2.1. Pulse Radiolysis, Spectra Resolution, and Concentration Profiles of Transients
The reaction of HO
● with compounds
1 and
2 was investigated in N
2O-saturated solutions of 0.2 mM
1 or
2, at natural pH (pH value of 7.0 was recorded) to mimic natural biological conditions. Transient absorption spectra in the range 270–700 nm recorded in the time range of 1 μs to 160 μs are presented in
Figure 1.
The transient spectrum obtained 1 μs after the electron pulse in N
2O-saturated aqueous solutions containing 0.2 mM of compound
1 at pH 7 and showed a dominant absorption band with λ
max = 330 nm (
Figure 1A). Based on our previous studies on dimethyl sulfide [
16], and various Met-containing derivatives [
11,
12,
17,
18], this band can be assigned unequivocally to the HO
• adduct at the sulfur atom
HOS•, established as the first step in the HO
•-induced oxidation of compound
1 (see
Scheme 4). However, as shown in
Figure 1A, the observed transient-spectral profile changed with time, indicating contributions from various transient species. The spectrum obtained 1 µs after the electron pulse underwent further changes and, at 3 µs after the electron pulse, had transformed into the spectrum showing two pronounced shoulders in the 380–400 nm and 450–500 nm ranges. The latter shoulder further developed into an absorption band with λ
max = 470 nm, which was clearly seen in the absorption spectrum obtained 10 μs after the pulse together with an absorption band with a maximum located around λ = 290 nm. These absorption bands reached their maximum absorbance values around 7 and 15 μs after the electron pulse, respectively (see upper inset in
Figure 1A). These absorption bands can be tentatively assigned to the intermolecular sulfur–sulfur three-electron bonded radical cations (
SS•+) and α-(alkylthio)alkyl radicals (
αS(1)• and/or
αS(2)•), respectively (
Scheme 4). Moreover, a distinct flat region between 320–400 nm was observed indicating the presence of various additional transient species. Based on our previous studies on Ac-Met-OMe [
11], the C
α-centered radicals (
αC(1)• and/or
αC(2)•) and intramolecular sulfur–nitrogen three-electron bonded radicals (
SN(1)• and/or
SN(2)•) can be considered as potential transients (
Scheme 4). It is noteworthy that the transient-absorption spectrum obtained 30 μs after the pulse is characterized by the distinct absorption band with λ
max = 290 nm and an even more pronounced flat region between 320–460 nm, which can be rationalized by the faster decay kinetics of
SS•+ compared to the decay kinetics of the remaining transients, i.e.,
αS• and
SN•. This fact is clearly confirmed by a comparison of the short- and long-time kinetic profiles recorded at four selected wavelengths (290, 340, 390, and 490 nm) corresponding to the absorption maxima of the four most abundant radicals present in the irradiated system, i.e.,
αS•,
HOS•,
SN•, and
SS•+ (see insets in
Figure 1A). The short-time kinetic traces look different, and they reached their maximum absorbance signal at various times after the pulse. Moreover, the long-time kinetic profiles in the time domain above 70 μs clearly illustrate the kinetics of radical–radical coupling reactions (cf. lower inset in
Figure 1A).
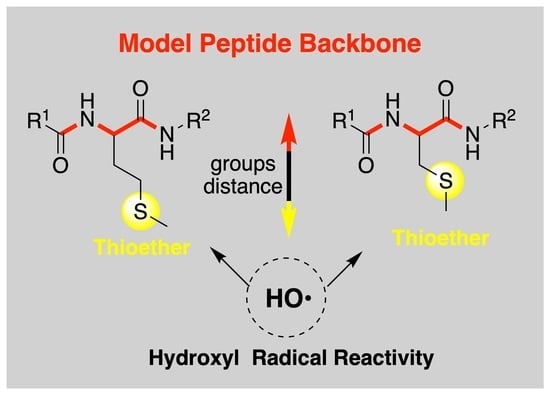
In order to check whether the thioether group distance from the peptide backbone has an influence on the character of intermediates resulting from the reactions of HO
• radicals, transient absorption spectra were recorded after the electron pulse in N
2O-saturated aqueous solutions containing 0.2 mM
2 at pH 7. The transient absorption spectra recorded in the time domain between 1 and 10 μs after the electron pulse are characterized by different features in comparison to the absorption spectra recorded in the case of
1 (cf.
