
Linking Late Endosomal Cholesterol with Cancer Progression and Anticancer Drug Resistance

 , , , and
, , , and 
Abstract
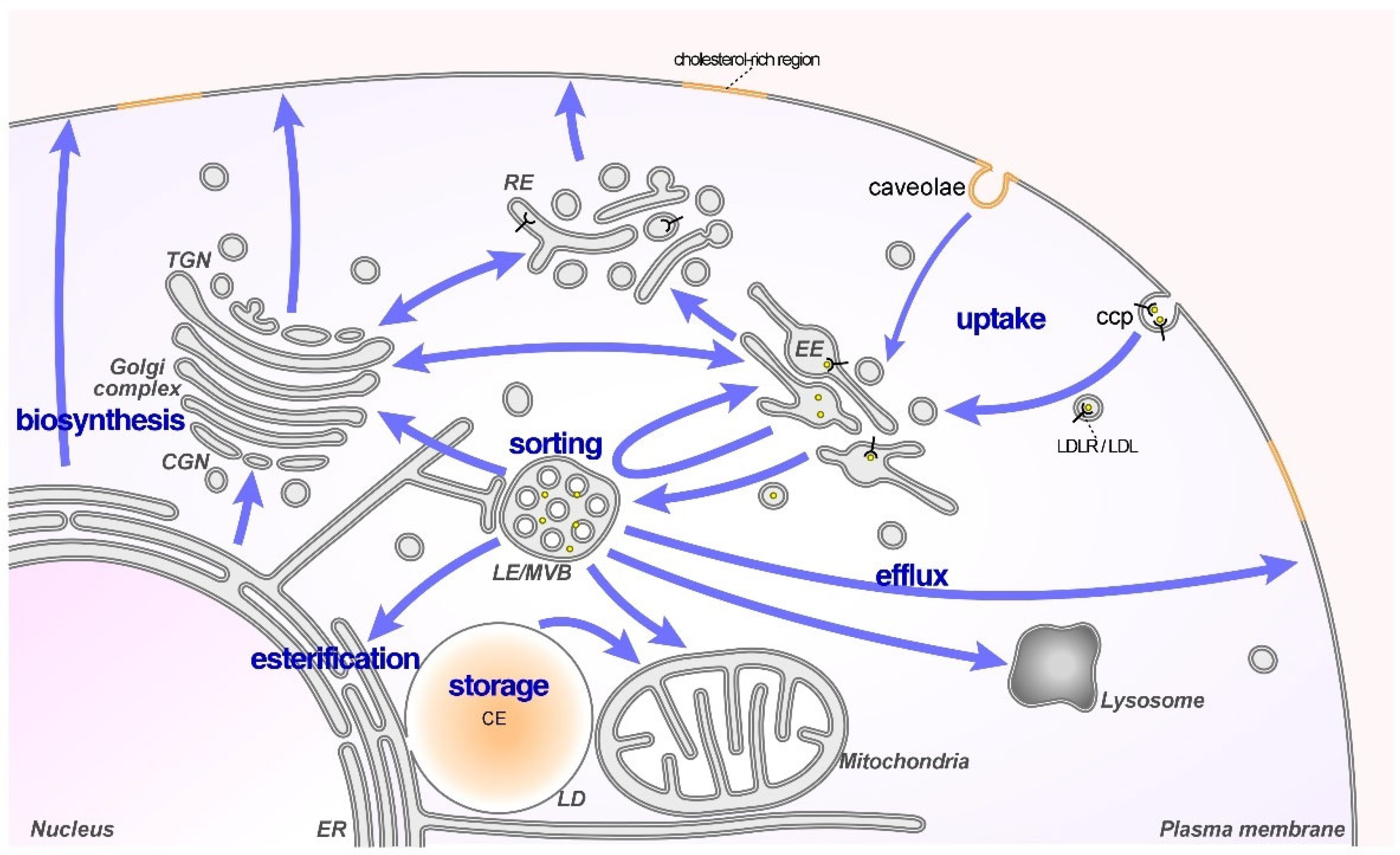
:1. Introduction
2. LDL-Cholesterol: A Risk Factor for Cancer
Increased LDL-Cholesterol Uptake Supports Cancer Growth and Progression
3. Cholesterol Transporters in LE/Lys Contribute to Cancer Cell Behavior
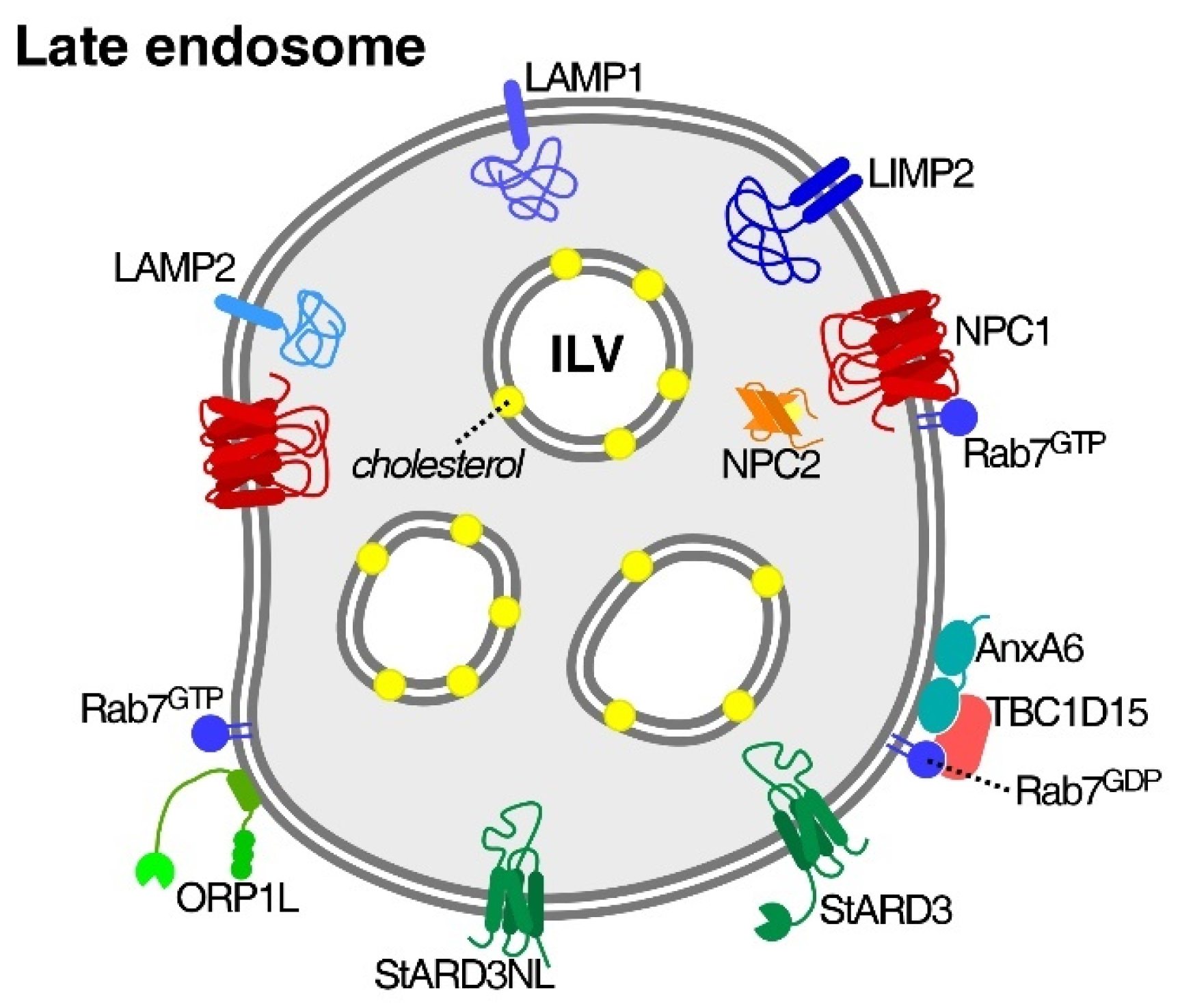
3.1. Niemann–Pick Type C1/2 (NPC1/2) Proteins
3.2. Cholesterol-Sensitive Mechanisms in LE/Lys That Influence Cancer Cell Activities
3.3. NPC1 and Cholesterol Transport Routes to Focal Adhesions
3.4. NPC1 Influences SNARE-Dependent Cell Surface Delivery of ECM Proteins and Integrins
4. Potential Roles of LE/Lys-Chol Transfer across Membrane Contact Sites for Cancer Cell Actions
4.1. StARD3
4.2. ORP Proteins
4.3. Rab Proteins
4.4. Annexin A6
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef] [Green Version]
- Hoy, A.J.; Nagarajan, S.R.; Butler, L.M. Tumour fatty acid metabolism in the context of therapy resistance and obesity. Nat. Rev. Cancer 2021, 21, 753–766. [Google Scholar] [CrossRef]
- Ding, X.; Zhang, W.; Li, S.; Yang, H. The role of cholesterol metabolism in cancer. Am. J. Cancer Res. 2019, 9, 219–227. [Google Scholar]
- Gabitova, L.; Gorin, A.; Astsaturov, I. Molecular pathways: Sterols and receptor signaling in cancer. Clin. Cancer Res. 2014, 20, 28–34. [Google Scholar] [CrossRef] [Green Version]
- Harayama, T.; Riezman, H. Understanding the diversity of membrane lipid composition. Nat. Rev. Mol. Cell Biol. 2018, 19, 281–296. [Google Scholar] [CrossRef]
- Maxfield, F.R.; van Meer, G. Cholesterol, the central lipid of mammalian cells. Curr. Opin. Cell Biol. 2010, 22, 422–449. [Google Scholar] [CrossRef] [Green Version]
- Brown, M.S.; Goldstein, J.L. A receptor-mediated pathway for cholesterol homeostasis. Science 1986, 232, 34–47. [Google Scholar] [CrossRef] [Green Version]
- Chang, T.Y.; Chang, C.C.; Ohgami, N.; Yamauchi, Y. Cholesterol sensing, trafficking, and esterification. Annu. Rev. Cell Dev. Biol. 2006, 22, 129–157. [Google Scholar] [CrossRef]
- Enrich, C.; Rentero, C.; Grewal, T.; Futter, C.E.; Eden, E.R. Cholesterol overload: Contact sites to the rescue! Contact 2019, 2, 2515256419893507. [Google Scholar] [CrossRef] [Green Version]
- Enrich, C.; Rentero, C.; Hierro, A.; Grewal, T. Role of cholesterol in snare-mediated trafficking on intracellular membranes. J. Cell Sci. 2015, 128, 1071–1081. [Google Scholar] [CrossRef] [Green Version]
- Ikonen, E. Mechanisms of cellular cholesterol compartmentalization: Recent insights. Curr. Opin. Cell Biol. 2018, 53, 77–83. [Google Scholar] [CrossRef]
- Meng, Y.; Heybrock, S.; Neculai, D.; Saftig, P. Cholesterol handling in lysosomes and beyond. Trends Cell Biol. 2020, 30, 452–466. [Google Scholar] [CrossRef]
- Raftopulos, N.L.; Washaya, T.C.; Niederprum, A.; Egert, A.; Hakeem-Sanni, M.F.; Varney, B.; Aishah, A.; Georgieva, M.L.; Olsson, E.; Dos Santos, D.Z.; et al. Prostate cancer cell proliferation is influenced by ldl-cholesterol availability and cholesteryl ester turnover. Cancer Metab. 2022, 10, 1. [Google Scholar] [CrossRef]
- Yan, A.; Jia, Z.; Qiao, C.; Wang, M.; Ding, X. Cholesterol metabolism in drugresistant cancer (review). Int. J. Oncol. 2020, 57, 1103–1115. [Google Scholar] [CrossRef]
- Huang, B.; Song, B.L.; Xu, C. Cholesterol metabolism in cancer: Mechanisms and therapeutic opportunities. Nat. Metab. 2020, 2, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Zhou, S.; Tang, Q.; Xia, H.; Bi, F. Cholesterol metabolism: New functions and therapeutic approaches in cancer. Biochim. Biophys. Acta Rev. Cancer 2020, 1874, 188394. [Google Scholar] [CrossRef]
- Johnson, K.E.; Siewert, K.M.; Klarin, D.; Damrauer, S.M.; Program, V.A.M.V.; Chang, K.M.; Tsao, P.S.; Assimes, T.L.; Maxwell, K.N.; Voight, B.F. The relationship between circulating lipids and breast cancer risk: A mendelian randomization study. PLoS Med. 2020, 17, e1003302. [Google Scholar] [CrossRef]
- Jeon, J.C.; Park, J.; Park, S.; Moon, K.H.; Cheon, S.H.; Park, S. Hypercholesterolemia is associated with a shorter time to castration-resistant prostate cancer in patients who have undergone androgen deprivation therapy. World J. Mens Health 2016, 34, 28–33. [Google Scholar] [CrossRef] [Green Version]
- Llaverias, G.; Danilo, C.; Mercier, I.; Daumer, K.; Capozza, F.; Williams, T.M.; Sotgia, F.; Lisanti, M.P.; Frank, P.G. Role of cholesterol in the development and progression of breast cancer. Am. J. Pathol. 2011, 178, 402–412. [Google Scholar] [CrossRef]
- Huang, P.; Nedelcu, D.; Watanabe, M.; Jao, C.; Kim, Y.; Liu, J.; Salic, A. Cellular cholesterol directly activates smoothened in hedgehog signaling. Cell 2016, 166, 1176–1187.e14. [Google Scholar] [CrossRef] [Green Version]
- Sheng, R.; Chen, Y.; Gee, H.Y.; Stec, E.; Melowic, H.R.; Blatner, N.R.; Tun, M.P.; Kim, Y.; Kallberg, M.; Fujiwara, T.K.; et al. Cholesterol modulates cell signaling and protein networking by specifically interacting with pdz domain-containing scaffold proteins. Nat. Commun. 2012, 3, 1249. [Google Scholar] [CrossRef] [Green Version]
- Castellano, M.B.; Thelen, A.M.; Moldavski, O.; Feltes, M.; van der Welle, R.E.; Mydock-McGrane, L.; Jiang, X.; van Eijkeren, R.J.; Davis, O.B.; Louie, S.M.; et al. Lysosomal cholesterol activates mtorc1 via an slc38a9-niemann-pick c1 signaling complex. Science 2017, 355, 1306–1311. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Ruiz, C.; de la Rosa, L.C.; Ribas, V.; Fernandez-Checa, J.C. Mitochondrial cholesterol and cancer. Semin. Cancer Biol. 2021, 73, 76–85. [Google Scholar] [CrossRef]
- Acier, A.; Godard, M.; Gassiot, F.; Finetti, P.; Rubis, M.; Nowak, J.; Bertucci, F.; Iovanna, J.L.; Tomasini, R.; Lecorche, P.; et al. Ldl receptor-peptide conjugate as in vivo tool for specific targeting of pancreatic ductal adenocarcinoma. Commun. Biol. 2021, 4, 987. [Google Scholar] [CrossRef]
- Guillaumond, F.; Bidaut, G.; Ouaissi, M.; Servais, S.; Gouirand, V.; Olivares, O.; Lac, S.; Borge, L.; Roques, J.; Gayet, O.; et al. Cholesterol uptake disruption, in association with chemotherapy, is a promising combined metabolic therapy for pancreatic adenocarcinoma. Proc. Natl. Acad. Sci. USA 2015, 112, 2473–2478. [Google Scholar] [CrossRef] [Green Version]
- Antalis, C.J.; Uchida, A.; Buhman, K.K.; Siddiqui, R.A. Migration of mda-mb-231 breast cancer cells depends on the availability of exogenous lipids and cholesterol esterification. Clin. Exp. Metastasis 2011, 28, 733–741. [Google Scholar] [CrossRef]
- Feldt, M.; Menard, J.; Rosendahl, A.H.; Lettiero, B.; Bendahl, P.O.; Belting, M.; Borgquist, S. The effect of statin treatment on intratumoral cholesterol levels and ldl receptor expression: A window-of-opportunity breast cancer trial. Cancer Metab. 2020, 8, 25. [Google Scholar] [CrossRef]
- Bhat, M.; Skill, N.; Marcus, V.; Deschenes, M.; Tan, X.; Bouteaud, J.; Negi, S.; Awan, Z.; Aikin, R.; Kwan, J.; et al. Decreased pcsk9 expression in human hepatocellular carcinoma. BMC Gastroenterol. 2015, 15, 176. [Google Scholar] [CrossRef] [Green Version]
- Yen, C.F.; Kalunta, C.I.; Chen, F.S.; Kaptein, J.S.; Lin, C.K.; Lad, P.M. Regulation of low-density lipoprotein receptors and assessment of their functional role in burkitt’s lymphoma cells. Biochim. Biophys. Acta 1995, 1257, 47–57. [Google Scholar] [CrossRef]
- Gueddari, N.; Favre, G.; Hachem, H.; Marek, E.; le Gaillard, F.; Soula, G. Evidence for up-regulated low density lipoprotein receptor in human lung adenocarcinoma cell line a549. Biochimie 1993, 75, 811–819. [Google Scholar] [CrossRef]
- Vitols, S.; Peterson, C.; Larsson, O.; Holm, P.; Aberg, B. Elevated uptake of low density lipoproteins by human lung cancer tissue in vivo. Cancer Res. 1992, 52, 6244–6247. [Google Scholar]
- Lum, D.F.; McQuaid, K.R.; Gilbertson, V.L.; Hughes-Fulford, M. Coordinate up-regulation of low-density lipoprotein receptor and cyclo-oxygenase-2 gene expression in human colorectal cells and in colorectal adenocarcinoma biopsies. Int. J. Cancer 1999, 83, 162–166. [Google Scholar] [CrossRef]
- Niendorf, A.; Nagele, H.; Gerding, D.; Meyer-Pannwitt, U.; Gebhardt, A. Increased ldl receptor mrna expression in colon cancer is correlated with a rise in plasma cholesterol levels after curative surgery. Int. J. Cancer 1995, 61, 461–464. [Google Scholar] [CrossRef]
- Daker, M.; Bhuvanendran, S.; Ahmad, M.; Takada, K.; Khoo, A.S. Deregulation of lipid metabolism pathway genes in nasopharyngeal carcinoma cells. Mol. Med. Rep. 2013, 7, 731–741. [Google Scholar] [CrossRef] [Green Version]
- Guo, D.; Reinitz, F.; Youssef, M.; Hong, C.; Nathanson, D.; Akhavan, D.; Kuga, D.; Amzajerdi, A.N.; Soto, H.; Zhu, S.; et al. An lxr agonist promotes glioblastoma cell death through inhibition of an egfr/akt/srebp-1/ldlr-dependent pathway. Cancer Discov. 2011, 1, 442–456. [Google Scholar] [CrossRef] [Green Version]
- Vitols, S.; Gahrton, G.; Ost, A.; Peterson, C. Elevated low density lipoprotein receptor activity in leukemic cells with monocytic differentiation. Blood 1984, 63, 1186–1193. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Guo, P.; Dong, K.; Guo, P.; Wang, H.; Lv, X. Identification of key biomarkers and potential molecular mechanisms in renal cell carcinoma by bioinformatics analysis. J. Comput. Biol. 2019, 26, 1278–1295. [Google Scholar] [CrossRef]
- Alexopoulos, C.G.; Blatsios, B.; Avgerinos, A. Serum lipids and lipoprotein disorders in cancer patients. Cancer 1987, 60, 3065–3070. [Google Scholar] [CrossRef]
