
CDK1 Promotes Epithelial–Mesenchymal Transition and Migration of Head and Neck Squamous Carcinoma Cells by Repressing ∆Np63α-Mediated Transcriptional Regulation

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
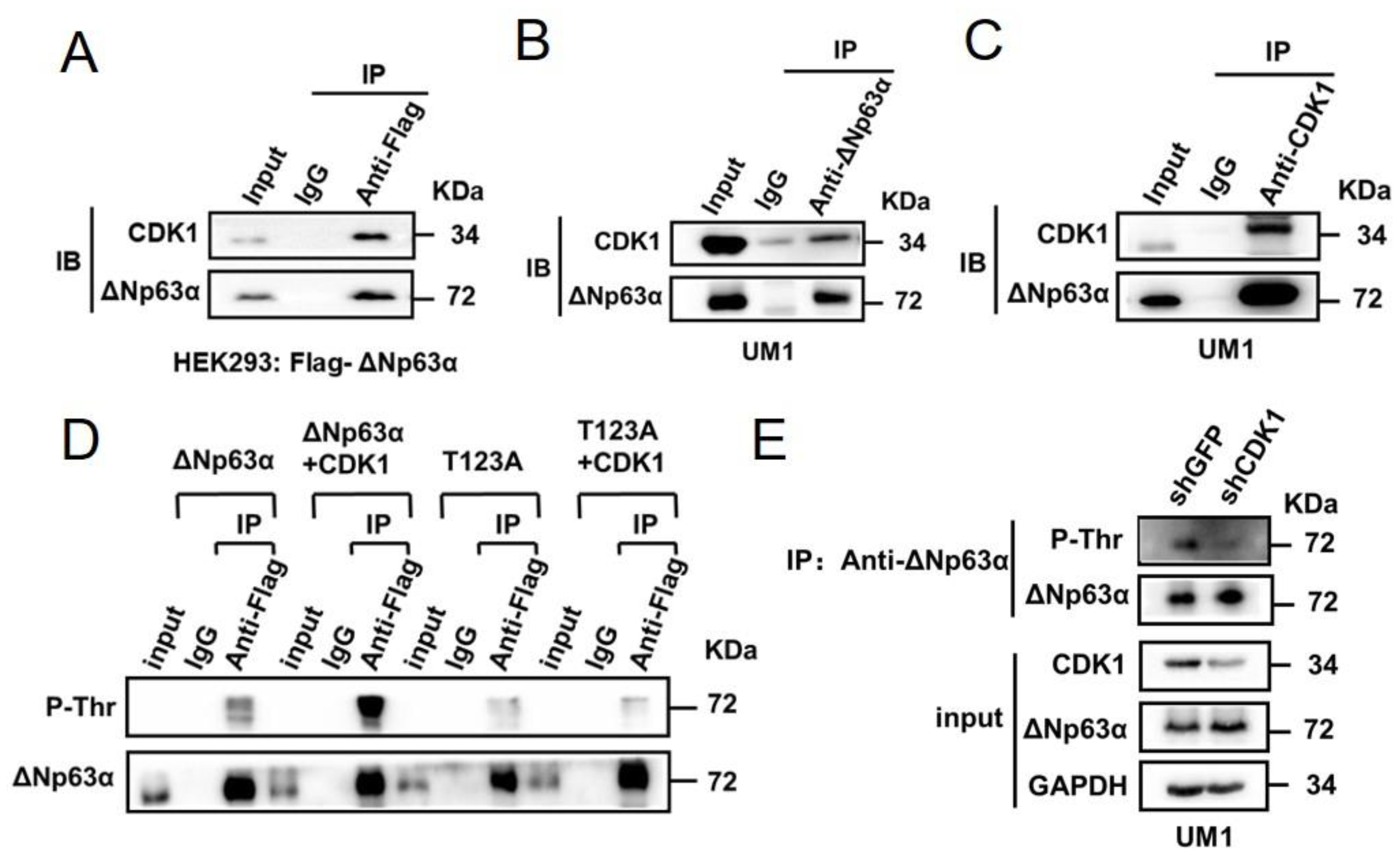
