
Immunomodulation of Melanoma by Chemo-Thermo-Immunotherapy Using Conjugates of Melanogenesis Substrate NPrCAP and Magnetite Nanoparticles: A Review


 ,
,  ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Synthesis of Novel Conjugates of NPrCAP and Magnetite Nanoparticles for Developing Melanoma-Targeted Chemo-Thermo-Immunotherapy (CTI Therapy)
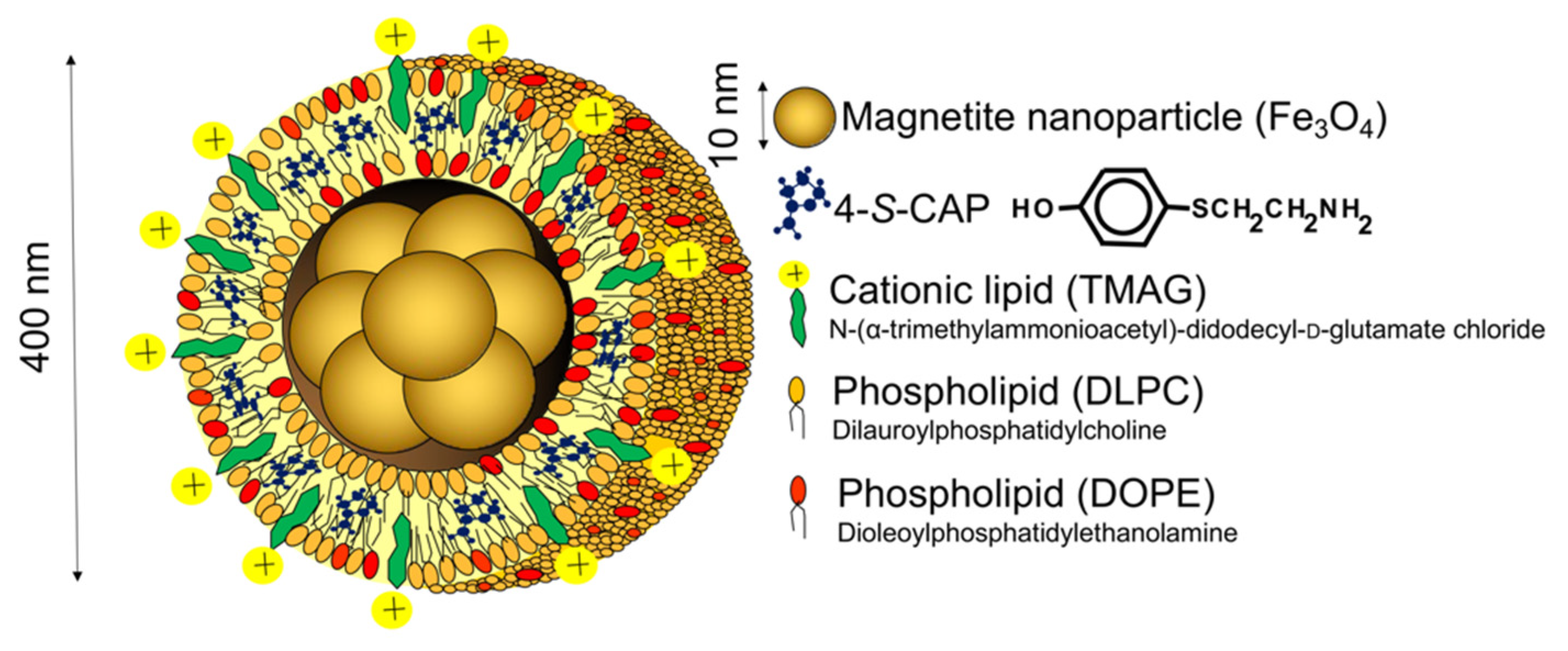
2.1. Preparation of 4-S-CAP-Loaded Magnetite Cationic L Iposomes and Measurement of In Vitro/In Vivo Antimelanoma Effects; Starting Rational Basis for Developing CTI Therapy
2.2. Synthetic Method of NPrCAP-SH for CTI Therapy
2.3. Synthesis of NPrCAP/PEG/APTES/DNM Bound to Dextran Nanomagnetite
2.4. Quantification of NPrCAP Bound to DNM
2.5. The Different Reactivities of NPrCAP/MNP and NPrCAP/PEG/APTES/DNM as Substrates for Tyrosinase
3. Selective Inhibition of Melanoma Growth by NPrCAP/MNP Conjugates in a Mouse Melanoma Model
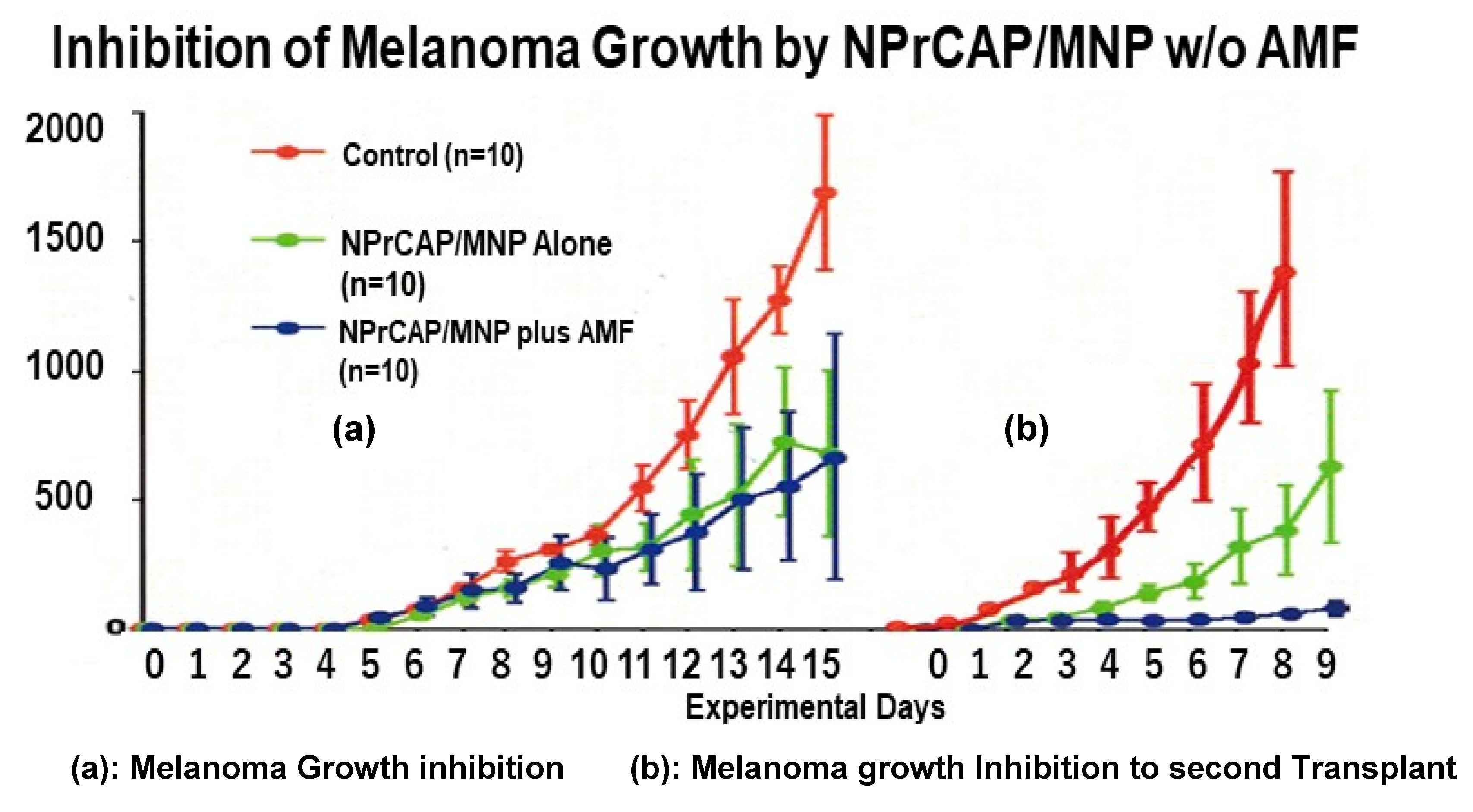
3.1. Melanoma-Targeting Drug Delivery and Growth Inhibition
3.2. Growth Inhibition of Re-Challenge Melanoma Transplant
3.3. Induction of NPrCAP-Mediated Melanoma Apoptosis
4. Specificity and Mechanism of Immunomodulation by CTI Therapy in Melanoma
4.1. NPrCAP as Neo-Antigen Producer
4.2. T-Cell Receptor Repertoires of Tumor-Infiltrating Lymphocytes
4.3. CTI Therapy as In Situ Peptide Vaccine Immunotherapy
5. Approach to Advanced Melanoma Patients
5.1. Scale-Up Production of NPrCAP/PEG/APTES/DNM for Clinical Application
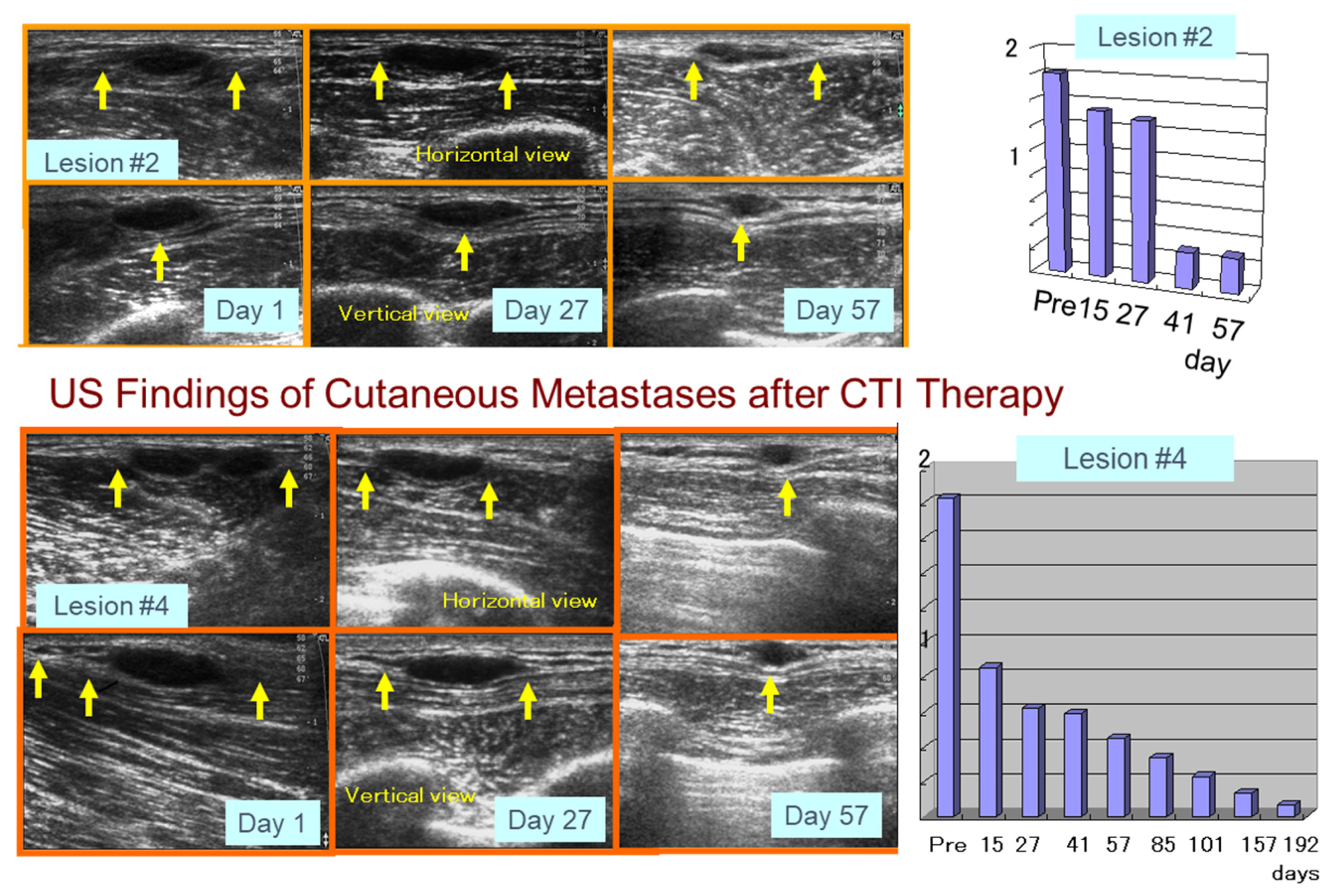
5.2. Preliminary Human Clinical Trial of CTI Therapy for Advanced Melanoma Patients
6. Summary and Perspective
- (1)
- Treatment of primary melanoma transplants with conjugates of the melanogenesis substrate NPrCAP and MNPs (NPrCAP/MNPs), together with AMF exposure, can result in the generation of cytotoxic T cells that inhibit the growth of re-challenge melanomas that are transplanted at the opposite site of the body from the primary tumors;
- (2)
- NPrCAP alone appears to generate some chemotherapeutic and immunotherapeutic properties through both apoptotic and non-apoptotic processes;
- (3)
- A unique melanogenesis cascade can be employed for developing a novel targeted chemo-thermo-immunologic strategy (CTI Therapy) for advanced melanoma patients. It is achieved by conjugating NPrCAP with magnetite nanoparticles (NPrCAP/MNPs) and, together with AMF exposure, NPrCAP/MNPs can induce cytotoxic T cells that inhibit the growth of re-challenge melanoma transplants at the opposite side of the body from the treated primary melanoma.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| AMF | alternating magnetic field |