- Budd, D.; Ginsberg, H. Hypocholesterolemia and acute myelogenous leukemia. Association between disease activity and plasma low-density lipoprotein cholesterol concentrations. Cancer 1986, 58, 1361–1365. [Google Scholar] [CrossRef]
- Vitols, S.; Gahrton, G.; Bjorkholm, M.; Peterson, C. Hypocholesterolaemia in malignancy due to elevated low-density-lipoprotein-receptor activity in tumour cells: Evidence from studies in patients with leukaemia. Lancet 1985, 2, 1150–1154. [Google Scholar] [CrossRef]
- Hu, J.; la Vecchia, C.; Negri, E.; de Groh, M.; Morrison, H.; Mery, L. The Canadian Cancer Registries Epidemiology Research. Macronutrient intake and stomach cancer. Cancer Causes Control 2015, 26, 839–847. [Google Scholar] [CrossRef] [PubMed]
- Moon, H.; Ruelcke, J.E.; Choi, E.; Sharpe, L.J.; Nassar, Z.D.; Bielefeldt-Ohmann, H.; Parat, M.O.; Shah, A.; Francois, M.; Inder, K.L.; et al. Diet-induced hypercholesterolemia promotes androgen-independent prostate cancer metastasis via iqgap1 and caveolin-1. Oncotarget 2015, 6, 7438–7453. [Google Scholar] [CrossRef] [Green Version]
- Munir, R.; Usman, H.; Hasnain, S.; Smans, K.; Kalbacher, H.; Zaidi, N. Atypical plasma lipid profile in cancer patients: Cause or consequence? Biochimie 2014, 102, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Chao, F.C.; Efron, B.; Wolf, P. The possible prognostic usefulness of assessing serum proteins and cholesterol in malignancy. Cancer 1975, 35, 1223–1229. [Google Scholar] [CrossRef]
- Henriksson, P.; Eriksson, M.; Ericsson, S.; Rudling, M.; Stege, R.; Berglund, L.; Angelin, B. Hypocholesterolaemia and increased elimination of low-density lipoproteins in metastatic cancer of the prostate. Lancet 1989, 2, 1178–1180. [Google Scholar] [CrossRef]
- Chen, Z.; Chen, L.; Sun, B.; Liu, D.; He, Y.; Qi, L.; Li, G.; Han, Z.; Zhan, L.; Zhang, S.; et al. Ldlr inhibition promotes hepatocellular carcinoma proliferation and metastasis by elevating intracellular cholesterol synthesis through the mek/erk signaling pathway. Mol. Metab. 2021, 51, 101230. [Google Scholar] [CrossRef] [PubMed]
- Stopsack, K.H.; Gerke, T.A.; Andren, O.; Andersson, S.O.; Giovannucci, E.L.; Mucci, L.A.; Rider, J.R. Cholesterol uptake and regulation in high-grade and lethal prostate cancers. Carcinogenesis 2017, 38, 806–811. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Feng, Y.; Zhang, G.; Xu, Y. The endonuclease ape1 processes mir-92b formation, thereby regulating expression of the tumor suppressor ldlr in cervical cancer cells. Ther. Adv. Med. Oncol. 2019, 11, 1758835919855859. [Google Scholar] [CrossRef] [Green Version]
- Jamalzei, B.; Tehrani, F.S.K.; Atri, M. Evaluation of ldl receptor and scavenger receptor, class b, type 1 in the malignant and benign breast tumors: The correlation with the expression of mir-199a-5p, mir-199b-5p and mir-455-5p. Gene 2020, 749, 144720. [Google Scholar] [CrossRef]
- Pires, L.A.; Hegg, R.; Freitas, F.R.; Tavares, E.R.; Almeida, C.P.; Baracat, E.C.; Maranhao, R.C. Effect of neoadjuvant chemotherapy on low-density lipoprotein (ldl) receptor and ldl receptor-related protein 1 (lrp-1) receptor in locally advanced breast cancer. Braz. J. Med. Biol. Res. 2012, 45, 557–564. [Google Scholar] [CrossRef] [Green Version]
- Ho, Y.K.; Smith, R.G.; Brown, M.S.; Goldstein, J.L. Low-density lipoprotein (ldl) receptor activity in human acute myelogenous leukemia cells. Blood 1978, 52, 1099–1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallagher, E.J.; Zelenko, Z.; Neel, B.A.; Antoniou, I.M.; Rajan, L.; Kase, N.; LeRoith, D. Elevated tumor ldlr expression accelerates ldl cholesterol-mediated breast cancer growth in mouse models of hyperlipidemia. Oncogene 2017, 36, 6462–6471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furuya, Y.; Sekine, Y.; Kato, H.; Miyazawa, Y.; Koike, H.; Suzuki, K. Low-density lipoprotein receptors play an important role in the inhibition of prostate cancer cell proliferation by statins. Prostate Int. 2016, 4, 56–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes-Fulford, M.; Chen, Y.; Tjandrawinata, R.R. Fatty acid regulates gene expression and growth of human prostate cancer pc-3 cells. Carcinogenesis 2001, 22, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Sekine, Y.; Koike, H.; Nakano, T.; Nakajima, K.; Takahashi, S.; Suzuki, K. Remnant lipoproteins induced proliferation of human prostate cancer cell, pc-3 but not lncap, via low density lipoprotein receptor. Cancer Epidemiol. 2009, 33, 16–23. [Google Scholar] [CrossRef]
- Floeth, M.; Elges, S.; Gerss, J.; Schwoppe, C.; Kessler, T.; Herold, T.; Wardelmann, E.; Berdel, W.E.; Lenz, G.; Mikesch, J.H.; et al. Low-density lipoprotein receptor (ldlr) is an independent adverse prognostic factor in acute myeloid leukaemia. Br. J. Haematol. 2021, 192, 494–503. [Google Scholar] [CrossRef]
- Gonias, S.L.; Karimi-Mostowfi, N.; Murray, S.S.; Mantuano, E.; Gilder, A.S. Expression of ldl receptor-related proteins (lrps) in common solid malignancies correlates with patient survival. PLoS ONE 2017, 12, e0186649. [Google Scholar] [CrossRef] [Green Version]
- Rudling, M.J.; Stahle, L.; Peterson, C.O.; Skoog, L. Content of low density lipoprotein receptors in breast cancer tissue related to survival of patients. Br. Med. J. Clin. Res. Ed. 1986, 292, 580–582. [Google Scholar] [CrossRef] [Green Version]
- Chang, W.C.; Wang, H.C.; Cheng, W.C.; Yang, J.C.; Chung, W.M.; Ho, Y.P.; Chen, L.; Hung, Y.C.; Ma, W.L. Ldlr-mediated lipidome-transcriptome reprogramming in cisplatin insensitivity. Endocr. Relat. Cancer 2020, 27, 81–95. [Google Scholar] [CrossRef]
- Li, J.; Qu, X.; Tian, J.; Zhang, J.T.; Cheng, J.X. Cholesterol esterification inhibition and gemcitabine synergistically suppress pancreatic ductal adenocarcinoma proliferation. PLoS ONE 2018, 13, e0193318. [Google Scholar] [CrossRef] [Green Version]
- Yue, S.; Li, J.; Lee, S.Y.; Lee, H.J.; Shao, T.; Song, B.; Cheng, L.; Masterson, T.A.; Liu, X.; Ratliff, T.L.; et al. Cholesteryl ester accumulation induced by pten loss and pi3k/akt activation underlies human prostate cancer aggressiveness. Cell Metab. 2014, 19, 393–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Infante, R.E.; Wang, M.L.; Radhakrishnan, A.; Kwon, H.J.; Brown, M.S.; Goldstein, J.L. Npc2 facilitates bidirectional transfer of cholesterol between npc1 and lipid bilayers, a step in cholesterol egress from lysosomes. Proc. Natl. Acad. Sci. USA 2008, 105, 15287–15292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.L.; Motamed, M.; Infante, R.E.; Abi-Mosleh, L.; Kwon, H.J.; Brown, M.S.; Goldstein, J.L. Identification of surface residues on niemann-pick c2 essential for hydrophobic handoff of cholesterol to npc1 in lysosomes. Cell Metab. 2010, 12, 166–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Y.; Peng, X.; Li, B.; Zhao, G. Development of autophagy signature-based prognostic nomogram for refined glioma survival prognostication. Biomed. Res. Int. 2020, 2020, 1872962. [Google Scholar] [CrossRef]
- Singh, V.; Singh, L.C.; Vasudevan, M.; Chattopadhyay, I.; Borthakar, B.B.; Rai, A.K.; Phukan, R.K.; Sharma, J.; Mahanta, J.; Kataki, A.C.; et al. Esophageal cancer epigenomics and integrome analysis of genome-wide methylation and expression in high risk northeast indian population. OMICS 2015, 19, 688–699. [Google Scholar] [CrossRef]
- Lund, R.R.; Leth-Larsen, R.; Caterino, T.D.; Terp, M.G.; Nissen, J.; Laenkholm, A.V.; Jensen, O.N.; Ditzel, H.J. Nadh-cytochrome b5 reductase 3 promotes colonization and metastasis formation and is a prognostic marker of disease-free and overall survival in estrogen receptor-negative breast cancer. Mol. Cell Proteom. 2015, 14, 2988–2999. [Google Scholar] [CrossRef] [Green Version]
- Du, X.; Zhang, Y.; Jo, S.R.; Liu, X.; Qi, Y.; Osborne, B.; Byrne, F.L.; Smith, G.C.; Turner, N.; Hoehn, K.L.; et al. Akt activation increases cellular cholesterol by promoting the proteasomal degradation of niemann-pick c1. Biochem. J. 2015, 471, 243–253. [Google Scholar] [CrossRef]
- Hoque, M.; Rentero, C.; Conway, J.R.; Murray, R.Z.; Timpson, P.; Enrich, C.; Grewal, T. The cross-talk of ldl-cholesterol with cell motility: Insights from the niemann pick type c1 mutation and altered integrin trafficking. Cell Adh. Migr. 2015, 9, 384–391. [Google Scholar] [CrossRef] [Green Version]
- Jose, J.; Hoque, M.; Engel, J.; Beevi, S.S.; Wahba, M.; Georgieva, M.I.; Murphy, K.J.; Hughes, W.E.; Cochran, B.J.; Lu, A.; et al. Annexin a6 and npc1 regulate ldl-inducible cell migration and distribution of focal adhesions. Sci. Rep. 2022, 12, 596. [Google Scholar] [CrossRef]
- Kanerva, K.; Uronen, R.L.; Blom, T.; Li, S.; Bittman, R.; Lappalainen, P.; Peranen, J.; Raposo, G.; Ikonen, E. Ldl cholesterol recycles to the plasma membrane via a rab8a-myosin5b-actin-dependent membrane transport route. Dev. Cell 2013, 27, 249–262. [Google Scholar] [CrossRef] [Green Version]