2.1. CDK1 Binds to ∆Np63α and Phosphorylates It on Residue T123
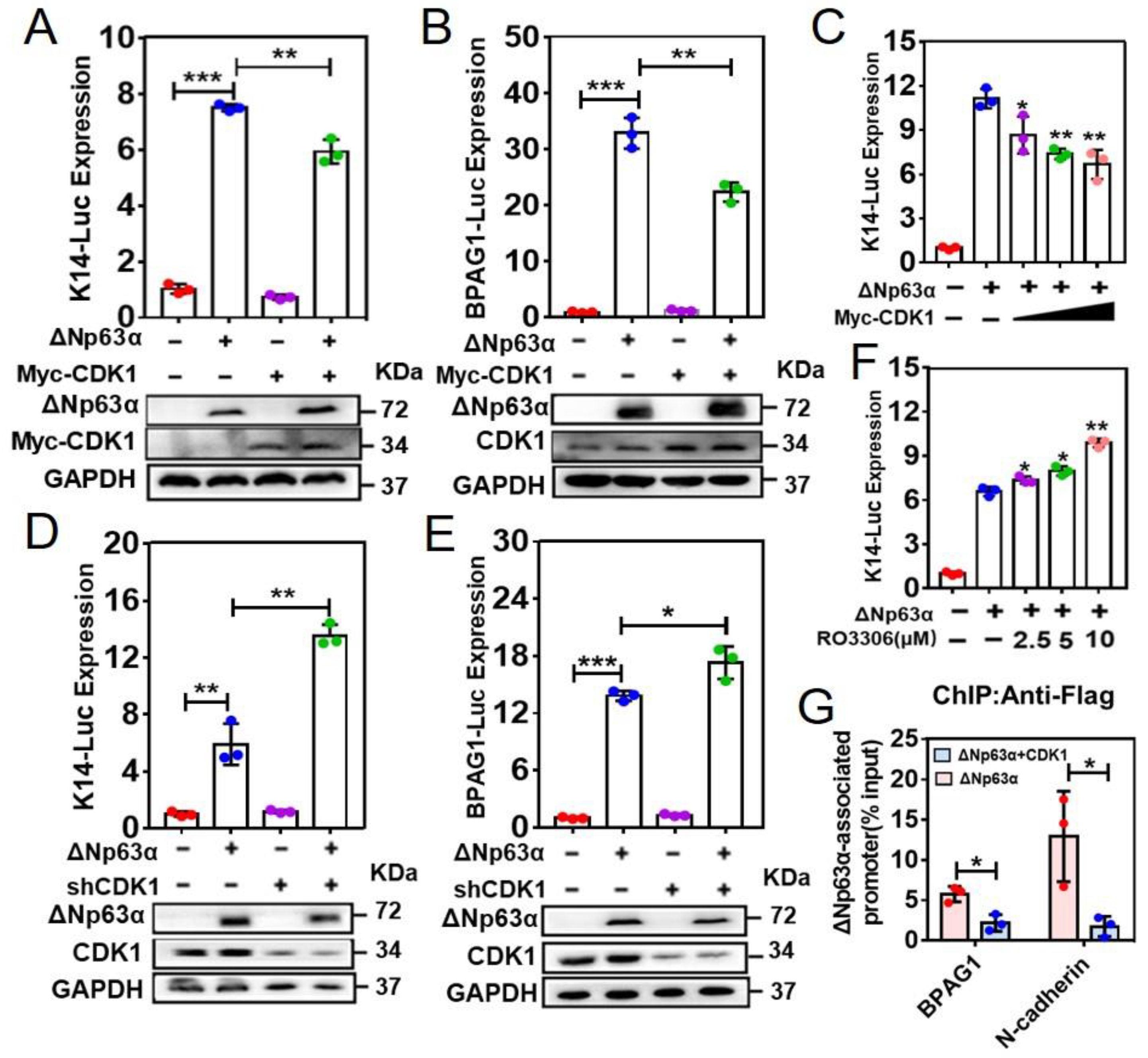
2.2. CDK1 Impairs Association of ∆Np63α with Its Downstream Gene Promoters to Inhibit ∆Np63α-Mediated Transcriptional Regulation