| APTES | 3-aminopropyltriethoxysilane |
| BSO | buthionine sulfoxide |
| CA-CysC | 5-S-cysteaminyl-3-S-cysteinylcatechol |
| 4-S-CAP | 4-S-cysteaminylphenol |
| CDR3 | third complementarity-determining region |
| CP | cysteinylphenol |
| CTI | chemo-thermo-immuno |
| CTL | cytotoxic T lymphocytes |
| DC | dendritic cell |
| DDS | drug delivery system |
| DNM | dextran nanomagnetite |
| DOPA | 3,4-dihydroxyphenylalanine |
| GMP | good manufacturing practice |
| HPLC | high-performance liquid chromatography |
| HSP | heat shock protein |
| ICI | immune checkpoint inhibitor |
| IFN | interferon |
| MCL | magnetite cationic liposome |
| MEK | mitogen-activated protein kinase/extracellular receptor kinase |
| MHC | major histocompatibility complex |
| MITF | microphthalmia transcription factor |
| MNP | magnetite nanoparticle(s) |
| MSH | melanocyte-stimulating hormone |
| MTT | 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide |
| NAcCAP | N-acetyl-4-S-CAP |
| NPCMD | N-propionyl-4-S-cysteaminylphenol/maleimide-dextran |
| NPrCAP-SH | N-(1-mercaptopropionyl)-4-S-cysteaminyl phenol |
| NPrCAP/MNP | N-propionyl-4-S-cysteaminylphenol/magnetite nanoparticle |
| NPrCAP/PEG/MNP | N-propionyl-4-S-cysteaminylphenol/polyethylene glycol/magnetite nanoparticle |
| NPrCAP/PEG/APTES/DNM | N-propionyl-4-S-cysteaminylphenol/polyethylene glycol/3-aminopropyltriethoxysilane/dextran nanomagnetite |
| NPrCAQ | N-propionyl-4-S-cysteaminyl-1,2-benzoquinone |
| ROS | reactive oxygen species |
| TCR | T-cell receptor |
| TIL | tumor-infiltrating lymphocyte |
| TRIL | TNF-related apoptosis-inducing ligand |
| TRPs | tyrosinase related proteins |
References
- Hida, T.; Kamiya, T.; Kawakami, A.; Ogino, J.; Sohma, H.; Uhara, H.; Jimbow, K. Elucidation of Melanogenesis Cascade for Identifying Pathophysiology and Therapeutic Approach of Pigmentary Disorders and Melanoma. Int. J. Mol. Sci. 2020, 21, 6129. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, A.; Fisher, D.E. The master role of microphthalmia-associated transcription factor in melanocyte and melanoma biology. Lab. Investig. 