- Reverter, M.; Rentero, C.; Garcia-Melero, A.; Hoque, M.; de Muga, S.V.; Alvarez-Guaita, A.; Conway, J.R.; Wood, P.; Cairns, R.; Lykopoulou, L.; et al. Cholesterol regulates syntaxin 6 trafficking at trans-golgi network endosomal boundaries. Cell Rep. 2014, 7, 883–897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, Y.; Duvvuri, M.; Duncan, M.B.; Liu, J.; Krise, J.P. Niemann-pick c1 protein facilitates the efflux of the anticancer drug daunorubicin from cells according to a novel vesicle-mediated pathway. J. Pharmacol. Exp. Ther. 2006, 316, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Naren, D.; Wu, J.; Gong, Y.; Yan, T.; Wang, K.; Xu, W.; Yang, X.; Shi, F.; Shi, R. Niemann-pick disease type c1(npc1) is involved in resistance against imatinib in the imatinib-resistant ph+ acute lymphoblastic leukemia cell line sup-b15/ri. Leuk. Res. 2016, 42, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Cheng, Y.; Abraham, J.M.; Yan, R.; Liu, X.; Chen, W.; Ibrahim, S.; Schroth, G.P.; Ke, X.; He, Y.; et al. Rna sequencing of esophageal adenocarcinomas identifies novel fusion transcripts, including npc1-melk, arising from a complex chromosomal rearrangement. Cancer 2017, 123, 3916–3924. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Gil, J.L.; Bianconi, S.E.; Farhat, N.; Kleiner, D.E.; Nelson, M.; Porter, F.D. Hepatocellular carcinoma as a complication of niemann-pick disease type c1. Am. J. Med. Genet. A 2021, 185, 3111–3117. [Google Scholar] [CrossRef]
- Moussay, E.; Kaoma, T.; Baginska, J.; Muller, A.; van Moer, K.; Nicot, N.; Nazarov, P.V.; Vallar, L.; Chouaib, S.; Berchem, G.; et al. The acquisition of resistance to tnfalpha in breast cancer cells is associated with constitutive activation of autophagy as revealed by a transcriptome analysis using a custom microarray. Autophagy 2011, 7, 760–770. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.J.; Kim, J.; Spaunhurst, K.; Montoya, J.; Khodosh, R.; Chandra, K.; Fu, T.; Gilliam, A.; Molgo, M.; Beachy, P.A.; et al. Open-label, exploratory phase ii trial of oral itraconazole for the treatment of basal cell carcinoma. J. Clin. Oncol. 2014, 32, 745–751. [Google Scholar] [CrossRef]
- Aftab, B.T.; Dobromilskaya, I.; Liu, J.O.; Rudin, C.M. Itraconazole inhibits angiogenesis and tumor growth in non-small cell lung cancer. Cancer Res. 2011, 71, 6764–6772. [Google Scholar] [CrossRef] [Green Version]
- Tsubamoto, H.; Sonoda, T.; Ikuta, S.; Tani, S.; Inoue, K.; Yamanaka, N. Combination chemotherapy with itraconazole for treating metastatic pancreatic cancer in the second-line or additional setting. Anticancer Res. 2015, 35, 4191–4196. [Google Scholar]
- Antonarakis, E.S.; Heath, E.I.; Smith, D.C.; Rathkopf, D.; Blackford, A.L.; Danila, D.C.; King, S.; Frost, A.; Ajiboye, A.S.; Zhao, M.; et al. Repurposing itraconazole as a treatment for advanced prostate cancer: A noncomparative randomized phase ii trial in men with metastatic castration-resistant prostate cancer. Oncologist 2013, 18, 163–173. [Google Scholar] [CrossRef]
- Shimazu, R.; Tanaka, G.; Tomiyama, R.; Kuratomi, Y.; Inokuchi, A. Cepharanthin effect on radiation-induced xerostomia and taste disorder in patients with head and neck cancer. Nihon Jibiinkoka Gakkai Kaiho 2009, 112, 648–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nomoto, S.; Imada, H.; Ohguri, T.; Yahara, K.; Kato, F.; Morioka, T.; Korogi, Y. Effect of cepharanthin in preventing radiation induced normal tissue damage in prostate cancer. Gan To Kagaku Ryoho 2004, 31, 1063–1066. [Google Scholar] [PubMed]
- Gowda, R.; Inamdar, G.S.; Kuzu, O.; Dinavahi, S.S.; Krzeminski, J.; Battu, M.B.; Voleti, S.R.; Amin, S.; Robertson, G.P. Identifying the structure-activity relationship of leelamine necessary for inhibiting intracellular cholesterol transport. Oncotarget 2017, 8, 28260–28277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuzu, O.F.; Gowda, R.; Noory, M.A.; Robertson, G.P. Modulating cancer cell survival by targeting intracellular cholesterol transport. Br. J. Cancer 2017, 117, 513–524. [Google Scholar] [CrossRef] [Green Version]
- Head, S.A.; Shi, W.Q.; Yang, E.J.; Nacev, B.A.; Hong, S.Y.; Pasunooti, K.K.; Li, R.J.; Shim, J.S.; Liu, J.O. Simultaneous targeting of npc1 and vdac1 by itraconazole leads to synergistic inhibition of mtor signaling and angiogenesis. ACS Chem. Biol. 2017, 12, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Dang, Y.; Ren, Y.R.; Liu, J.O. Cholesterol trafficking is required for mtor activation in endothelial cells. Proc. Natl. Acad. Sci. USA 2010, 107, 4764–4769. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Li, J.; Zhang, T.; Zou, L.; Chen, Y.; Wang, K.; Lei, Y.; Yuan, K.; Li, Y.; Lan, J.; et al. Itraconazole suppresses the growth of glioblastoma through induction of autophagy: Involvement of abnormal cholesterol trafficking. Autophagy 2014, 10, 1241–1255. [Google Scholar] [CrossRef] [Green Version]
- Lyu, J.; Yang, E.J.; Head, S.A.; Ai, N.; Zhang, B.; Wu, C.; Li, R.J.; Liu, Y.; Chakravarty, H.; Zhang, S.; et al. Astemizole inhibits mtor signaling and angiogenesis by blocking cholesterol trafficking. Int. J. Biol. Sci. 2018, 14, 1175–1185. [Google Scholar] [CrossRef]
- Davis, O.B.; Shin, H.R.; Lim, C.Y.; Wu, E.Y.; Kukurugya, M.; Maher, C.F.; Perera, R.M.; Ordonez, M.P.; Zoncu, R. Npc1-mtorc1 signaling couples cholesterol sensing to organelle homeostasis and is a targetable pathway in niemann-pick type c. Dev. Cell 2021, 56, 260–276.e7. [Google Scholar] [CrossRef]
- Eid, W.; Dauner, K.; Courtney, K.C.; Gagnon, A.; Parks, R.J.; Sorisky, A.; Zha, X. Mtorc1 activates srebp-2 by suppressing cholesterol trafficking to lysosomes in mammalian cells. Proc. Natl. Acad. Sci. USA 2017, 114, 7999–8004. [Google Scholar] [CrossRef] [Green Version]
- Lim, C.Y.; Davis, O.B.; Shin, H.R.; Zhang, J.; Berdan, C.A.; Jiang, X.; Counihan, J.L.; Ory, D.S.; Nomura, D.K.; Zoncu, R. Er-lysosome contacts enable cholesterol sensing by mtorc1 and drive aberrant growth signalling in niemann-pick type c. Nat. Cell Biol. 2019, 21, 1206–1218. [Google Scholar] [CrossRef] [PubMed]
- Eskelinen, E.L.; Schmidt, C.K.; Neu, S.; Willenborg, M.; Fuertes, G.; Salvador, N.; Tanaka, Y.; Lullmann-Rauch, R.; Hartmann, D.; Heeren, J.; et al. Disturbed cholesterol traffic but normal proteolytic function in lamp-1/lamp-2 double-deficient fibroblasts. Mol. Biol. Cell 2004, 15, 3132–3145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Pfeffer, S.R. Lysosomal membrane glycoproteins bind cholesterol and contribute to lysosomal cholesterol export. eLife 2016, 5, e21635. [Google Scholar] [CrossRef] [PubMed]
- Schneede, A.; Schmidt, C.K.; Holtta-Vuori, M.; Heeren, J.; Willenborg, M.; Blanz, J.; Domanskyy, M.; Breiden, B.; Brodesser, S.; Landgrebe, J.; et al. Role for lamp-2 in endosomal cholesterol transport. J. Cell Mol. Med. 2011, 15, 280–295. [Google Scholar] [CrossRef] [Green Version]
- Alessandrini, F.; Pezze, L.; Ciribilli, Y. Lamps: Shedding light on cancer biology. Semin. Oncol. 2017, 44, 239–253. [Google Scholar] [CrossRef]
- Piao, S.; Amaravadi, R.K. Targeting the lysosome in cancer. Ann. N. Y. Acad. Sci. 2016, 1371, 45–54. [Google Scholar] [CrossRef] [Green Version]
- Saftig, P.; Klumperman, J. Lysosome biogenesis and lysosomal membrane proteins: Trafficking meets function. Nat. Rev. Mol. Cell Biol. 2009, 10, 623–635. [Google Scholar] [CrossRef]
- Binker, M.G.; Cosen-Binker, L.I.; Terebiznik, M.R.; Mallo, G.V.; McCaw, S.E.; Eskelinen, E.L.; Willenborg, M.; Brumell, J.H.; Saftig, P.; Grinstein, S.; et al. Arrested maturation of neisseria-containing phagosomes in the absence of the lysosome-associated membrane proteins, lamp-1 and lamp-2. Cell Microbiol. 2007, 9, 2153–2166. [Google Scholar] [CrossRef]
- Huynh, K.K.; Eskelinen, E.L.; Scott, C.C.; Malevanets, A.; Saftig, P.; Grinstein, S. Lamp proteins are required for fusion of lysosomes with phagosomes. EMBO J. 2007, 26, 313–324. [Google Scholar] [CrossRef]
- Jager, S.; Bucci, C.; Tanida, I.; Ueno, T.; Kominami, E.; Saftig, P.; Eskelinen, E.L. Role for rab7 in maturation of late autophagic vacuoles. J. Cell Sci. 2004, 117, 4837–4848. [Google Scholar] [CrossRef] [Green Version]
- Ding, Z.B.; Fu, X.T.; Shi, Y.H.; Zhou, J.; Peng, Y.F.; Liu, W.R.; Shi, G.M.; Gao, Q.; Wang, X.Y.; Song, K.; et al. Lamp2a is required for tumor growth and promotes tumor recurrence of hepatocellular carcinoma. Int. J. Oncol. 2016, 49, 2367–2376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furuta, K.; Ikeda, M.; Nakayama, Y.; Nakamura, K.; Tanaka, M.; Hamasaki, N.; Himeno, M.; Hamilton, S.R.; August, J.T. Expression of lysosome-associated membrane proteins in human colorectal neoplasms and inflammatory diseases. Am. J. Pathol. 2001, 159, 449–455. [Google Scholar] [CrossRef] [Green Version]
- Kon, M.; Kiffin, R.; Koga, H.; Chapochnick, J.; Macian, F.; Varticovski, L.; Cuervo, A.M. Chaperone-mediated autophagy is required for tumor growth. Sci. Transl. Med. 2011, 3, 109ra17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morell, C.; Bort, A.; Vara-Ciruelos, D.; Ramos-Torres, A.; Altamirano-Dimas, M.; Diaz-Laviada, I.; Rodriguez-Henche, N. Up-regulated expression of lamp2 and autophagy activity during neuroendocrine differentiation of prostate cancer lncap cells. PLoS ONE 2016, 11, e0162977. [Google Scholar] [CrossRef] [Green Version]
- Saha, T. Lamp2a overexpression in breast tumors promotes cancer cell survival via chaperone-mediated autophagy. Autophagy 2012, 8, 1643–1656. [Google Scholar] [CrossRef] [Green Version]
- Zheng, L.; Terman, A.; Hallbeck, M.; Dehvari, N.; Cowburn, R.F.; Benedikz, E.; Kagedal, K.; Cedazo-Minguez, A.; Marcusson, J. Macroautophagy-generated increase of lysosomal amyloid beta-protein mediates oxidant-induced apoptosis of cultured neuroblastoma cells. Autophagy 2011, 7, 1528–1545. [Google Scholar] [CrossRef] [Green Version]
- Bao, L.; Lv, L.; Feng, J.; Chen, Y.; Wang, X.; Han, S.; Zhao, H. Mir-487b-5p regulates temozolomide resistance of lung cancer cells through lamp2-medicated autophagy. DNA Cell Biol. 2016, 35, 385–392. [Google Scholar] [CrossRef]