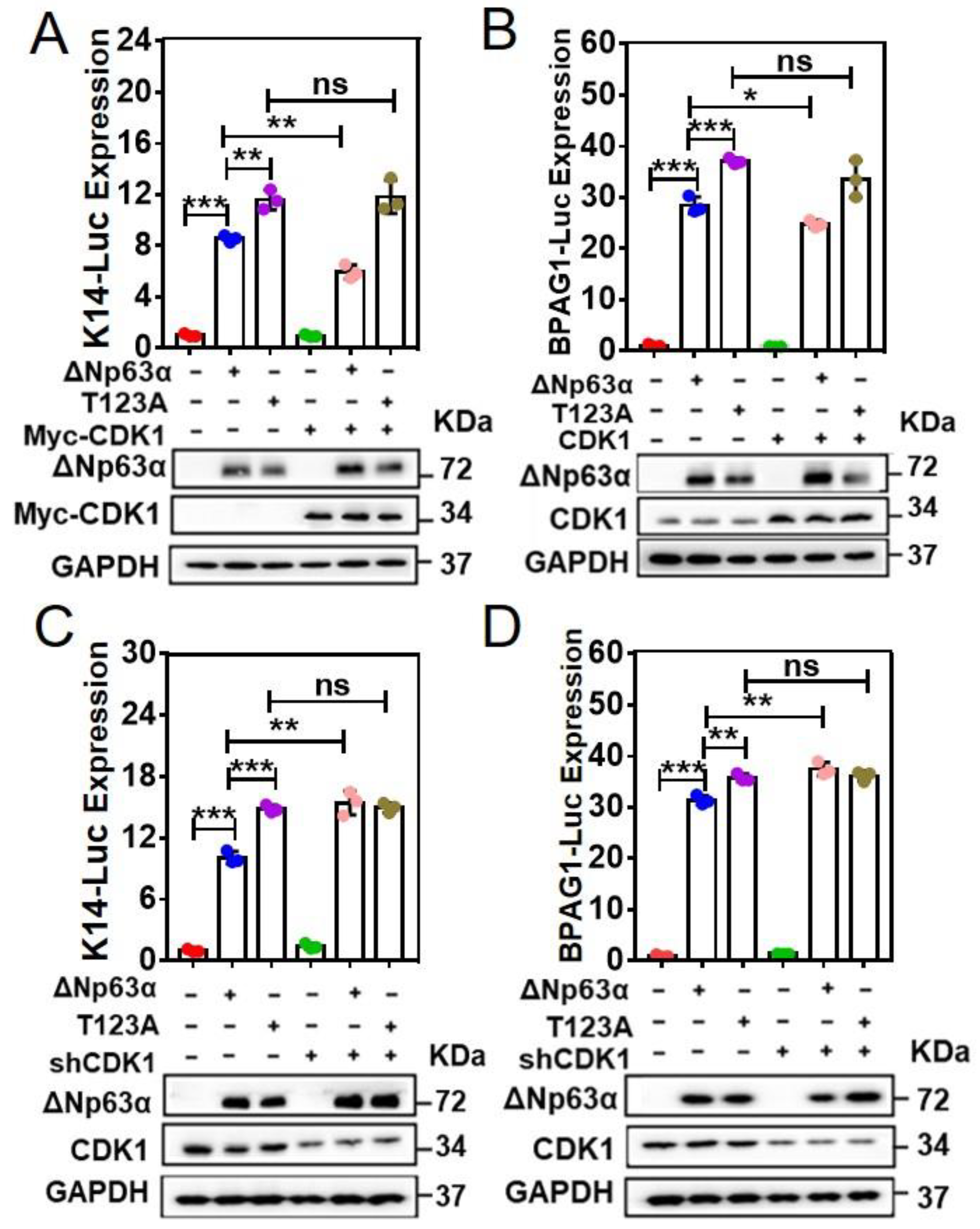
2.3. T123A Mutation in ∆Np63α Abrogates the Inhibitory Effect of CDK1 on ∆Np63α-Mediated Transcriptional Regulation
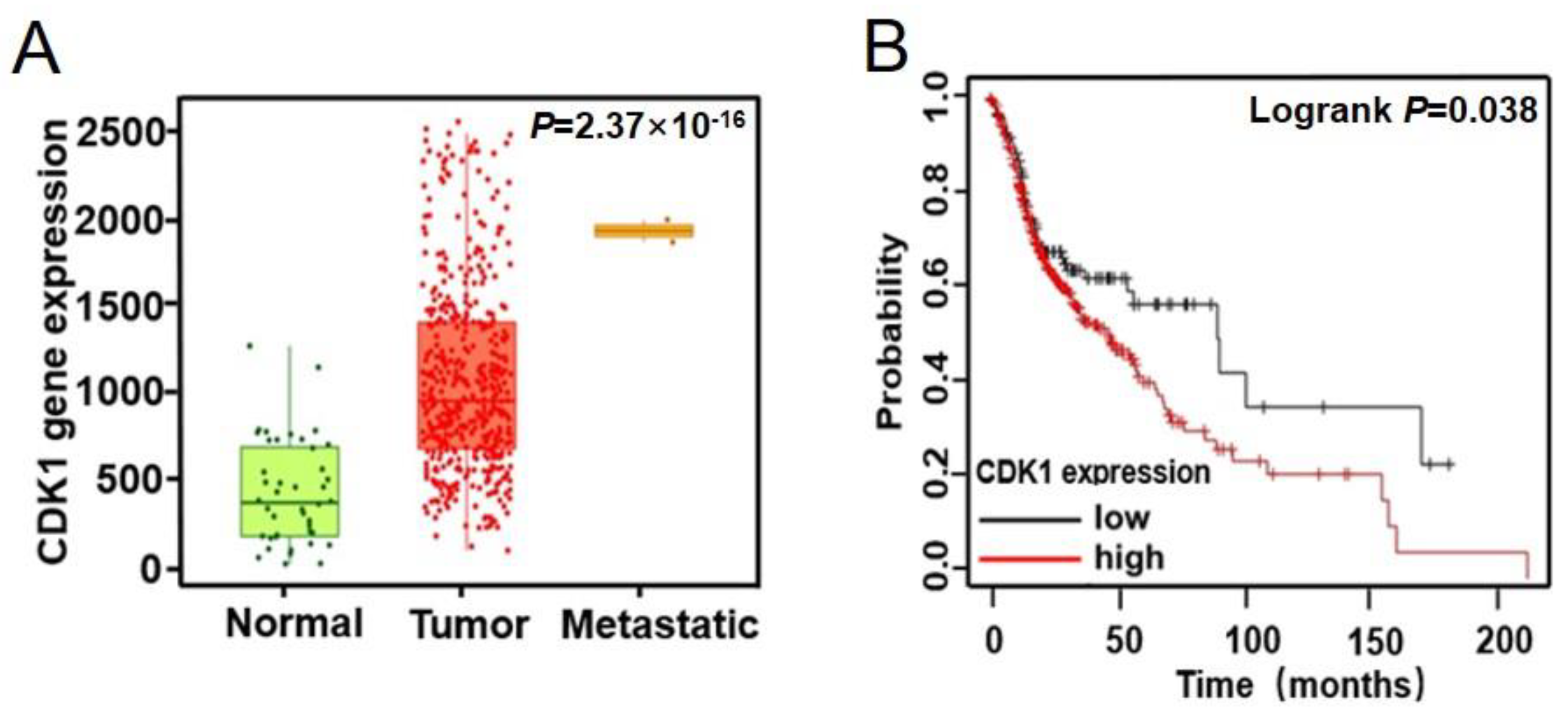
2.4. CDK1 Is Upregulated in Head and Neck Squamous Cell Carcinomas (HNSCCs) and