2017, 97, 649–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jimbow, K.; Park, J.S.; Kato, F.; Hirosaki, K.; Toyofuku, K.; Hua, C.; Yamashita, T. Assembly, target-signaling and intracellular transport of tyrosinase gene family protein in the initial stage of melanosome biogenesis. Pigment Cell Res. 2000, 13, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Jimbow, K.; Prota, G.; Quevedo, W.C.; Fitzpatrick, T.B. Biology of Melanocytes. In Fitzpatrick’s Dermatology in General Medicine, 5th ed.; Freedburg, I.M., Eisen, A.Z., Wolff, E., Austen, K.F., Goldsmith, L.A., Katz, S.I., Fitzpatrick, T.B., Eds.; McGraw-Hill: New York, NY, USA, 1998; pp. 192–220. [Google Scholar]
- Jimbow, K. N-acetyl-4-S-cystenylphenol as a new type of depigmenting agent for the melanoderma of patients with melasma. Arch. Dermatol. 1991, 127, 1528–1534. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.; Hou, Z.; Wang, M.; Li, C.; Lin, J. Recent advances in hyperthermia therapy-based synergistic immunotherapy. Adv. Mater. 2021, 33, e2004788. [Google Scholar] [CrossRef]
- Li, W.; Yang, J.; Luo, L.; Jiang, M.; Qin, B.; Yin, H.; Zhu, C.; Yuan, X.; Zhang, J.; Luo, Z.; et al. Targeting photodynamic and photothermal therapy to the endoplasmic reticulum enhances immunogenic cancer cell death. Nat. Commun. 2019, 10, 3349. [Google Scholar] [CrossRef] [Green Version]
- Valcourt, D.M.; Dang, M.N.; Day, E.S. IR820-loaded PLGA nanoparticles for photothermal therapy of triple-negative breast cancer. J. Biomed. Mater. Res. Part A 2019, 107, 1702–1712. [Google Scholar] [CrossRef]
- Riley, R.S.; Day, E.S. Gold nanoparticle-mediated photothermal therapy: Applications and opportunities for multimodal cancer treatment. WIREs Nanomed. Nanobiotechnol. 2017, 9, e1449. [Google Scholar] [CrossRef]
- Kobayashi, T.; Kakimi, K.; Nakayama, E.; Jimbow, K. Antitumor immunity by magnetic nanoparticle-mediated hyperthermia. Nanomedicine 2014, 9, 1715–1726. [Google Scholar] [CrossRef] [Green Version]
- Algan, O.; Fosmire, H.; Hynynen, K.; Dalkin, B.; Cui, H.; Drach, G.; Stea, B.; Cassady, J.R. External beam radiotherapy and hyperthermia in the treatment of patients with locally advanced prostate carcinoma. Cancer 2000, 89, 399–403. [Google Scholar] [CrossRef]
- Sato, M.; Yamashita, T.; Ohkura, M.; Osai, Y.; Sato, A.; Takada, T.; Matsusaka, H.; Ono, I.; Tamura, Y.; Sato, N.; et al. N-propionyl-cysteaminylphenol-magnetite conjugate (NPrCAP/M) is a nanoparticle for the targeted growth suppression of melanoma cells. J. Investig. Dermatol. 2009, 129, 2233–2241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takada, T.; Yamashita, T.; Sato, M.; Sato, A.; Ono, I.; Tamura, Y.; Sato, N.; Miyamoto, A.; Ito, A.; Honda, H.; et al. Growth inhibition of re-challenge B16 melanoma transplant by conjugates of melanogenesis substrate and magnetite nanoparticles as the basis for developing melanoma-targeted chemo-thermo-immunotherapy. J. Biomed. Biotechnol. 2009, 2009, 457936. [Google Scholar] [CrossRef] [PubMed]