- Heybrock, S.; Kanerva, K.; Meng, Y.; Ing, C.; Liang, A.; Xiong, Z.J.; Weng, X.; Kim, Y.A.; Collins, R.; Trimble, W.; et al. Lysosomal integral membrane protein-2 (limp-2/scarb2) is involved in lysosomal cholesterol export. Nat. Commun. 2019, 10, 3521. [Google Scholar] [CrossRef]
- Das, A.; Brown, M.S.; Anderson, D.D.; Goldstein, J.L.; Radhakrishnan, A. Three pools of plasma membrane cholesterol and their relation to cholesterol homeostasis. eLife 2014, 3, e02882. [Google Scholar] [CrossRef]
- Trinh, M.N.; Brown, M.S.; Goldstein, J.L.; Han, J.; Vale, G.; McDonald, J.G.; Seemann, J.; Mendell, J.T.; Lu, F. Last step in the path of ldl cholesterol from lysosome to plasma membrane to er is governed by phosphatidylserine. Proc. Natl. Acad. Sci. USA 2020, 117, 18521–18529. [Google Scholar] [CrossRef]
- Caswell, P.T.; Vadrevu, S.; Norman, J.C. Integrins: Masters and slaves of endocytic transport. Nat. Rev. Mol. Cell Biol. 2009, 10, 843–853. [Google Scholar] [CrossRef] [PubMed]
- Alanko, J.; Ivaska, J. Endosomes: Emerging platforms for integrin-mediated fak signalling. Trends Cell Biol. 2016, 26, 391–398. [Google Scholar] [CrossRef]
- Echarri, A.; del Pozo, M.A. Caveolae internalization regulates integrin-dependent signaling pathways. Cell Cycle 2006, 5, 2179–2182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaus, K.; le Lay, S.; Balasubramanian, N.; Schwartz, M.A. Integrin-mediated adhesion regulates membrane order. J. Cell Biol. 2006, 174, 725–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, J.M.; Zheleznyak, A.; Chung, J.; Lindberg, F.P.; Sarfati, M.; Frazier, W.A.; Brown, E.J. Role of cholesterol in formation and function of a signaling complex involving alphavbeta3, integrin-associated protein (cd47), and heterotrimeric g proteins. J. Cell Biol. 1999, 146, 673–682. [Google Scholar] [CrossRef]
- Ramprasad, O.G.; Srinivas, G.; Rao, K.S.; Joshi, P.; Thiery, J.P.; Dufour, S.; Pande, G. Changes in cholesterol levels in the plasma membrane modulate cell signaling and regulate cell adhesion and migration on fibronectin. Cell Motil. Cytoskelet. 2007, 64, 199–216. [Google Scholar] [CrossRef]
- Wang, R.; Bi, J.; Ampah, K.K.; Ba, X.; Liu, W.; Zeng, X. Lipid rafts control human melanoma cell migration by regulating focal adhesion disassembly. Biochim. Biophys. Acta 2013, 1833, 3195–3205. [Google Scholar] [CrossRef] [Green Version]
- Freed-Pastor, W.A.; Mizuno, H.; Zhao, X.; Langerod, A.; Moon, S.H.; Rodriguez-Barrueco, R.; Barsotti, A.; Chicas, A.; Li, W.; Polotskaia, A.; et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 2012, 148, 244–258. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Kanerva, K.; Vanharanta, L.; Almeida-Souza, L.; Lietha, D.; Olkkonen, V.M.; Ikonen, E. Orp2 couples ldl-cholesterol transport to fak activation by endosomal cholesterol/pi(4,5)p2 exchange. EMBO J. 2021, 40, e106871. [Google Scholar] [CrossRef]
- Timpson, P.; Jones, G.E.; Frame, M.C.; Brunton, V.G. Coordination of cell polarization and migration by the rho family gtpases requiRes. src tyrosine kinase activity. Curr. Biol. 2001, 11, 1836–1846. [Google Scholar] [CrossRef] [Green Version]
- Tu, C.; Ortega-Cava, C.F.; Winograd, P.; Stanton, M.J.; Reddi, A.L.; Dodge, I.; Arya, R.; Dimri, M.; Clubb, R.J.; Naramura, M.; et al. Endosomal-sorting complexes required for transport (escrt) pathway-dependent endosomal traffic regulates the localization of active src at focal adhesions. Proc. Natl. Acad. Sci. USA 2010, 107, 16107–16112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dozynkiewicz, M.A.; Jamieson, N.B.; Macpherson, I.; Grindlay, J.; van den Berghe, P.V.; von Thun, A.; Morton, J.P.; Gourley, C.; Timpson, P.; Nixon, C.; et al. Rab25 and clic3 collaborate to promote integrin recycling from late endosomes/lysosomes and drive cancer progression. Dev. Cell 2012, 22, 131–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steffen, A.; le Dez, G.; Poincloux, R.; Recchi, C.; Nassoy, P.; Rottner, K.; Galli, T.; Chavrier, P. Mt1-mmp-dependent invasion is regulated by ti-vamp/vamp7. Curr. Biol. 2008, 18, 926–931. [Google Scholar] [CrossRef] [PubMed]
- Urano, Y.; Watanabe, H.; Murphy, S.R.; Shibuya, Y.; Geng, Y.; Peden, A.A.; Chang, C.C.; Chang, T.Y. Transport of ldl-derived cholesterol from the npc1 compartment to the er involves the trans-golgi network and the snare protein complex. Proc. Natl. Acad. Sci. USA 2008, 105, 16513–16518. [Google Scholar] [CrossRef] [Green Version]
- Choudhury, A.; Sharma, D.K.; Marks, D.L.; Pagano, R.E. Elevated endosomal cholesterol levels in niemann-pick cells inhibit rab4 and perturb membrane recycling. Mol. Biol. Cell 2004, 15, 4500–4511. [Google Scholar] [CrossRef]
- Cubells, L.; de Muga, S.V.; Tebar, F.; Bonventre, J.V.; Balsinde, J.; Pol, A.; Grewal, T.; Enrich, C. Annexin a6-induced inhibition of cytoplasmic phospholipase a2 is linked to caveolin-1 export from the golgi. J. Biol. Chem. 2008, 283, 10174–10183. [Google Scholar] [CrossRef] [Green Version]
- Cubells, L.; de Muga, S.V.; Tebar, F.; Wood, P.; Evans, R.; Ingelmo-Torres, M.; Calvo, M.; Gaus, K.; Pol, A.; Grewal, T.; et al. Annexin a6-induced alterations in cholesterol transport and caveolin export from the golgi complex. Traffic 2007, 8, 1568–1589. [Google Scholar] [CrossRef]
- Ketteler, J.; Klein, D. Caveolin-1, cancer and therapy resistance. Int. J. Cancer 2018, 143, 2092–2104. [Google Scholar] [CrossRef]
- Nassar, Z.D.; Parat, M.O. Caveola-forming proteins and prostate cancer. Cancer Metastasis Rev 2020, 39, 415–433. [Google Scholar] [CrossRef]
- Nwosu, Z.C.; Ebert, M.P.; Dooley, S.; Meyer, C. Caveolin-1 in the regulation of cell metabolism: A cancer perspective. Mol. Cancer 2016, 15, 71. [Google Scholar] [CrossRef] [Green Version]
- Pol, A.; Martin, S.; Fernandez, M.A.; Ingelmo-Torres, M.; Ferguson, C.; Enrich, C.; Parton, R.G. Cholesterol and fatty acids regulate dynamic caveolin trafficking through the golgi complex and between the cell surface and lipid bodies. Mol. Biol. Cell 2005, 16, 2091–2105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez-Guaita, A.; de Muga, S.V.; Owen, D.M.; Williamson, D.; Magenau, A.; Garcia-Melero, A.; Reverter, M.; Hoque, M.; Cairns, R.; Cornely, R.; et al. Evidence for annexin a6-dependent plasma membrane remodelling of lipid domains. Br. J. Pharmacol. 2015, 172, 1677–1690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganley, I.G.; Espinosa, E.; Pfeffer, S.R. A syntaxin 10-snare complex distinguishes two distinct transport routes from endosomes to the trans-golgi in human cells. J. Cell Biol. 2008, 180, 159–172. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Melero, A.; Reverter, M.; Hoque, M.; Meneses-Salas, E.; Koese, M.; Conway, J.R.; Johnsen, C.H.; Alvarez-Guaita, A.; Morales-Paytuvi, F.; Elmaghrabi, Y.A.; et al. Annexin a6 and late endosomal cholesterol modulate integrin recycling and cell migration. J. Biol. Chem. 2016, 291, 1320–1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hulce, J.J.; Cognetta, A.B.; Niphakis, M.J.; Tully, S.E.; Cravatt, B.F. Proteome-wide mapping of cholesterol-interacting proteins in mammalian cells. Nat. Methods 2013, 10, 259–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reverter, M.; Rentero, C.; de Muga, S.V.; Alvarez-Guaita, A.; Mulay, V.; Cairns, R.; Wood, P.; Monastyrskaya, K.; Pol, A.; Tebar, F.; et al. Cholesterol transport from late endosomes to the golgi regulates t-snare trafficking, assembly, and function. Mol. Biol. Cell 2011, 22, 4108–4123. [Google Scholar] [CrossRef]
- Tiwari, A.; Jung, J.J.; Inamdar, S.M.; Brown, C.O.; Goel, A.; Choudhury, A. Endothelial cell migration on fibronectin is regulated by syntaxin 6-mediated alpha5beta1 integrin recycling. J. Biol. Chem. 2011, 286, 36749–36761. [Google Scholar] [CrossRef] [Green Version]
- Gorshtein, G.; Grafinger, O.; Coppolino, M.G. Targeting snare-mediated vesicle transport to block invadopodium-based cancer cell invasion. Front. Oncol. 2021, 11, 679955. [Google Scholar] [CrossRef]
- Riggs, K.A.; Hasan, N.; Humphrey, D.; Raleigh, C.; Nevitt, C.; Corbin, D.; Hu, C. Regulation of integrin endocytic recycling and chemotactic cell migration by syntaxin 6 and vamp3 interaction. J. Cell Sci. 2012, 125, 3827–3839. [Google Scholar] [CrossRef] [Green Version]
- Williams, K.C.; Coppolino, M.G. Phosphorylation of membrane type 1-matrix metalloproteinase (mt1-mmp) and its vesicle-associated membrane protein 7 (vamp7)-dependent trafficking facilitate cell invasion and migration. J. Biol. Chem. 2011, 286, 43405–43416. [Google Scholar] [CrossRef] [Green Version]
- Williams, K.C.; McNeilly, R.E.; Coppolino, M.G. Snap23, syntaxin4, and vesicle-associated membrane protein 7 (vamp7) mediate trafficking of membrane type 1-matrix metalloproteinase (mt1-mmp) during invadopodium formation and tumor cell invasion. Mol. Biol. Cell 2014, 25, 2061–2070. [Google Scholar] [CrossRef] [PubMed]
- Powelka, A.M.; Sun, J.; Li, J.; Gao, M.; Shaw, L.M.; Sonnenberg, A.; Hsu, V.W. Stimulation-dependent recycling of integrin beta1 regulated by arf6 and rab11. Traffic 2004, 5, 20–36. [Google Scholar] [CrossRef] [PubMed]
- Holtta-Vuori, M.; Tanhuanpaa, K.; Mobius, W.; Somerharju, P.; Ikonen, E. Modulation of cellular cholesterol transport and homeostasis by rab11. Mol. Biol. Cell 2002, 13, 3107–3122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choudhury, A.; Dominguez, M.; Puri, V.; Sharma, D.K.; Narita, K.; Wheatley, C.L.; Marks, D.L.; Pagano, R.E. Rab proteins mediate golgi transport of caveola-internalized glycosphingolipids and correct lipid trafficking in niemann-pick c cells. J. Clin. Invest. 2002, 109, 1541–1550. [Google Scholar] [CrossRef]