Is Correlated to Malignant Progression of HNSCCs
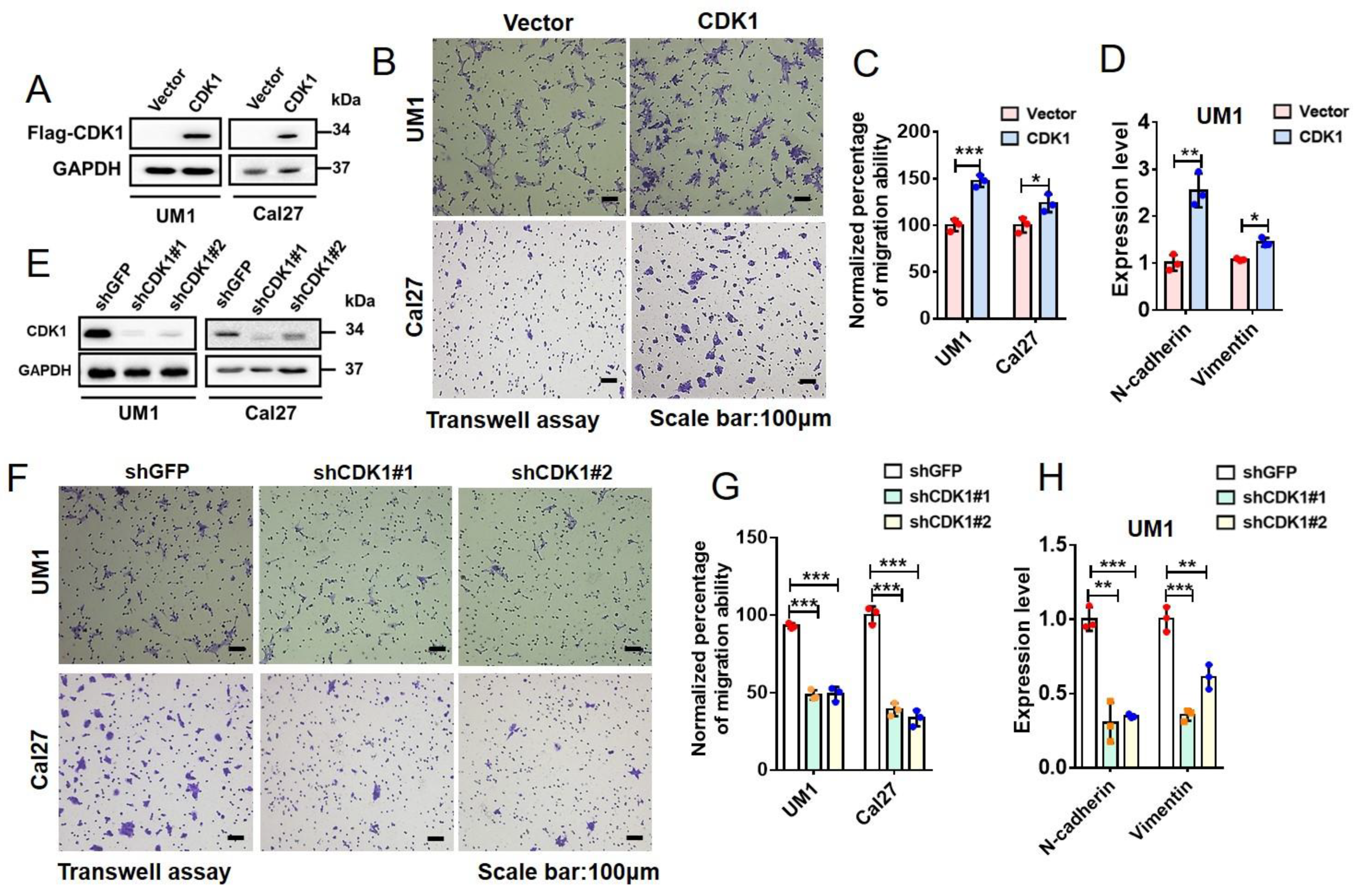
2.5. CDK1 Promotes Epithelial–Mesenchymal Transition (EMT) and Migration of HNSCC Cells
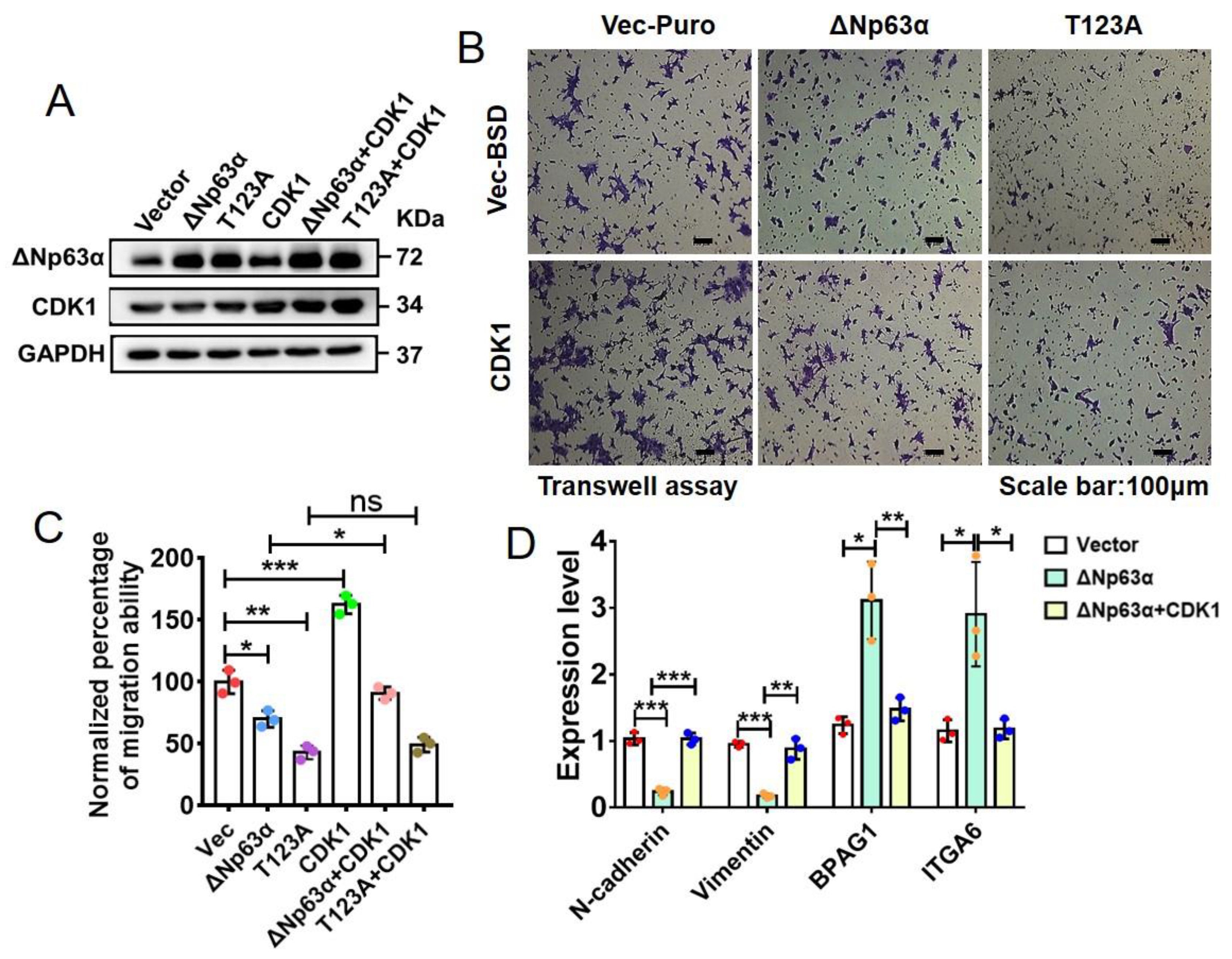