Figure 1A,B). They consisted of two absorption bands with maxima located at λ = 290 and 400 nm. The formation of these bands was fully developed within the range of 3 μs. However, the kinetic time profiles recorded in the time domain >3 μs at 290 nm and 400 nm were different (
Figure 1B, upper insert). While the absorption at 400 nm started to decay, the absorption at 290 nm was stable. Consequently, at 3 μs the absorption band with λ
max = 400 nm was more intense than the absorption band with λ
max = 290 nm, whereas at 10 μs the intensity of these absorption bands reversed. These absorption bands can be tentatively assigned to α-(alkylthio)alkyl radicals (
αS•) and intramolecular sulfur–oxygen three-electron bonded radical cations (
SO•+) (
Scheme 5). Interestingly, no distinct absorption bands with λ
max = 330 and 470 nm were observed, which could be assigned to
HOS• and
SS•+, respectively (
Figure 1B). With the lapse of time to when the 400 nm absorption band disappeared completely (
Figure 1B, lower inset), the absorption spectrum recorded after 160 μs is characterized only by a pronounced absorption band with λ
max = 290 nm.
The purpose of this subsection is to provide arguments, based on spectral and kinetic analyses, for the presence of potential transients, which are formed during the HO
•-induced oxidation of
1 and
2 in aqueous solutions. Resolution of the absorption spectra recorded at any desired time delay, following the electron pulse ranging from 200 ns to 80 μs, into contributions from these transients allows for the quantification (as
G-values) and extraction of their concentration profiles. This is a key issue that enables justification for the mechanistic assumptions reported in
Scheme 4 and
Scheme 5, also explaining the formation of stable products (see
Section 2.2).
Based on our previous work on Ac-Met-OMe [
11], the following transient species
HOS•,
αC(1)•,
αS(1)•,
αS(2)•,
SS•+,
SN(1)•, and
SN(2)• in
Scheme 4 were taken into account in spectral resolutions (see also
Figure S1 in Supplementary Materials). Since the amide nitrogen atom is much more effective in activating the methyl group for H-atom abstraction than the oxygen atom in the ester group [
19], the additional C-centered radical
αC(2)• was also considered in these spectral resolutions. The transient absorption spectrum recorded 1 μs after the electron pulse was resolved into contributions from the three components
HOS•,
αC(1)•, and
SS•+ with the respective
G-values 0.27, 0.08, and 0.02 µmol J
−1 that correspond to 73%, 22%, and 5% of all radicals present at that time (
Table 1). The calculated total
G-value of 0.37 μmol J
−1 does not constitute the expected
G-value of HO
• (0.56 μmol J
−1) available for the reaction of
1 at pH 7 and the concentration (0.2 mM) of
1. This difference in
G-values can be easily understood since at this time, the reactions of HO
• with
1 via all pathways presented at
Scheme 4 are not completed yet. However, their percentage contributions confirm the domination of the
HOS• in the transient absorption spectrum and rationalizes its spectral features (cf.
Figure 1A). From the concentration profiles obtained from the resolution of the spectra recorded at various times after the electron pulse, one can see that at 1.4 μs after the pulse
HOS• reaches its maximum
G-value = 0.33 μmol J
−1 (
Figure 2A). It is still the most abundant radical in the irradiated solution containing
1, and its concentration constitutes 72% of all radicals present at that time (
Table 1). However, there are additional spectral components that start to contribute to this spectrum and include the following radicals:
αC(1)•,
αS•,
SS•+, and
SN• with the respective
G-values: 0.08, 0.01, 0.03, and 0.01 μmol J
−1 (
Table 1). These intermediates were previously identified during HO
•-induced oxidation of Ac–Met–OMe under similar experimental conditions [
11]. The calculated total
G-value of 0.46 μmol J
−1 does not constitute yet the expected
G-value of HO
• radicals available for the reaction of
1. This difference in
G-values can be rationalized similarly as for the earlier recording time equal to 1 μs. However, the appearance of
SS•+ and
SN• also indicates an involvement of secondary reactions with the participation of
HOS• (
Scheme 4).