- Cabukusta, B.; Neefjes, J. Mechanisms of lysosomal positioning and movement. Traffic 2018, 19, 761–769. [Google Scholar] [CrossRef]
- Ridgway, N.D.; Zhao, K. Cholesterol transfer at endosomal-organelle membrane contact sites. Curr. Opin. Lipidol. 2018, 29, 212–217. [Google Scholar] [CrossRef]
- Ballabio, A.; Bonifacino, J.S. Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat. Rev. Mol. Cell Biol. 2020, 21, 101–118. [Google Scholar] [CrossRef]
- Gil-Hernandez, A.; Arroyo-Campuzano, M.; Simoni-Nieves, A.; Zazueta, C.; Gomez-Quiroz, L.E.; Silva-Palacios, A. Relevance of membrane contact sites in cancer progression. Front. Cell Dev. Biol. 2020, 8, 622215. [Google Scholar] [CrossRef]
- Jain, A.; Zoncu, R. Organelle transporters and inter-organelle communication as drivers of metabolic regulation and cellular homeostasis. Mol. Metab. 2022, 60, 101481. [Google Scholar] [CrossRef]
- Peretti, D.; Kim, S.; Tufi, R.; Lev, S. Lipid transfer proteins and membrane contact sites in human cancer. Front. Cell Dev. Biol. 2019, 7, 371. [Google Scholar] [CrossRef] [Green Version]
- Alpy, F.; Rousseau, A.; Schwab, Y.; Legueux, F.; Stoll, I.; Wendling, C.; Spiegelhalter, C.; Kessler, P.; Mathelin, C.; Rio, M.C.; et al. Stard3 or stard3nl and vap form a novel molecular tether between late endosomes and the er. J. Cell Sci. 2013, 126, 5500–5512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alpy, F.; Stoeckel, M.E.; Dierich, A.; Escola, J.M.; Wendling, C.; Chenard, M.P.; Vanier, M.T.; Gruenberg, J.; Tomasetto, C.; Rio, M.C. The steroidogenic acute regulatory protein homolog mln64, a late endosomal cholesterol-binding protein. J. Biol. Chem. 2001, 276, 4261–4269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holtta-Vuori, M.; Alpy, F.; Tanhuanpaa, K.; Jokitalo, E.; Mutka, A.L.; Ikonen, E. Mln64 is involved in actin-mediated dynamics of late endocytic organelles. Mol. Biol. Cell 2005, 16, 3873–3886. [Google Scholar] [CrossRef] [Green Version]
- Liapis, A.; Chen, F.W.; Davies, J.P.; Wang, R.; Ioannou, Y.A. Mln64 transport to the late endosome is regulated by binding to 14-3-3 via a non-canonical binding site. PLoS ONE 2012, 7, e34424. [Google Scholar] [CrossRef]
- Wilhelm, L.P.; Wendling, C.; Vedie, B.; Kobayashi, T.; Chenard, M.P.; Tomasetto, C.; Drin, G.; Alpy, F. Stard3 mediates endoplasmic reticulum-to-endosome cholesterol transport at membrane contact sites. EMBO J. 2017, 36, 1412–1433. [Google Scholar] [CrossRef]
- Zhang, M.; Liu, P.; Dwyer, N.K.; Christenson, L.K.; Fujimoto, T.; Martinez, F.; Comly, M.; Hanover, J.A.; Blanchette-Mackie, E.J.; Strauss, J.F., 3rd. Mln64 mediates mobilization of lysosomal cholesterol to steroidogenic mitochondria. J. Biol. Chem. 2002, 277, 33300–33310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meneses-Salas, E.; Garcia-Melero, A.; Kanerva, K.; Blanco-Munoz, P.; Morales-Paytuvi, F.; Bonjoch, J.; Casas, J.; Egert, A.; Beevi, S.S.; Jose, J.; et al. Annexin a6 modulates tbc1d15/rab7/stard3 axis to control endosomal cholesterol export in npc1 cells. Cell Mol. Life Sci. 2020, 77, 2839–2857. [Google Scholar] [CrossRef] [Green Version]
- Borthwick, F.; Allen, A.M.; Taylor, J.M.; Graham, A. Overexpression of stard3 in human monocyte/macrophages induces an anti-atherogenic lipid phenotype. Clin. Sci. 2010, 119, 265–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balboa, E.; Castro, J.; Pinochet, M.J.; Cancino, G.I.; Matias, N.; Saez, P.J.; Martinez, A.; Alvarez, A.R.; Garcia-Ruiz, C.; Fernandez-Checa, J.C.; et al. Mln64 induces mitochondrial dysfunction associated with increased mitochondrial cholesterol content. Redox Biol. 2017, 12, 274–284. [Google Scholar] [CrossRef]
- Hoglinger, D.; Burgoyne, T.; Sanchez-Heras, E.; Hartwig, P.; Colaco, A.; Newton, J.; Futter, C.E.; Spiegel, S.; Platt, F.M.; Eden, E.R. Npc1 regulates er contacts with endocytic organelles to mediate cholesterol egress. Nat. Commun. 2019, 10, 4276. [Google Scholar] [CrossRef] [Green Version]
- Charman, M.; Kennedy, B.E.; Osborne, N.; Karten, B. Mln64 mediates egress of cholesterol from endosomes to mitochondria in the absence of functional niemann-pick type c1 protein. J. Lipid Res. 2010, 51, 1023–1034. [Google Scholar] [CrossRef] [Green Version]
- Vassilev, B.; Sihto, H.; Li, S.; Holtta-Vuori, M.; Ilola, J.; Lundin, J.; Isola, J.; Kellokumpu-Lehtinen, P.L.; Joensuu, H.; Ikonen, E. Elevated levels of star-related lipid transfer protein 3 alter cholesterol balance and adhesiveness of breast cancer cells: Potential mechanisms contributing to progression of her2-positive breast cancers. Am. J. Pathol. 2015, 185, 987–1000. [Google Scholar] [CrossRef] [PubMed]
- Alpy, F.; Boulay, A.; Moog-Lutz, C.; Andarawewa, K.L.; Degot, S.; Stoll, I.; Rio, M.C.; Tomasetto, C. Metastatic lymph node 64 (mln64), a gene overexpressed in breast cancers, is regulated by sp/klf transcription factors. Oncogene 2003, 22, 3770–3780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budczies, J.; Pfarr, N.; Stenzinger, A.; Treue, D.; Endris, V.; Ismaeel, F.; Bangemann, N.; Blohmer, J.U.; Dietel, M.; Loibl, S.; et al. Ioncopy: A novel method for calling copy number alterations in amplicon sequencing data including significance assessment. Oncotarget 2016, 7, 13236–13247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dressman, M.A.; Baras, A.; Malinowski, R.; Alvis, L.B.; Kwon, I.; Walz, T.M.; Polymeropoulos, M.H. Gene expression profiling detects gene amplification and differentiates tumor types in breast cancer. Cancer Res. 2003, 63, 2194–2199. [Google Scholar]
- Fararjeh, A.F.S.; al Khader, A.; Kaddumi, E.; Obeidat, M.; Al-Fawares, O. Differential expression and prognostic significance of stard3 gene in breast carcinoma. Int. J. Mol. Cell Med. 2021, 10, 34–41. [Google Scholar] [CrossRef]
- Kauraniemi, P.; Barlund, M.; Monni, O.; Kallioniemi, A. New amplified and highly expressed genes discovered in the erbb2 amplicon in breast cancer by cdna microarrays. Cancer Res. 2001, 61, 8235–8240. [Google Scholar]
- Moog-Lutz, C.; Tomasetto, C.; Regnier, C.H.; Wendling, C.; Lutz, Y.; Muller, D.; Chenard, M.P.; Basset, P.; Rio, M.C. Mln64 exhibits homology with the steroidogenic acute regulatory protein (star) and is over-expressed in human breast carcinomas. Int. J. Cancer 1997, 71, 183–191. [Google Scholar] [CrossRef]
- Cai, W.; Ye, L.; Sun, J.; Mansel, R.E.; Jiang, W.G. Expression of mln64 influences cellular matrix adhesion of breast cancer cells, the role for focal adhesion kinase. Int. J. Mol. Med. 2010, 25, 573–580. [Google Scholar]
- Peiro, G.; Ortiz-Martinez, F.; Gallardo, A.; Perez-Balaguer, A.; Sanchez-Paya, J.; Ponce, J.J.; Tibau, A.; Lopez-Vilaro, L.; Escuin, D.; Adrover, E.; et al. Src, a potential target for overcoming trastuzumab resistance in her2-positive breast carcinoma. Br. J. Cancer 2014, 111, 689–695. [Google Scholar] [CrossRef] [Green Version]
- Vu, T.; Claret, F.X. Trastuzumab: Updated mechanisms of action and resistance in breast cancer. Front. Oncol. 2012, 2, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asif, K.; Memeo, L.; Palazzolo, S.; Frion-Herrera, Y.; Parisi, S.; Caligiuri, I.; Canzonieri, V.; Granchi, C.; Tuccinardi, T.; Rizzolio, F. Stard3: A prospective target for cancer therapy. Cancers 2021, 13, 4693. [Google Scholar] [CrossRef] [PubMed]
- Maslyanko, M.; Harris, R.D.; Mu, D. Connecting cholesterol efflux factors to lung cancer biology and therapeutics. Int. J. Mol. Sci. 2021, 22, 7209. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, L.P.; Tomasetto, C.; Alpy, F. Touche! Stard3 and stard3nl tether the er to endosomes. Biochem. Soc. Trans. 2016, 44, 493–498. [Google Scholar] [CrossRef]
- Kao, J.; Pollack, J.R. Rna interference-based functional dissection of the 17q12 amplicon in breast cancer reveals contribution of coamplified genes. Genes Chromosomes Cancer 2006, 45, 761–769. [Google Scholar] [CrossRef]
- Sahlberg, K.K.; Hongisto, V.; Edgren, H.; Makela, R.; Hellstrom, K.; Due, E.U.; Vollan, H.K.M.; Sahlberg, N.; Wolf, M.; Borresen-Dale, A.L.; et al. The her2 amplicon includes several genes required for the growth and survival of her2 positive breast cancer cells. Mol. Oncol. 2013, 7, 392–401. [Google Scholar] [CrossRef]
- Kwon, M.J.; Kim, R.N.; Song, K.; Jeon, S.; Jeong, H.M.; Kim, J.S.; Han, J.; Hong, S.; Oh, E.; Choi, J.S.; et al. Genes co-amplified with erbb2 or met as novel potential cancer-promoting genes in gastric cancer. Oncotarget 2017, 8, 92209–92226. [Google Scholar] [CrossRef] [Green Version]
- Qiu, Y.; Zhang, Z.Y.; Du, W.D.; Ye, L.; Xu, S.; Zuo, X.B.; Zhou, F.S.; Chen, G.; Ma, X.L.; Schneider, M.E.; et al. Association analysis of erbb2 amplicon genetic polymorphisms and stard3 expression with risk of gastric cancer in the chinese population. Gene 2014, 535, 225–232. [Google Scholar] [CrossRef]
- De Marco, C.; Zoppoli, P.; Rinaldo, N.; Morganella, S.; Morello, M.; Zuccala, V.; Carriero, M.V.; Malanga, D.; Chirillo, R.; Bruni, P.; et al. Genome-wide analysis of copy number alterations led to the characterisation of pdcd10 as oncogene in ovarian cancer. Transl. Oncol. 2021, 14, 101013. [Google Scholar] [CrossRef]
- Stigliano, A.; Gandini, O.; Cerquetti, L.; Gazzaniga, P.; Misiti, S.; Monti, S.; Gradilone, A.; Falasca, P.; Poggi, M.; Brunetti, E.; et al. Increased metastatic lymph node 64 and cyp17 expression are associated with high stage prostate cancer. J. Endocrinol. 2007, 194, 55–61. [Google Scholar] [CrossRef] [Green Version]