- Ishii-Osai, Y.; Yamashita, T.; Tamura, Y.; Sato, N.; Ito, A.; Honda, H.; Wakamatsu, K.; Ito, S.; Nakayama, E.; Okura, M.; et al. N-propionyl-4-S-cysteaminylphenol induces apoptosis in B16F1 cells and mediates tumor-specific T-cell immune responses in a mouse melanoma model. J. Dermatol. Sci. 2012, 67, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Sato, A.; Tamura, Y.; Sato, N.; Yamashita, T.; Takada, T.; Sato, M.; Osai, Y.; Okura, M.; Ono, I.; Ito, A.; et al. Melanoma-targeted chemo-thermo-immuno (CTI)-therapy using N-propionyl-4-S-cysteaminylphenol-magnetite nanoparticles elicits CTL response via heat shock protein-peptide complex release. Cancer Sci. 2010, 101, 1939–1946. [Google Scholar] [CrossRef]
- Ito, A.; Yamaguchi, M.; Okamoto, N.; Sanematsu, Y.; Kawabe, Y.; Wakamatsu, K.; Ito, S.; Honda, H.; Kobayashi, T.; Nakayama, E.; et al. T-cell receptor repertoires of tumor-infiltrating lymphocytes after hyperthermia using functionalized magnetite nanoparticles. Nanomedicine 2013, 8, 891–902. [Google Scholar] [CrossRef]
- Miura, S.; Ueda, T.; Jimbow, K.; Ito, S.; Fujita, K. Synthesis of cysteinylphenol, cysteaminylphenol, and related compounds, and In Vivo evaluation of antimelanoma effect. Arch. Dermatol. Res. 1987, 279, 219–225. [Google Scholar] [CrossRef]
- Yamada, I.; Seki, S.; Matsubara, O.; Ito, S.; Suzuki, S.; Kasuga, T. The cytotoxicity of cysteinylcatechols and related compounds to human melanoma cells In Vitro. J. Investig. Dermatol. 1987, 88, 538–540. [Google Scholar] [CrossRef] [Green Version]
- Shinkai, M.; Yanase, M.; Honda, H.; Wakabayashi, T.; Yoshida, J.; Kobayashi, T. Intracellular hyperthermia for cancer using magnetite cationic liposomes: In vitro Study. Jpn. J. Cancer Res. 1996, 87, 1179–1183. [Google Scholar] [CrossRef]
- Ito, A.; Fujioka, M.; Yoshida, T.; Wakamatsu, K.; Ito, S.; Yamashita, T.; Jimbow, K.; Honda, H. 4-S-Cysteaminylphenol-loaded magnetite cationic liposomes for combination therapy of hyperthermia with chemotherapy against malignant melanoma. Cancer Sci. 2007, 98, 424–430. [Google Scholar] [CrossRef]