Using the same spectral resolution approach, the transient spectrum recorded 4 µs after the electron pulse was resolved into contributions from the same components, however, resulting in different fitted
G-values: 0.12, 0.09, 0.14, 0.10, and 0.08 μmol J
−1, respectively (
Table 1), and additionally from
αC(2)● with the
G-value = 0.03. The total
G-value of all radicals present (0.56 μmol J
−1) is in excellent agreement with the expected yield of HO
• (vide supra). This is the time point when all HO
● have reacted with
1, but not all
HOS• have yet undergone transformations into the secondary transients.
The next key time point is 10 µs after the electron pulse when all
HOS● have undergone full transformations into the secondary products. The spectrum was resolved into contributions from the species
αC(1)•,
αC(2)•,
αS•,
SS•+, and
SN•, with fitted
G-values: 0.07, 0.08, 0.23, 0.12, and 0.10 μmol J
−1, respectively (
Table 1). The sum of all component spectra with their respective radiation chemical yields (
G-values) resulted in a good fit (⊗ symbols in inset of
Figure 2A) to the experimental spectrum. Their percentage contributions to the transient absorption spectrum rationalizes nicely its spectral features (
Figure 1A). The total
G-value of all radicals present (0.60 μmol J
−1) slightly exceeds the expected yield of HO
• (vide supra). However, this can be rationalized by the fact that H-atoms formed during radiolysis of water (Equation (1)) can also form
αC(1)•,
αC(2)•,
αS(1)•, and
αS(2)• by direct H-atom abstraction from
1. It is worthy to note that the most abundant radicals present at this time are
αS(1)• and
αS(2)•, which constitute nearly 40% of all radicals (
Table 1). The excellent material balance of all radicals identified in the system equal to the
G-value of HO
● available for the reaction with
1 proves that they are precursors of the corresponding end-products. At this point, it has to be stressed that after 10 μs, following the electron pulse, the total
G-values of the radicals began to decrease slowly, reaching the value of 0.54, 0.44, and 0.37 μmol J
−1 at 30, 80, and 170 μs, respectively (
Table 1). This suggests that in this time domain 40% of all of the radicals formed in the system were consumed in radical–radical processes leading to the final products. Moreover, over this time domain
αS(1)● and
αS(2)● remain the most abundant radicals, constituting nearly 50% of all of the radicals on the timescale of hundreds of microseconds.
Based on our previous work on S-methyl-glutathione (a tripeptide containing an internal S-methyl-cysteine residue) [
20], the following species of
HOS●,
αS●,
SS●+, and
SO●+ were taken into account in spectral resolutions (
Scheme 5 and
Figure S1). Additional support for taking intramolecular sulfur–oxygen three-electron-bonded radicals (
SO●+) into the spectral resolutions came from the photosensitized oxidation studies of
two where
SO●+ was identified as one of the transient products [
10]. Since the amide nitrogen atom is much more effective in activating the methyl group for H-atom abstraction than the oxygen atom of the ester group [
19], the
αC(2)● was also taken into account in the current spectral resolutions.
The first significant difference between compounds
1 and
2 concerns the concentration profiles of
HOS• radicals. In solutions containing
2,
HOS• reach their maximum
G-value = 0.16 μmol J
−1 at 0.5 μs after the electron pulse, compared to 1.4 μs for
1 (cf.