- Vinatzer, U.; Dampier, B.; Streubel, B.; Pacher, M.; Seewald, M.J.; Stratowa, C.; Kaserer, K.; Schreiber, M. Expression of her2 and the coamplified genes grb7 and mln64 in human breast cancer: Quantitative real-time reverse transcription-pcr as a diagnostic alternative to immunohistochemistry and fluorescence in situ hybridization. Clin. Cancer Res. 2005, 11, 8348–8357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lapillo, M.; Salis, B.; Palazzolo, S.; Poli, G.; Granchi, C.; Minutolo, F.; Rotondo, R.; Caligiuri, I.; Canzonieri, V.; Tuccinardi, T.; et al. First-of-its-kind stard3 inhibitor: In silico identification and biological evaluation as anticancer agent. ACS Med. Chem. Lett. 2019, 10, 475–480. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Zhou, D.; Sun, Y.; Zhu, J.; Ghoneim, D.; Wu, C.; Yao, Q.; Gamazon, E.R.; Cox, N.J.; Wu, L. A transcriptome-wide association study identifies candidate susceptibility genes for pancreatic cancer risk. Cancer Res. 2020, 80, 4346–4354. [Google Scholar] [CrossRef]
- Yun, S.M.; Yoon, K.; Lee, S.; Kim, E.; Kong, S.H.; Choe, J.; Kang, J.M.; Han, T.S.; Kim, P.; Choi, Y.; et al. Ppp1r1b-stard3 chimeric fusion transcript in human gastric cancer promotes tumorigenesis through activation of pi3k/akt signaling. Oncogene 2014, 33, 5341–5347. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.; Land, H. Anticancer activity of the cholesterol exporter abca1 gene. Cell Rep. 2012, 2, 580–590. [Google Scholar] [CrossRef] [Green Version]
- Olkkonen, V.M.; Ikonen, E. Cholesterol transport in the late endocytic pathway: Roles of orp family proteins. J. Steroid Biochem. Mol. Biol. 2022, 216, 106040. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.; Lehto, M.; Tanhuanpaa, K.; Cover, T.L.; Olkkonen, V.M. The oxysterol-binding protein homologue orp1l interacts with rab7 and alters functional properties of late endocytic compartments. Mol. Biol. Cell 2005, 16, 5480–5492. [Google Scholar] [CrossRef]
- Johansson, M.; Rocha, N.; Zwart, W.; Jordens, I.; Janssen, L.; Kuijl, C.; Olkkonen, V.M.; Neefjes, J. Activation of endosomal dynein motors by stepwise assembly of rab7-rilp-p150glued, orp1l, and the receptor betalll spectrin. J. Cell Biol. 2007, 176, 459–471. [Google Scholar] [CrossRef] [Green Version]
- Van der Kant, R.; Zondervan, I.; Janssen, L.; Neefjes, J. Cholesterol-binding molecules mln64 and orp1l mark distinct late endosomes with transporters abca3 and npc1. J. Lipid Res. 2013, 54, 2153–2165. [Google Scholar] [CrossRef] [Green Version]
- Rocha, N.; Kuijl, C.; van der Kant, R.; Janssen, L.; Houben, D.; Janssen, H.; Zwart, W.; Neefjes, J. Cholesterol sensor orp1l contacts the er protein vap to control rab7-rilp-p150 glued and late endosome positioning. J. Cell Biol. 2009, 185, 1209–1225. [Google Scholar] [CrossRef] [Green Version]
- Van der Kant, R.; Fish, A.; Janssen, L.; Janssen, H.; Krom, S.; Ho, N.; Brummelkamp, T.; Carette, J.; Rocha, N.; Neefjes, J. Late endosomal transport and tethering are coupled processes controlled by rilp and the cholesterol sensor orp1l. J. Cell Sci. 2013, 126, 3462–3474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vihervaara, T.; Uronen, R.L.; Wohlfahrt, G.; Bjorkhem, I.; Ikonen, E.; Olkkonen, V.M. Sterol binding by osbp-related protein 1l regulates late endosome motility and function. Cell Mol. Life Sci. 2011, 68, 537–551. [Google Scholar] [CrossRef] [PubMed]
- Wijdeven, R.H.; Janssen, H.; Nahidiazar, L.; Janssen, L.; Jalink, K.; Berlin, I.; Neefjes, J. Cholesterol and orp1l-mediated er contact sites control autophagosome transport and fusion with the endocytic pathway. Nat. Commun. 2016, 7, 11808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eden, E.R.; Sanchez-Heras, E.; Tsapara, A.; Sobota, A.; Levine, T.P.; Futter, C.E. Annexin a1 tethers membrane contact sites that mediate er to endosome cholesterol transport. Dev. Cell 2016, 37, 473–483. [Google Scholar] [CrossRef] [Green Version]
- Kobuna, H.; Inoue, T.; Shibata, M.; Gengyo-Ando, K.; Yamamoto, A.; Mitani, S.; Arai, H. Multivesicular body formation requiRes. osbp-related proteins and cholesterol. PLoS Genet 2010, 6, e1001055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, K.; Ridgway, N.D. Oxysterol-binding protein-related protein 1l regulates cholesterol egress from the endo-lysosomal system. Cell Rep. 2017, 19, 1807–1818. [Google Scholar] [CrossRef] [Green Version]
- Cianciola, N.L.; Greene, D.J.; Morton, R.E.; Carlin, C.R. Adenovirus ridalpha uncovers a novel pathway requiring orp1l for lipid droplet formation independent of npc1. Mol. Biol. Cell 2013, 24, 3309–3325. [Google Scholar] [CrossRef]
- Kentala, H.; Koponen, A.; Kivela, A.M.; Andrews, R.; Li, C.; Zhou, Y.; Olkkonen, V.M. Analysis of orp2-knockout hepatocytes uncovers a novel function in actin cytoskeletal regulation. FASEB J. 2018, 32, 1281–1295. [Google Scholar] [CrossRef] [Green Version]
- Kentala, H.; Koponen, A.; Vihinen, H.; Pirhonen, J.; Liebisch, G.; Pataj, Z.; Kivela, A.; Li, S.; Karhinen, L.; Jaaskelainen, E.; et al. Osbp-related protein-2 (orp2): A novel akt effector that controls cellular energy metabolism. Cell Mol. Life Sci. 2018, 75, 4041–4057. [Google Scholar] [CrossRef] [Green Version]
- Du, X.; Kumar, J.; Ferguson, C.; Schulz, T.A.; Ong, Y.S.; Hong, W.; Prinz, W.A.; Parton, R.G.; Brown, A.J.; Yang, H. A role for oxysterol-binding protein-related protein 5 in endosomal cholesterol trafficking. J. Cell Biol. 2011, 192, 121–135. [Google Scholar] [CrossRef] [Green Version]
- Charman, M.; Colbourne, T.R.; Pietrangelo, A.; Kreplak, L.; Ridgway, N.D. Oxysterol-binding protein (osbp)-related protein 4 (orp4) is essential for cell proliferation and survival. J. Biol. Chem. 2014, 289, 15705–15717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koga, Y.; Ishikawa, S.; Nakamura, T.; Masuda, T.; Nagai, Y.; Takamori, H.; Hirota, M.; Kanemitsu, K.; Baba, Y.; Baba, H. Oxysterol binding protein-related protein-5 is related to invasion and poor prognosis in pancreatic cancer. Cancer Sci. 2008, 99, 2387–2394. [Google Scholar] [CrossRef] [PubMed]
- Nagano, K.; Imai, S.; Zhao, X.; Yamashita, T.; Yoshioka, Y.; Abe, Y.; Mukai, Y.; Kamada, H.; Nakagawa, S.; Tsutsumi, Y.; et al. Identification and evaluation of metastasis-related proteins, oxysterol binding protein-like 5 and calumenin, in lung tumors. Int. J. Oncol. 2015, 47, 195–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strating, J.R.; van der Linden, L.; Albulescu, L.; Bigay, J.; Arita, M.; Delang, L.; Leyssen, P.; van der Schaar, H.M.; Lanke, K.H.; Thibaut, H.J.; et al. Itraconazole inhibits enterovirus replication by targeting the oxysterol-binding protein. Cell Rep. 2015, 10, 600–615. [Google Scholar] [CrossRef] [Green Version]
- Meutiawati, F.; Bezemer, B.; Strating, J.; Overheul, G.J.; Zusinaite, E.; van Kuppeveld, F.J.M.; van Cleef, K.W.R.; van Rij, R.P. Posaconazole inhibits dengue virus replication by targeting oxysterol-binding protein. Antiviral Res. 2018, 157, 68–79. [Google Scholar] [CrossRef]
- Pfeffer, S.R. Rab gtpases: Master regulators that establish the secretory and endocytic pathways. Mol. Biol. Cell 2017, 28, 712–715. [Google Scholar] [CrossRef]
- Ganley, I.G.; Pfeffer, S.R. Cholesterol accumulation sequesters rab9 and disrupts late endosome function in npc1-deficient cells. J. Biol. Chem. 2006, 281, 17890–17899. [Google Scholar] [CrossRef] [Green Version]
- Linder, M.D.; Uronen, R.L.; Holtta-Vuori, M.; van der Sluijs, P.; Peranen, J.; Ikonen, E. Rab8-dependent recycling promotes endosomal cholesterol removal in normal and sphingolipidosis cells. Mol. Biol. Cell 2007, 18, 47–56. [Google Scholar] [CrossRef] [Green Version]
- Walter, M.; Davies, J.P.; Ioannou, Y.A. Telomerase immortalization upregulates rab9 expression and restoRes. ldl cholesterol egress from niemann-pick c1 late endosomes. J. Lipid Res. 2003, 44, 243–253. [Google Scholar] [CrossRef] [Green Version]
- Lebrand, C.; Corti, M.; Goodson, H.; Cosson, P.; Cavalli, V.; Mayran, N.; Faure, J.; Gruenberg, J. Late endosome motility depends on lipids via the small gtpase rab7. EMBO J. 2002, 21, 1289–1300. [Google Scholar] [CrossRef] [Green Version]
- Ferro, E.; Bosia, C.; Campa, C.C. Rab11-mediated trafficking and human cancers: An updated review. Biology 2021, 10, 26. [Google Scholar] [CrossRef]
- Porther, N.; Barbieri, M.A. The role of endocytic rab gtpases in regulation of growth factor signaling and the migration and invasion of tumor cells. Small GTPases 2015, 6, 135–144. [Google Scholar] [CrossRef] [Green Version]
- Tang, B.L.; Ng, E.L. Rabs and cancer cell motility. Cell Motil. Cytoskelet. 2009, 66, 365–370. [Google Scholar] [CrossRef]
- Van den Boomen, D.J.H.; Sienkiewicz, A.; Berlin, I.; Jongsma, M.L.M.; van Elsland, D.M.; Luzio, J.P.; Neefjes, J.J.C.; Lehner, P.J. A trimeric rab7 gef controls npc1-dependent lysosomal cholesterol export. Nat. Commun. 2020, 11, 5559. [Google Scholar] [CrossRef]
- Rink, J.; Ghigo, E.; Kalaidzidis, Y.; Zerial, M. Rab conversion as a mechanism of progression from early to late endosomes. Cell 2005, 122, 735–749. [Google Scholar] [CrossRef] [Green Version]
- Meneses-Salas, E.; Garcia-Melero, A.; Blanco-Munoz, P.; Jose, J.; Brenner, M.S.; Lu, A.; Tebar, F.; Grewal, T.; Rentero, C.; Enrich, C. Selective degradation permits a feedback loop controlling annexin a6 and cholesterol levels in endolysosomes of npc1 mutant cells. Cells 2020, 9, 1152. [Google Scholar] [CrossRef]