- Hasegawa, K.; Ito, S.; Inoue, S.; Wakamatsu, K.; Ozeki, H.; Ishiguro, I. Dihydro-1,4-benzothiazine-6,7-dione, the ultimate toxic metabolite of 4-S-cysteinylphenol and 4-S-cysteaminylcatechol. Biochem. Pharmacol. 1997, 53, 1435–1444. [Google Scholar] [CrossRef]
- Katschinski, D.M.; Boos, K.; Schindler, S.G.; Fandrey, J. Pivotal role of reactive oxygen species as intracellular mediators of hyperthermia-induced apoptosis. J. Biol. Chem. 2000, 275, 21094–21098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padgette, S.R.; Herman, H.H.; Han, J.H.; Pollock, S.H.; May, S.W. Antihypertensive activities of phenyl aminoethyl sulfides, a class of synthetic substrates for dopamine β-hydroxylase. J. Med. Chem. 1984, 27, 1354–1357. [Google Scholar] [CrossRef] [PubMed]
- Jimbow, K.; Takada, T.; Osai, Y.; Thoma, P.D.; Sato, M.; Sato, A.; Kamiya, T.; Ono, I.; Tamura, Y.; Sato, N.; et al. Melanogenesis exploitation and melanoma nanomedicine: Utilization melanogenesis substrate, NPrCAP for exploiting melanoma-targeting drug and its conjugation with magnetite nanoparticles for developing melanoma chemo-thermo-immunotherapy. Open Conf. Proc. J. 2011, 2, 5–16. [Google Scholar] [CrossRef] [Green Version]
- Jimbow, K.; Tamura, Y.; Yoneta, A.; Kamiya, T.; Ono, I.; Yamashita, T.; Ito, A.; Honda, H.; Wakamatsu, K.; Ito, S.; et al. Conjugation of magnetic nanoparticle with melanogenesis substrate, NPrCAP provides melanoma targeted, in situ peptide vaccine immunotherapy through HSP production by chemo-thermo-therapy. J. Biomater. Nanobiotechnol. 2012, 3, 140–153. [Google Scholar] [CrossRef]
- Mizote, Y.; Wakamatsu, K.; Ito, S.; Uenaka, A.; Ohue, Y.; Kurose, K.; Isobe, M.; Ito, A.; Tamura, Y.; Honda, H.; et al. TLR4 and NLRP3 inflammasome activation in monocytes by N-propionyl cysteaminylphenol-maleimide-dextran (NPCMD). J. Dermatol. Sci. 2014, 73, 209–215. [Google Scholar] [CrossRef]
- Ito, S.; Kato, T.; Ishikawa, K.; Kasuga, T.; Jimbow, K. Mechanism of selective toxicity of 4-S-cysteinylphenol and 4-S-cysteaminylphenol to melanocyte. Biochem. Pharmacol. 1987, 36, 2007–2011. [Google Scholar] [CrossRef]
- Ito, S.; Nishigaki, A.; Ishii-Osai, Y.; Ojika, M.; Wakamatsu, K.; Yamashita, T.; Tamura, Y.; Ito, A.; Honda, H.; Nakayama, E.; et al. Mechanism of putative neo-antigen formation from N-propionyl-4-S-cysteaminylphenol, a tyrosinase substrate, in melanoma models. Biochem. Pharmacol. 2012, 84, 646–653. [Google Scholar] [CrossRef]