Figure 2A,B). Moreover, this maximum
G-value is slightly over two-fold lower than that observed for
1. Simultaneously, at the same time a comparable
G-value = 0.12 μmol J
−1 for
SO•+ was obtained (
Table 2). This substantial decrease in this
HOS• G-value can be rationalized by the occurrence of an additional very fast
HOS• decay process leading to
SO•+ formation at the expense of the measured
G(
HOS•). This decay channel is not effective in
1. In principle,
HOS• has been shown to decay along three different pathways: (i) a spontaneous unimolecular dissociation into
S•+ and HO
−, (ii) a proton-assisted elimination of HO
− leading to
S•+ and H
2O, and (iii) a displacement of HO
− by a second thioether molecule leading to
SS•+. Typical values of the rate constants for these processes are: (5–7) × 10
5 s
−1, 1 × 10
10 M
−1 s
−1 and 1 × 10
8 M
−1 s
−1, respectively. Since the experimental conditions (pH = 7 and concentration of
1 and
2 = 0.2 mM) were similar, these three reactions should occur with the similar rates for both compounds. Moreover, taking into account the experimental conditions, the last two reaction pathways responsible for the decay of
HOS• are negligible for both compounds
1 and
2. At this point the following question arises: what is the nature of this additional process present in
2 that can compete with the spontaneous dissociation of
HOS• (
Scheme 5)? Based on the known radical chemistry coming from research on the oxidation of (carboxyalkyl)thiopropionic acid derivatives [
21] and β-, γ-, and δ-hydroxyalkyl sulfides [
22], an intramolecular carbonyl-assisted decay of
HOS• radicals can occur that directly leads to the five-membered
SO•+ intermediate (
Scheme 5).
Compatible with the above observations is the time point equal to 2.25 μs after the electron pulse (
Table 2) when all HO
• radicals have reacted with
2, but not all
HOS• have yet undergone transformations into the secondary products. The transient spectrum was resolved into contributions from the four components:
HOS•,
αC(2)•,
αS•, and
SO•+ using the respective fitted
G-values: 0.10, 0.03, 0.16, and 0.27 μmol J
−1. Moreover, the most abundant transient species at that time is
SO•+ contrary to the system containing
1, where
HOS• was still the most abundant radical. For comparison, this specific time point for
1 was equal to 4 μs (
Table 1 for 4 μs).
The next key time point is 10 µs after the electron pulse (similar as for 1) when all
HOS• have undergone full transformations into the secondary products. The spectrum was resolved into contributions from the following components
αC(2)•,
αS•,
SS•+, and
SO•+ (vide inset in
Figure 2B), with the following fitted
G-values: 0.15, 0.23, 0.01, and 0.22 μmol J
−1, respectively (
Table 2). The sum of all component spectra with their respective radiation chemical yields (
G-values) resulted in a good fit (⊗ symbols in inset of
Figure 2B) to the experimental spectrum. The total
G-value of all transient species present (0.61 μmol J
−1) can be similarly rationalized as for
1 (vide supra). The excellent material balance, that all of the radicals identified in the system are equal to the
G-value of HO
• available for the reaction with
2, proves that the identified radical transients are precursors of the corresponding end products. At this point, it has to be emphasized that, at 10 μs after the electron pulse, the
αC(2)• radicals reached their highest
G-values for both compounds
1 and
2 equal to 0.07 and 0.15 μmol J
−1, respectively (cf.
Table 1 and
Table 2). This is the second feature differentiating these compounds. Therefore, the next questions arise: what is the reason for the observed differences in the
G-values of
αC(2)• formed in
1 and
2, and why was the formation of
αC(2)• more efficient in
2?
In order to address these two questions, we propose the following explanation. The
αC(2)• radicals in
1 and
2 are formed via two pathways: via direct H-atom abstraction from the methyl group attached to the amide nitrogen atom by HO
•, or indirectly by a sequence of reactions involving
SN(2)• in the case of
1, and
SO•+ in the case of
2 as precursors of
αC(2)• (cf.
Scheme 4 and
Scheme 5). The rate constants of all primary reactions of HO
• radicals with
1 and
2 leading to
HOS•,
αS(1)• and
αS(2)•,
αC(1)•, and
αC(2)• are similar for both compounds. Therefore, the sequences of reactions leading to
SN(2)• for
1 and
SO•+ for
2 seem to be responsible for the observed differences in radiation chemical yields (
G-values) of
αC(2)• radicals.
