19. Int. J. Mol. Sci. 2022, 23, 6196. https://doi.org/10.3390/ijms23116196
Six I, Guillaume N, Jacob V, Mentaverri R, Kamel S, Boullier A, Slama M. The Endothelium and COVID-19: An Increasingly Clear Link Brief Title: Endotheliopathy in COVID-19. International Journal of Molecular Sciences. 2022; 23(11):6196. https://doi.org/10.3390/ijms23116196
Chicago/Turabian StyleSix, Isabelle, Nicolas Guillaume, Valentine Jacob, Romuald Mentaverri, Said Kamel, Agnès Boullier, and Michel Slama. 2022. "The Endothelium and COVID-19: An Increasingly Clear Link Brief Title: Endotheliopathy in COVID-19" International Journal of Molecular Sciences 23, no. 11: 6196. https://doi.org/10.3390/ijms23116196
APA StyleSix, I., Guillaume, N., Jacob, V., Mentaverri, R., Kamel, S., Boullier, A., & Slama, M. (2022). The Endothelium and COVID-19: An Increasingly Clear Link Brief Title: Endotheliopathy in COVID-19. International Journal of Molecular Sciences, 23(11), 6196. https://doi.org/10.3390/ijms23116196