- Jimbow, K.; Szabo, G.; Fitzpatrick, T.B. Ultrastructural investigation of autophagocytosis of melanosomes and programmed death of melanocytes in White Leghorn feathers: A study of morphogenetic events leading to hypomelanosis. Dev. Biol. 1974, 36, 8–23. [Google Scholar] [CrossRef]
- Ito, Y.; Jimbow, K. Selective cytotoxicity of 4-S-cysteaminylphenol on follicular melanocytes of the black mouse: Rational basis for its application on melanosome chemotherapy. Cancer Res. 1987, 47, 3278–3284. [Google Scholar]
- Pankovich, J.M.; Jimbow, K. Tyrosine transport in a human melanoma cell line as a basis for selective transport of cytotoxic analogues. Biochem. J. 1991, 280, 721–725. [Google Scholar] [CrossRef]
- Jimbow, K.; Iwashina, T.; Alena, F.; Yamada, K.; Pankovich, J.; Umemura, T. Exploitation of pigment biosynthesis pathway as a selective chemotherapeutic approach for malignant melanoma. J. Investig. Dermatol. 1993, 100 (Suppl. S2), S231–S238. [Google Scholar] [CrossRef] [Green Version]
- Alena, F.; Ishikawa, T.; Gili, A.; Jimbow, K. Selective In Vivo accumulation of N-acetyl-4-S-cysteaminylphenol in B16F10 murine melanoma and enhancement of its In Vitro and In Vivo antimelanoma effect by combination of buthionine sulfoximine. Cancer Res. 1994, 54, 2661–2666. [Google Scholar] [PubMed]
- Reszka, K.; Jimbow, K. Electron donor and acceptor properties of melanin pigments in the skin. In Oxidative Stress in Dermatology; Fuchs, J., Packer, L., Eds.; Marcel Dekker: New York, NY, USA, 1993; pp. 287–320. [Google Scholar]
- Miura, T.; Jimbow, K.; Ito, S. The In Vivo antimelanoma effect of 4-S-cysteaminylphenol and its n-acetyl derivative. Int. J. Cancer 1990, 46, 931–934. [Google Scholar] [CrossRef] [PubMed]
- Tandon, M.; Thomas, P.D.; Shokravi, M.; Singh, S.; Samra, S.; Chang, D.; Jimbow, K. Synthesis and antitumor effect of the melanogenesis-based antimelanoma agent N-propionyl-4-S-cysteaminylphenol. Biochem. Pharmacol. 1998, 15, 2023–2029. [Google Scholar] [CrossRef]
- Thomas, P.D.; Kishi, H.; Cao, H.; Ota, M.; Yamashita, T.; Singh, S.; Jimbow, K. Selective incorporation and specific cytocidal effect as the cellular basis for the antimelanoma action of sulphur containing tyrosine analogs. J. Investig. Dermatol. 1999, 113, 928–934. [Google Scholar] [CrossRef] [Green Version]
- Westerhof, W.; Manini, P.; Napolitano, A.; d’Ischia, M. The haptenation theory of vitiligo and melanoma rejection: A close-up. Exp. Dermatol. 2011, 20, 92–96. [Google Scholar] [CrossRef]