In the case of compound
1, the common precursor for
SN(1)•,
SN(2)•,
αS(1)•, and
αS(2)• is the monomeric sulfur radical cation (
S•+), which is formed directly from
HOS•. Comparing the
G-values of transients at 10 μs (when all
HOS• have undergone full transformations into the secondary products), it is clearly visible that
αS• are formed with the largest efficiency, which means that the deprotonation of
S•+ is more efficient than the formation of
SN• (
Table 1). Moreover, the formation of the six-membered
SN(2)• is kinetically less favorable than the formation of the five-membered
SN(1)•. As a consequence, the smallest amount of
S●+ can be converted to
SN(2)•. It has to be also noted that not all
SN(2)● radicals can be quantitatively converted into
αC(2)• since
SN(2)• exists in an equilibrium with
S•+ (cf.
Scheme 1). In the case of
2, the formation of
SO•+ does not exclusively occur via
S•+. A substantial amount of five-membered
SO•+ is formed directly from
HOS•, which is the reason for a more efficient formation of
αC(2)• in
2 (
Scheme 5). Contrary to
1, at 10 μs the
G-value of a direct precursor of
αC(2)• in
2 is comparable with the
G-value of
αS(1)• and
αS(2)• (cf.
Table 1 and
Table 2).
The third difference between these two compounds that needs to be explained is the negligible
SS•+ contribution in transient absorption spectra recorded in solutions containing
2 (vide
Figure 1B and
Table 2). There are several possible scenarios that can be taken into account. The formation of
SO•+ directly from
HOS•, excluding
S•+, which is also a direct precursor of
SS•+, can only partly explain this observation. Furthermore, based on our previous studies on S-methylglutathione [
20], where the rate constant of fast formation of five-membered
SO•+ directly from
S•+ was estimated with the lower limit as >6 × 10
7 s
−1, the slow formation of
SS•+ (taking into account the low concentration of
2) is the next possible rationalization. In the case of
1, the rate constant of five-membered
SN(1)• formation directly from
S•+ was not measured earlier, however, it must be lower in comparison to the rate constant of the analogous reaction leading to
SO•+ (vide supra).
The fourth difference between
1 and
2 is the lack of
αC(1)• formation in the case of
2 (vide
Table 2). This observation can be rationalized by the unimportance of the SN
● formation, in particular of SN(1)
●, due to its unstable four-membered structure. Moreover, an additional possible potential reaction pathway leading to
αC(1)• formation via direct H-abstraction seems to be also unimportant. The lack of
αC(1)• formation was further confirmed by the lack of final products involving their contribution (cf.
Table 3 and
Table 4) (vide discussion in 2.2 addressing the issue of
αC(1)• absence in
2).
2.2. γ-Radiolysis and Product Analysis
In addition to the reactive species e
aq−, HO
•, and H
•, the radiolysis of neutral water leads also to H
+ (0.28) and H
2O
2 (0.07); in parenthesis:
G in µmol J
−1 [
23]. In N
2O-saturated solution, the
G(HO
•) = 0.56 µmol J
−1, therefore HO
• and H
• account for 90% and 10%, respectively, of the reactive species (cf. Reactions (1) and (2)).
N
2O-saturated solutions containing compounds
1 or
2 (1.0 mM) at natural pH were irradiated for 800 Gy under stationary state conditions with a dose rate of 46.7 Gy min
−1 followed by LC–MS and high-resolution MS/MS analysis. Representative HPLC runs of irradiated samples are shown in
Figure 3 and
Figure 4 for compounds
1 and
2, respectively.
It is well documented from previous studies on Met derivatives that the sulfoxide
3 is formed from the in-situ generation of hydrogen peroxide [
11,
12], whereas the H
• addition to the sulfur center (
k = 1.7 × 10
9 M
−1 s
−1) with the formation of a sulfuranyl radical intermediate affords compound
4 and the CH
3S
• radical (
Scheme 6) [
24,
25]. In analogy, we expected the same reactions with starting material
2 giving
5 and
6, respectively. It is worth underlining that these reactions are not detectable in the pulse-radiolysis study, therefore their existence is based on the final-product identifications.
Figure 3 shows the HPLC chromatogram obtained by the injection of the crude reaction mixture of compound
1 with a separation of 24 peaks including the starting material
1 (peak 3), whereas
Figure 4 shows the chromatogram obtained from the reaction of compound
2 that contains 15 peaks including the starting material
2 (peak 3). All peaks were identified, and their chemical structures assigned by examination of their high-resolution mass data and characteristic fragmentation patterns (see
Supporting Materials). In both cases, to the left of the starting material
1 or
2 we could identify the presence of sulfoxides
3 or
5 (peak 1) and the desulfurization compounds
4 or
6 (peak 2), respectively (cf.