- Guerra, F.; Bucci, C. Multiple roles of the small gtpase rab7. Cells 2016, 5, 34. [Google Scholar] [CrossRef]
- Snider, M.D. A role for rab7 gtpase in growth factor-regulated cell nutrition and apoptosis. Mol. Cell 2003, 12, 796–797. [Google Scholar] [CrossRef]
- Croizet-Berger, K.; Daumerie, C.; Couvreur, M.; Courtoy, P.J.; van den Hove, M.F. The endocytic catalysts, rab5a and rab7, are tandem regulators of thyroid hormone production. Proc. Natl. Acad. Sci. USA 2002, 99, 8277–8282. [Google Scholar] [CrossRef] [Green Version]
- Davidson, B.; Zhang, Z.; Kleinberg, L.; Li, M.; Florenes, V.A.; Wang, T.L.; Ie, M.S. Gene expression signatuRes. differentiate ovarian/peritoneal serous carcinoma from diffuse malignant peritoneal mesothelioma. Clin. Cancer Res. 2006, 12, 5944–5950. [Google Scholar] [CrossRef] [Green Version]
- Zhao, T.; Ding, X.; Yan, C.; Du, H. Endothelial rab7 gtpase mediates tumor growth and metastasis in lysosomal acid lipase-deficient mice. J. Biol. Chem. 2017, 292, 19198–19208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, X.; Zhang, W.; Zhao, T.; Yan, C.; Du, H. Rab7 gtpase controls lipid metabolic signaling in myeloid-derived suppressor cells. Oncotarget 2017, 8, 30123–30137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flinn, R.J.; Yan, Y.; Goswami, S.; Parker, P.J.; Backer, J.M. The late endosome is essential for mtorc1 signaling. Mol. Biol. Cell 2010, 21, 833–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margiotta, A.; Progida, C.; Bakke, O.; Bucci, C. Rab7a regulates cell migration through rac1 and vimentin. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 367–381. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Zhang, M.; Ma, Z.; Guo, K.; Tergaonkar, V.; Zeng, Q.; Hong, W. A role of rab7 in stabilizing egfr-her2 and in sustaining akt survival signal. J. Cell Physiol. 2012, 227, 2788–2797. [Google Scholar] [CrossRef]
- Steffan, J.J.; Dykes, S.S.; Coleman, D.T.; Adams, L.K.; Rogers, D.; Carroll, J.L.; Williams, B.J.; Cardelli, J.A. Supporting a role for the gtpase rab7 in prostate cancer progression. PLoS ONE 2014, 9, e87882. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Zhang, H.; Liu, S.; Kim, C.K.; Xu, Y.; Hurley, L.A.; Nishikawa, R.; Nagane, M.; Hu, B.; Stegh, A.H.; et al. Internalized cd44s splice isoform attenuates egfr degradation by targeting rab7a. Proc. Natl. Acad. Sci. USA 2017, 114, 8366–8371. [Google Scholar] [CrossRef] [Green Version]
- Laviolette, L.A.; Mermoud, J.; Calvo, I.A.; Olson, N.; Boukhali, M.; Steinlein, O.K.; Roider, E.; Sattler, E.C.; Huang, D.; The, B.T.; et al. Negative regulation of egfr signalling by the human folliculin tumour suppressor protein. Nat. Commun. 2017, 8, 15866. [Google Scholar] [CrossRef]
- Okon, I.S.; Coughlan, K.A.; Zhang, C.; Moriasi, C.; Ding, Y.; Song, P.; Zhang, W.; Li, G.; Zou, M.H. Protein kinase lkb1 promotes rab7-mediated neuropilin-1 degradation to inhibit angiogenesis. J. Clin. Invest. 2014, 124, 4590–4602. [Google Scholar] [CrossRef] [Green Version]
- Guerra, F.; Paiano, A.; Migoni, D.; Girolimetti, G.; Perrone, A.M.; de Iaco, P.; Fanizzi, F.P.; Gasparre, G.; Bucci, C. Modulation of rab7a protein expression determines resistance to cisplatin through late endocytic pathway impairment and extracellular vesicular secretion. Cancers 2019, 11, 52. [Google Scholar] [CrossRef] [Green Version]
- Alonso-Curbelo, D.; Riveiro-Falkenbach, E.; Perez-Guijarro, E.; Cifdaloz, M.; Karras, P.; Osterloh, L.; Megias, D.; Canon, E.; Calvo, T.G.; Olmeda, D.; et al. Rab7 controls melanoma progression by exploiting a lineage-specific wiring of the endolysosomal pathway. Cancer Cell 2014, 26, 61–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, Y.; Gao, J.X.; Tian, H.; Yearsley, K.; Lange, A.R.; Robertson, F.M.; Barsky, S.H. Early to intermediate steps of tumor embolic formation involve specific proteolytic processing of e-cadherin regulated by rab7. Mol. Cancer Res. 2012, 10, 713–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, M.; Koseki, J.; Takahashi, H.; Miyoshi, N.; Nishida, N.; Nishimura, J.; Hata, T.; Matsuda, C.; Mizushima, T.; Yamamoto, H.; et al. Disruption of endolysosomal rab5/7 efficiently eliminates colorectal cancer stem cells. Cancer Res. 2019, 79, 1426–1437. [Google Scholar] [CrossRef] [Green Version]
- Kou, X.; Yang, Y.; Jiang, X.; Liu, H.; Sun, F.; Wang, X.; Liu, L.; Liu, H.; Lin, Z.; Jiang, L. Vorinostat and simvastatin have synergistic effects on triple-negative breast cancer cells via abrogating rab7 prenylation. Eur. J. Pharmacol. 2017, 813, 161–171. [Google Scholar] [CrossRef]
- Arildsen, N.S.; Hedenfalk, I. Simvastatin is a potential candidate drug in ovarian clear cell carcinomas. Oncotarget 2020, 11, 3660–3674. [Google Scholar] [CrossRef]
- Enrich, C.; Rentero, C.; de Muga, S.V.; Reverter, M.; Mulay, V.; Wood, P.; Koese, M.; Grewal, T. Annexin a6-linking ca(2+) signaling with cholesterol transport. Biochim. Biophys. Acta 2011, 1813, 935–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rentero, C.; Blanco-Munoz, P.; Meneses-Salas, E.; Grewal, T.; Enrich, C. Annexins-coordinators of cholesterol homeostasis in endocytic pathways. Int. J. Mol. Sci. 2018, 19, 1444. [Google Scholar] [CrossRef] [Green Version]
- Grewal, T.; Rentero, C.; Enrich, C.; Wahba, M.; Raabe, C.A.; Rescher, U. Annexin animal models-from fundamental principles to translational research. Int. J. Mol. Sci. 2021, 22, 3439. [Google Scholar] [CrossRef]
- Korolkova, O.Y.; Widatalla, S.E.; Williams, S.D.; Whalen, D.S.; Beasley, H.K.; Ochieng, J.; Grewal, T.; Sakwe, A.M. Diverse roles of annexin a6 in triple-negative breast cancer diagnosis, prognosis and egfr-targeted therapies. Cells 2020, 9, 1855. [Google Scholar] [CrossRef]
- Grewal, T.; Evans, R.; Rentero, C.; Tebar, F.; Cubells, L.; de Diego, I.; Kirchhoff, M.F.; Hughes, W.E.; Heeren, J.; Rye, K.A.; et al. Annexin a6 stimulates the membrane recruitment of p120gap to modulate ras and raf-1 activity. Oncogene 2005, 24, 5809–5820. [Google Scholar] [CrossRef] [Green Version]
- Koese, M.; Rentero, C.; Kota, B.P.; Hoque, M.; Cairns, R.; Wood, P.; de Muga, S.V.; Reverter, M.; Alvarez-Guaita, A.; Monastyrskaya, K.; et al. Annexin a6 is a scaffold for pkcalpha to promote egfr inactivation. Oncogene 2013, 32, 2858–2872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vila de Muga, S.; Timpson, P.; Cubells, L.; Evans, R.; Hayes, T.E.; Rentero, C.; Hegemann, A.; Reverter, M.; Leschner, J.; Pol, A.; et al. Annexin a6 inhibits ras signalling in breast cancer cells. Oncogene 2009, 28, 363–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grewal, T.; Enrich, C. Annexins--modulators of egf receptor signalling and trafficking. Cell Signal 2009, 21, 847–858. [Google Scholar] [CrossRef] [PubMed]
- Qi, H.; Liu, S.; Guo, C.; Wang, J.; Greenaway, F.T.; Sun, M.Z. Role of annexin a6 in cancer. Oncol. Lett. 2015, 10, 1947–1952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoque, M.; Elmaghrabi, Y.A.; Kose, M.; Beevi, S.S.; Jose, J.; Meneses-Salas, E.; Blanco-Munoz, P.; Conway, J.R.W.; Swarbrick, A.; Timpson, P.; et al. Annexin a6 improves anti-migratory and anti-invasive properties of tyrosine kinase inhibitors in egfr overexpressing human squamous epithelial cells. FEBS J. 2020, 287, 2961–2978. [Google Scholar] [CrossRef]
- Sakwe, A.M.; Koumangoye, R.; Guillory, B.; Ochieng, J. Annexin a6 contributes to the invasiveness of breast carcinoma cells by influencing the organization and localization of functional focal adhesions. Exp. Cell Res. 2011, 317, 823–837. [Google Scholar] [CrossRef] [Green Version]
- Koumangoye, R.B.; Nangami, G.N.; Thompson, P.D.; Agboto, V.K.; Ochieng, J.; Sakwe, A.M. Reduced annexin a6 expression promotes the degradation of activated epidermal growth factor receptor and sensitizes invasive breast cancer cells to egfr-targeted tyrosine kinase inhibitors. Mol. Cancer 2013, 12, 167. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Zhang, S.; Zhang, J.; Lam, E.; Liu, X.; Sun, J.; Feng, L.; Lu, H.; Yu, J.; Jin, H. Annexin a6 is down-regulated through promoter methylation in gastric cancer. Am J. Transl. Res. 2013, 5, 555–562. [Google Scholar]
- Lomnytska, M.I.; Becker, S.; Hellman, K.; Hellstrom, A.C.; Souchelnytskyi, S.; Mints, M.; Hellman, U.; Andersson, S.; Auer, G. Diagnostic protein marker patterns in squamous cervical cancer. Proteomics Clin. Appl. 2010, 4, 17–31. [Google Scholar] [CrossRef]
- Meier, E.M.; Rein-Fischboeck, L.; Pohl, R.; Wanninger, J.; Hoy, A.J.; Grewal, T.; Eisinger, K.; Krautbauer, S.; Liebisch, G.; Weiss, T.S.; et al. Annexin a6 protein is downregulated in human hepatocellular carcinoma. Mol. Cell Biochem. 2016, 418, 81–90. [Google Scholar] [CrossRef]
- Lomnytska, M.I.; Becker, S.; Bodin, I.; Olsson, A.; Hellman, K.; Hellstrom, A.C.; Mints, M.; Hellman, U.; Auer, G.; Andersson, S. Differential expression of anxa6, hsp27, prdx2, ncf2, and tpm4 during uterine cervix carcinogenesis: Diagnostic and prognostic value. Br. J. Cancer 2011, 104, 110–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francia, G.; Mitchell, S.D.; Moss, S.E.; Hanby, A.M.; Marshall, J.F.; Hart, I.R. Identification by differential display of annexin-vi, a gene differentially expressed during melanoma progression. Cancer Res. 1996, 56, 3855–3858. [Google Scholar]
- Noreen, S.; Gardner, Q.A.; Fatima, I.; Sadaf, S.; Akhtar, M.W. Upregulated expression of calcium-dependent annexin a6: A potential biomarker of ovarian carcinoma. Proteom. Clin. Appl. 2020, 14, e1900078. [Google Scholar] [CrossRef] [PubMed]