- Manini, P.; Napolitano, A.; Westerhof, W.; Riley, P.A.; D’Ischia, M. A reactive orthoquinone generated by tyrosinase-catalyzed oxidation of the skin depigmenting agent monobenzone: Self-coupling and thiol-conjugation reactions and possible implications for melanocyte toxicity. Chem. Res. Toxicol. 2009, 22, 1398–1405. [Google Scholar] [CrossRef]
- Van den Boon, J.G.; Picavet, D.I.; van Swieten, P.F.; van Veen, H.A.; Konijnenberg, D.; van Veelen, P.A.; van Capel, T.; de Jong, E.C.; Reits, E.A.; Drijfhout, J.W.; et al. Skin-depigmenting agent monobenzone induces potent T-cell autoimmunity toward pigmented cells by tyrosinase haptenation and melanosome autophagy. J. Investig. Dermatol. 2011, 131, 1240–1251. [Google Scholar] [CrossRef] [Green Version]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pagès, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [Green Version]
- Davis, M.M.; Bjorkman, P.J. T-cell antigen receptor genes and T-cell recognition. Nature 1988, 334, 395–402. [Google Scholar] [CrossRef]
- Harada, M.; Tamada, K.; Abe, K.; Li, T.; Onoe, Y.; Tada, H.; Tatsugami, K.; Ando, T.; Kimura, G.; Nomoto, K. Characterization of B16 melanoma-specific cytotoxic T lymphocytes. Cancer Immunol. Immunother. 1998, 47, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Ji, Q.; Feigenbaum, L.; Leighty, R.M.; Hurwitz, A.A. Melanoma progression despite infiltration by in vivo-primed TRP-2-specific T cells. J. Immunother. 2009, 32, 129–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanase, M.; Shinkai, M.; Honda, H.; Wakabayashi, T.; Yoshida, J.; Kobayashi, T. Antitumor immunity induction by intracellular hyperthermia using magnetite cationic liposomes. Jpn. J. Cancer Res. 1998, 89, 775–782. [Google Scholar] [CrossRef] [PubMed]
- Ito, A.; Tanaka, K.; Kondo, K.; Shinkai, M.; Honda, H.; Matsumoto, K.; Saida, T.; Kobayashi, T. Tumor regression by combined immunotherapy and hyperthermia using magnetic nanoparticles in an experimental subcutaneous murine melanoma. Cancer Sci. 2003, 94, 308–313. [Google Scholar] [CrossRef] [PubMed]
- Ito, A.; Honda, H.; Kobayashi, T. Cancer immunotherapy based on intracellular hyperthermia using magnetite nanoparticles: A novel concept of “heat-controlled necrosis” with heat shock protein expression. Cancer Immunol. Immunother. 2006, 55, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Kawai, N.; Futakuchi, M.; Yoshida, T.; Ito, A.; Sato, S.; Naiki, T.; Honda, H.; Shirai, T.; Kohri, K. Effect of heat therapy using magnetic nanoparticles conjugated with cationic liposomes on prostate tumor in bone. Prostate 2008, 68, 784–792. [Google Scholar] [CrossRef]