Figures S3 and S7). The desulfurization process with the formation of the thiyl radical (CH
3S
•) depicted in
Scheme 6 is extremely important, because some of the products derive from the combination of CH
3S
• with carbon-centered radicals.
From the pulse radiolysis studies described above, the reactions of HO
• with
1 or
2 were depicted following several paths. In
Scheme 4 and
Scheme 5 the formation of the adduct radical (
HOS•) is the main path, whereas four minor ones were identified as follows: the H-atom abstraction from the CH
2-S-CH
3 moiety to give the two
αS• radicals and the H-atom abstraction from the N-CH-CO or NHCH
3 moiety to give the two
αC• radicals. The
HOS• follows a first-order decay (
kd = 5.6 x 10
5 s
−1) by HO
− elimination to give the sulfide radical cation, which is at the crossroad of various possible reactions affording the intermediates
SS•+,
αS•,
SN•, and
αC• for compound
1, and
SS•+,
αS•,
SO•+, and
αC• for compound
2.
We suggest that in both starting materials the formation of the disulfide radical cation (
SS•+), which is in equilibrium with the sulfide radical cation (S
•+) and the starting material, is followed by the fragmentation of
SS•+ to afford the observed disulfide
7 or
8 (
Scheme 7, cf.
Figures S3 and S7). Analogous products are reported in radiolytic studies of other methionine derivatives [
10,
11]. Such an asymmetrical disulfide was found to be the major product in the tripeptide Gly-Met-Gly transformation [
12].
All the remaining peaks in both cases derived from the combination of two radicals.
Table 3 and
Table 4 collect the product formations as well as the precursor radicals in each case. The reaction of CH
3S
•, generated from the reaction of H
• with the substrates, can combine with various carbon-centered radicals to give the corresponding sulfides. Indeed, CH
3S
• reacts with
αS(1)•,
αS(2)•,
αC(1)•, and
αC(2)• for compound
1 (
Table 3) and with
αS(1)•,
αS(2)•, and
αC(2)• for compound
2 (
Table 4). Both tables report also the relative ratios based on the intensity of the peaks in the chromatograms. We suggest that the CH
3S
• adduct is like a footprint of the relative concentrations of the four carbon-centered radicals. Based on this rationalization, the relative concentrations of
αS(2)•,
αS(1)•,
αC(1)•, and
αC(2)• are 15.4/9.4/1.4/1.0 for the methionine derivative (
Table 3) whereas the relative concentrations of
αS(2)•,
αS(1)•, and
αC(2)• are 72.2/1.0/2.3 for the cysteine-methylated derivative (
Table 4). The absence of
αC(1)• in the pulse radiolysis simulation of the transients derived from compound
2 can be attributed to fast and well documented β-fragmentation of thiyl radical (
Scheme 8). Since no evidence of formation of compound
9 is obtained by LC–MS and high-resolution MS/MS analysis, we concluded that this path is unimportant. This confirms the unimportance of SN
• formation in the radiation chemistry of compound
2 (cf.
Scheme 4 and
Scheme 5). Although
αS• radicals were the most abundant in both experiments (
αC• was at least an order of magnitude smaller than
αS•), the relative percentage of the two isomers changed profoundly going from compound
1 to
2, i.e., the ratio
αS(2)•/
αS(1)• is 15.4/9.4 and 72.2/1.0, respectively.
It is expected for an
αS(2)• radical to be found at a higher concentration than
αS(1)• and in line with the higher stability of secondary vs. primary alkyl radical due to favorable deprotonation from the precursor sulfide radical cation. In the case of compound
1, the analysis of high-resolution MS/MS spectra was in favor of
αS(2)•/
αS(1)• by 15.4/9.4 (see
Scheme S1). For compound
2 on the other hand, the major peak fits very well with both αS(2)—SCH
3 and αS(1)—SCH
3 (see
Scheme S2); since the number of observed diastereoisomers fits only with higher concentrations of
αS(2)• radicals, we assigned the ratio 72.2/1.0 to
αS(2)•/
αS(1)•.