- Keklikoglou, I.; Cianciaruso, C.; Guc, E.; Squadrito, M.L.; Spring, L.M.; Tazzyman, S.; Lambein, L.; Poissonnier, A.; Ferraro, G.B.; Baer, C.; et al. Chemotherapy elicits pro-metastatic extracellular vesicles in breast cancer models. Nat. Cell Biol. 2019, 21, 190–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leca, J.; Martinez, S.; Lac, S.; Nigri, J.; Secq, V.; Rubis, M.; Bressy, C.; Serge, A.; Lavaut, M.N.; Dusetti, N.; et al. Cancer-associated fibroblast-derived annexin a6+ extracellular vesicles support pancreatic cancer aggressiveness. J. Clin. Invest. 2016, 126, 4140–4156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Sullivan, D.; Dowling, P.; Joyce, H.; McAuley, E.; McCann, A.; Henry, M.; McGovern, B.; Barham, P.; Kelleher, F.C.; Murphy, J.; et al. A novel inhibitory anti-invasive mab isolated using phenotypic screening highlights anxa6 as a functionally relevant target protein in pancreatic cancer. Br. J. Cancer 2017, 117, 1326–1335. [Google Scholar] [CrossRef]
- Lee, H.S.; Kang, Y.; Tae, K.; Bae, G.U.; Park, J.Y.; Cho, Y.H.; Yang, M. Proteomic biomarkers for bisphenol a-early exposure and women’s thyroid cancer. Cancer Res. Treat. 2018, 50, 111–117. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.C.; Sudhof, T.C.; Anderson, R.G. Annexin vi is required for budding of clathrin-coated pits. Cell 1992, 70, 283–291. [Google Scholar] [CrossRef]
- Uchihara, T.; Miyake, K.; Yonemura, A.; Komohara, Y.; Itoyama, R.; Koiwa, M.; Yasuda, T.; Arima, K.; Harada, K.; Eto, K.; et al. Extracellular vesicles from cancer-associated fibroblasts containing annexin a6 induces fak-yap activation by stabilizing beta1 integrin, enhancing drug resistance. Cancer Res. 2020, 80, 3222–3235. [Google Scholar] [CrossRef]
- Zaidi, A.H.; Gopalakrishnan, V.; Kasi, P.M.; Zeng, X.; Malhotra, U.; Balasubramanian, J.; Visweswaran, S.; Sun, M.; Flint, M.S.; Davison, J.M.; et al. Evaluation of a 4-protein serum biomarker panel-biglycan, annexin-a6, myeloperoxidase, and protein s100-a9 (b-amp)-for the detection of esophageal adenocarcinoma. Cancer 2014, 120, 3902–3913. [Google Scholar] [CrossRef]
- Whalen, D.S.; Widatalla, S.E.; Korolkova, O.Y.; Nangami, G.S.; Beasley, H.K.; Williams, S.D.; Virgous, C.; Lehmann, B.D.; Ochieng, J.; Sakwe, A.M. Implication of calcium activated rasgrf2 in annexin a6-mediated breast tumor cell growth and motility. Oncotarget 2019, 10, 133–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grewal, T.; Heeren, J.; Mewawala, D.; Schnitgerhans, T.; Wendt, D.; Salomon, G.; Enrich, C.; Beisiegel, U.; Jackle, S. Annexin vi stimulates endocytosis and is involved in the trafficking of low density lipoprotein to the prelysosomal compartment. J. Biol. Chem. 2000, 275, 33806–33813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamal, A.; Ying, Y.; Anderson, R.G. Annexin vi-mediated loss of spectrin during coated pit budding is coupled to delivery of ldl to lysosomes. J. Cell Biol. 1998, 142, 937–947. [Google Scholar] [CrossRef] [PubMed]
- Pons, M.; Grewal, T.; Rius, E.; Schnitgerhans, T.; Jackle, S.; Enrich, C. Evidence for the involvement of annexin 6 in the trafficking between the endocytic compartment and lysosomes. Exp. Cell Res. 2001, 269, 13–22. [Google Scholar] [CrossRef] [PubMed]
- De Diego, I.; Schwartz, F.; Siegfried, H.; Dauterstedt, P.; Heeren, J.; Beisiegel, U.; Enrich, C.; Grewal, T. Cholesterol modulates the membrane binding and intracellular distribution of annexin 6. J. Biol. Chem. 2002, 277, 32187–32194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Te Vruchte, D.; Lloyd-Evans, E.; Veldman, R.J.; Neville, D.C.; Dwek, R.A.; Platt, F.M.; van Blitterswijk, W.J.; Sillence, D.J. Accumulation of glycosphingolipids in niemann-pick c disease disrupts endosomal transport. J. Biol. Chem. 2004, 279, 26167–26175. [Google Scholar] [CrossRef] [Green Version]
- Widatalla, S.E.; Korolkova, O.Y.; Whalen, D.S.; Goodwin, J.S.; Williams, K.P.; Ochieng, J.; Sakwe, A.M. Lapatinib-induced annexin a6 upregulation as an adaptive response of triple-negative breast cancer cells to egfr tyrosine kinase inhibitors. Carcinogenesis 2019, 40, 998–1009. [Google Scholar] [CrossRef] [Green Version]
- Ahras, M.; Naing, T.; McPherson, R. Scavenger receptor class b type i localizes to a late endosomal compartment. J. Lipid Res. 2008, 49, 1569–1576. [Google Scholar] [CrossRef] [Green Version]
- Neufeld, E.B.; O’Brien, K.; Walts, A.D.; Stonik, J.A.; Demosky, S.J.; Malide, D.; Combs, C.A.; Remaley, A.T. Cellular localization and trafficking of the human abcg1 transporter. Biology 2014, 3, 781–800. [Google Scholar] [CrossRef] [Green Version]
- Neufeld, E.B.; Stonik, J.A.; Demosky, S.J., Jr.; Knapper, C.L.; Combs, C.A.; Cooney, A.; Comly, M.; Dwyer, N.; Blanchette-Mackie, J.; Remaley, A.T.; et al. The abca1 transporter modulates late endocytic trafficking: Insights from the correction of the genetic defect in tangier disease. J. Biol. Chem. 2004, 279, 15571–15578. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez-Pajares, J.L.; Hassen, C.B.; Chevalier, S.; Frank, P.G. Sr-bi: Linking cholesterol and lipoprotein metabolism with breast and prostate cancer. Front. Pharmacol. 2016, 7, 338. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| LDLR Upregulation and Tumorigenic Outcomes | Cancer Types |
|---|---|
| Elevated LDLR expression | breast cancer [26,49,50], colorectal cancer [32,33], glioblastoma [35], HCC [28], lung cancer [30,31], leukemia [36,51], lymphoma [29], nasopharyngeal carcinoma [34], renal cancer [37], PDAC [24,25] |
| Promoting proliferation, migration and invasion | breast cancer [52], colorectal cancer [38], nasopharyngeal cancer [34], PDAC [25], prostate cancer [53,54,55] |
| Poor prognosis and clinical outcomes | AML [56], breast cancer [52], cervical cancer [48], HCC [46], PDAC [57,58], Prostate cancer [47] |
| Chemoresistance | breast cancer [52], ovarian cancer [59], PDAC [25] |
| Contribution of NPC1 to Tumorigenic Outcomes | Cancer Types |
|---|---|
| Upregulation and cancer risk | esophageal cancer [65,74], HCC [75] |
| Promoting proliferation, migration and invasion | A431 squamous carcinoma [68,69,70,71], cervical cancer [67] |
| Poor prognosis | ER-negative breast cancer [66], glioma [64] |
| Chemoresistance | breast cancer [76], leukemia [70,73] |
| Therapeutic target | Itraconazole: basal cell carcinoma [77], non-small cell lung cancer [78], pancreatic cancer [79], prostate cancer [80] Cepharanthine: head and neck cancer [81], prostate cancer [82] Leelamine: metastatic melanoma [83,84] |
| Contribution of StARD3 to Tumorigenic Functions | Cancer Types |
|---|---|
| StARD3 expression and cancer risk | breast cancer [166], gastric cancer [177,178], ovarian cancer [179] |
| StARD3 expression and poor prognosis | breast cancer [169], ER-positive and triple-negative breast cancer [166], gastric cancer [177], prostate cancer [180] |
| Predictor for chemotherapy response | breast cancer [169,181] |
| Therapeutic target | compound VS1: breast and colon cancer [182] |
| Contribution of Rab7 to Tumorigenic Outcomes | Cancer Types |
|---|---|
| Tumor promotor | A431 squamous carcinoma [69,225], lung cancer [224], breast cancer [225], cervical carcinoma [140], ovarian cancer [220], peritoneal serous carcinoma [220], thyroid cancer [219] |
| Tumor suppressor | A549 lung cancer [229], glioblastoma [227], thyroid cancer [228] |
| Oncojanus | inflammatory breast cancer [232], melanoma [231] |
| Cisplatin chemoresistance | cervical cancer [230] |
| Therapeutic target | mefloquine hydrochloride: colorectal cancer stem cells [233] |
| Statins: TNBC, epithelial ovarian cancer cell lines [234,235]. CID-1067700: epithelial ovarian cancer cell lines [235]. |
| Contribution of AnxA6 to Tumorigenic Outcomes | Cancer Types |
|---|---|
| Tumor promotor | breast cancer [246], cervical cancer [251], esophageal cancer [252], melanoma [252], ovarian cancer [253], pancreatic cancer [254,255,256], women’s thyroid cancer [257] |
| Tumor suppressor | A431 epithelial carcinoma [240,241,242,245], breast cancer (TNBC, EGFR overexpressing and ER-negative) [239,240,241,246], cervical cancer [249], gastric cancer [248], HCC [250] |
| Chemotherapy response | TNBC [239,247,258], gastric cancer [259] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, M.K.L.; Jose, J.; Wahba, M.; Bernaus-Esqué, M.; Hoy, A.J.; Enrich, C.; Rentero, C.; Grewal, T. Linking Late Endosomal Cholesterol with Cancer Progression and Anticancer Drug Resistance. Int. J. Mol. Sci. 2022, 23, 7206. https://doi.org/10.3390/ijms23137206
Nguyen MKL, Jose J, Wahba M, Bernaus-Esqué M, Hoy AJ, Enrich C, Rentero C, Grewal T. Linking Late Endosomal Cholesterol with Cancer Progression and Anticancer Drug Resistance. International Journal of Molecular Sciences. 2022; 23(13):7206. https://doi.org/10.3390/ijms23137206
Chicago/Turabian StyleNguyen, Mai K. L., Jaimy Jose, Mohamed Wahba, Marc Bernaus-Esqué, Andrew J. Hoy, Carlos Enrich, Carles Rentero, and Thomas Grewal. 2022. "Linking Late Endosomal Cholesterol with Cancer Progression and Anticancer Drug Resistance" International Journal of Molecular Sciences 23, no. 13: 7206. https://doi.org/10.3390/ijms23137206
APA StyleNguyen, M. K. L., Jose, J., Wahba, M., Bernaus-Esqué, M., Hoy, A. J., Enrich, C., Rentero, C., & Grewal, T. (2022). Linking Late Endosomal Cholesterol with Cancer Progression and Anticancer Drug Resistance. International Journal of Molecular Sciences, 23(13), 7206. https://doi.org/10.3390/ijms23137206