- Tamura, Y.; Peng, P.; Liu, K.; Daou, M.; Srivastava, P.K. Immunotherapy of tumors with autologous tumor-derived heat shock protein preparations. Science 1997, 278, 117–120. [Google Scholar] [CrossRef]
- Srivastava, P.K.; Menoret, A.; Basu, S.; Binder, R.J.; McQuade, K.L. Heat shock proteins come of age: Primitive functions acquire new roles in an adaptive world. Immunity 1998, 8, 657–665. [Google Scholar] [CrossRef] [Green Version]
- Reimer, P.; Balzer, T. Ferucarbotran (Resovist): A new clinically approved RES-specific contrast agent for contrast-enhanced MRI of the liver: Properties, clinical development, and applications. Eur. Radiol. 2003, 13, 1266–1276. [Google Scholar] [CrossRef]
- Skudalski, L.A.; Waldman, R.; Kerr, P.E.; Grant-Kels, J.M. Melanoma: An update on systemic therapies. J. Am. Acad. Dermatol. 2022, 86, 515–524. [Google Scholar] [CrossRef]



















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tamura, Y.; Ito, A.; Wakamatsu, K.; Kamiya, T.; Torigoe, T.; Honda, H.; Yamashita, T.; Uhara, H.; Ito, S.; Jimbow, K. Immunomodulation of Melanoma by Chemo-Thermo-Immunotherapy Using Conjugates of Melanogenesis Substrate NPrCAP and Magnetite Nanoparticles: A Review. Int. J. Mol. Sci. 2022, 23, 6457. https://doi.org/10.3390/ijms23126457
Tamura Y, Ito A, Wakamatsu K, Kamiya T, Torigoe T, Honda H, Yamashita T, Uhara H, Ito S, Jimbow K. Immunomodulation of Melanoma by Chemo-Thermo-Immunotherapy Using Conjugates of Melanogenesis Substrate NPrCAP and Magnetite Nanoparticles: A Review. International Journal of Molecular Sciences. 2022; 23(12):6457. https://doi.org/10.3390/ijms23126457
Chicago/Turabian StyleTamura, Yasuaki, Akira Ito, Kazumasa Wakamatsu, Takafumi Kamiya, Toshihiko Torigoe, Hiroyuki Honda, Toshiharu Yamashita, Hisashi Uhara, Shosuke Ito, and Kowichi Jimbow. 2022. "Immunomodulation of Melanoma by Chemo-Thermo-Immunotherapy Using Conjugates of Melanogenesis Substrate NPrCAP and Magnetite Nanoparticles: A Review" International Journal of Molecular Sciences 23, no. 12: 6457. https://doi.org/10.3390/ijms23126457
APA StyleTamura, Y., Ito, A., Wakamatsu, K., Kamiya, T., Torigoe, T., Honda, H., Yamashita, T., Uhara, H., Ito, S., & Jimbow, K. (2022). Immunomodulation of Melanoma by Chemo-Thermo-Immunotherapy Using Conjugates of Melanogenesis Substrate NPrCAP and Magnetite Nanoparticles: A Review. International Journal of Molecular Sciences, 23(12), 6457. https://doi.org/10.3390/ijms23126457