It has to be stressed that the above observations cannot be directly confirmed by the pulse-radiolysis study described above. It was only seen that αS• are formed with slightly different efficiencies in compounds 1 and 2 (cf. 0.28 μM vs. 0.23 μM, respectively). Unfortunately, it was not possible to distinguish between the two αS(2)• and αS(1)• radicals and as a consequence to obtain directly their respective G-values in both compounds and to extract directly the kinetics of their formation.
However, these interesting observations need some comments and explanations. Since the main source of
αS• is the α-deprotonation of
S●+ [
26], it seems obvious that the deprotonation of
S•+ to form
αS(2)• is much faster for
2 than for
1. As it was stated earlier [
27], the efficiency of α-deprotonation strongly depends on the molecular structure of S
•+ and, to be more specific, on the relative alignment of the singly occupied sulfur p-orbital with the C−H σ-bond to be cleaved. This fact was nicely illustrated by deprotonation kinetics on the example of the monomeric sulfur radical cations derived from di-isopropyl sulfide (
i-(C
3H
7)
2S) and from 9-thia-bicyclo-[3.3.1]-nonane. In the first case a high probability of parallel alignment of the sulfur p-orbital and the C−H σ-orbitals, facilitated by the steric influence of the two methyl groups in combination with the free rotation around the C–S bond, accelerates α-deprotonation of
i-(C
3H
7)
2S
●+. On the other hand, in second case, the neighboring C−H bonds are fixed in a configuration perpendicular to the sulfur p-orbital, which practically prevents deprotonation [
2]. Perhaps, for compound
2 exists the more favorable conformational arrangement in the
S●+, than for compound
1, for deprotonation leading to
α-S(2)•. This can be facilitated, similarly as in
i-(C
3H
7)
2S
●+, by the steric influence of the peptide backbone that is located closer in
2 affording a better parallel alignment of the sulfur p-orbital and the C−H σ-orbitals in the CH
2 group in comparison to
1.
The data from the high-resolution MS/MS spectra showed 16 dimeric compounds that are derived from the radiolytic study of starting material
1 (
Figure S5). The accurate masses of these products (
m/
z 407.1781) correspond to a molecular weight MH
+ equivalent to two radicals (
αS• and/or
αC•) (see
Table 3). In comparison, eight dimeric compounds derived starting from compound
2 with MH
+ m/
z 379.1468, equivalent to two radicals (
αS• and/or
αC•) (
Figure S9). In all cases, the fragmentation patterns are not diagnostic, all of them showing an initial fragmentation step with loss of CH
3NH
2.
Further structural information may be obtained of all these dimers by taking into consideration the relative ratio of CH
3S
• adducts and from the analysis of potential diasteroisomers (
Table 3 and
Table 4). For example, the dimer αS(2)—αS(2) is expected to be the most favorable formed since the α
S(2)• radical is in a higher concentration.
Figure 5 and
Figure 6 show that αS(2)—αS(2) has four stereocenters, two are from the starting material fixed at the S configuration whereas two new stereocenters generated from the self-termination can be
R or
S. In a total of four products, two of them are identical, therefore we expect to have
SSSS,
SRSS, and
SRRS diastereoisomers. The α
S(2)• radical is likely to combine also with
αS(1)•,
αC(1)•, and
αC(2)•. In analogy, the αS(2)—αC(1) is expected to be formed as four diastereoisomers (
Figure 5), whereas both αS(2)—αS(1) and αS(2)—αC(2) afford only two
SSS and
SRS isomers (
Figure 5 and
Figure 6). Due to the relative high concentration of the
αS(1)• radical in the experiment with compound
1, a combination of products of
αS(1)• with
αS(1)•,
αC(1)•, and
αC(2)• are also expected (
Table 3—total four diastereoisomers).


 ,
, 


 e−aq (0.28), HO• (0.28), H• (0.06)
e−aq (0.28), HO• (0.28), H• (0.06)















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}