2.6. CDK1 Impairs Inhibitions of HNSCC Cell EMT and Migration Induced by Wild-Type ∆Np63α but Not Those by Its T123A Mutant
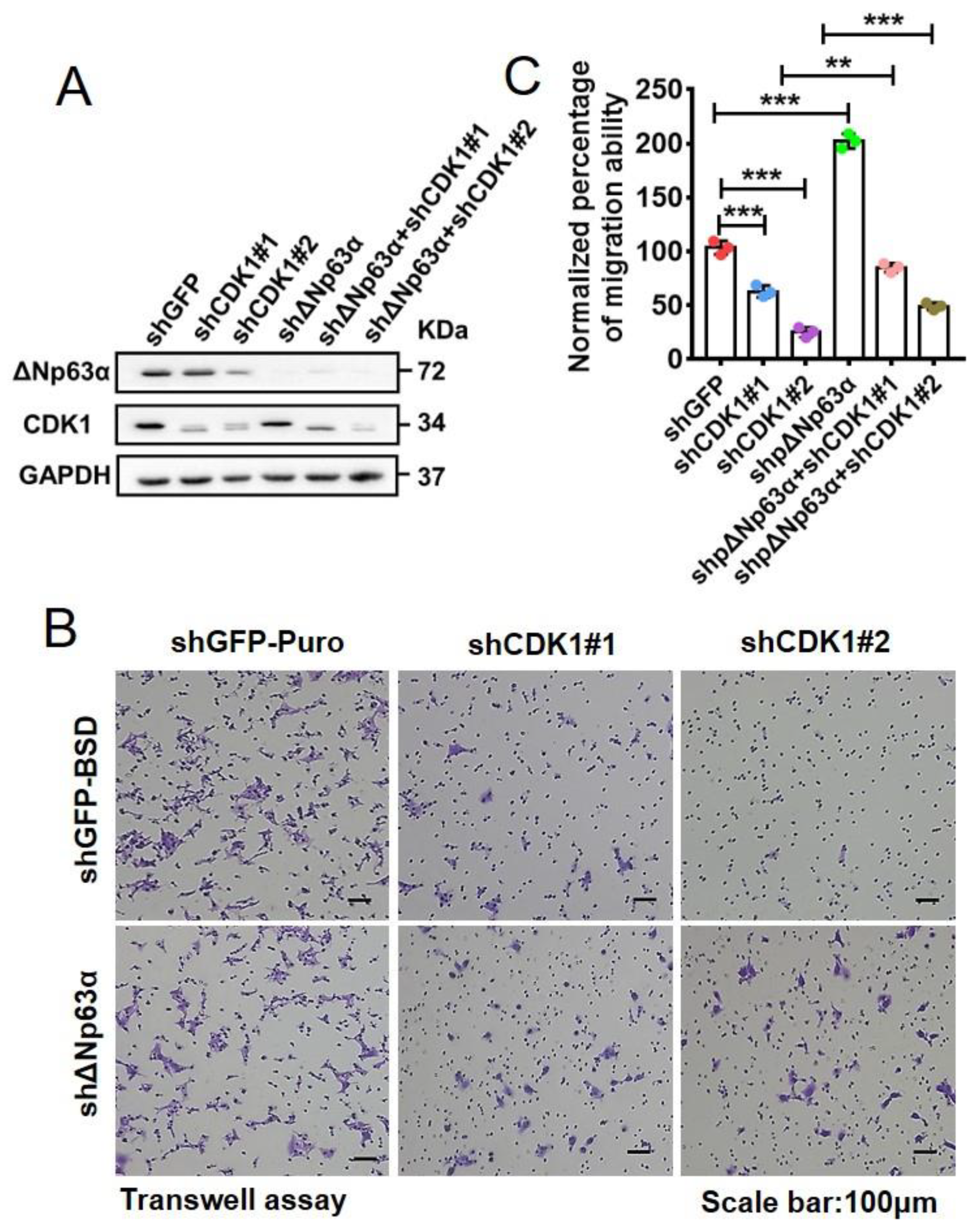
2.7. Knockdown of ∆Np63α Significantly Rescues Migration Inhibition Induced by CDK1 Depletion in HNSCC Cells
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Cell Culture
4.2. Lentivirus Packaging and Infection
4.3. Bioinformatics Analyses
4.4. Immunoblot (IB) Analysis
4.5. Reverse Transcription Quantitative PCR (RT-qPCR) Analysis
4.6. Immunoprecipitation (IP)
4.7. Chromatin Immunoprecipitation (ChIP)
4.8. Transwell Migration Assay
4.9. Luciferase Reporter Assay
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Johnson, D.E.; Burtness, B.; Leemans, C.R.; Lui, V.W.Y.; Bauman, J.E.; Grandis, J.R. Head and neck squamous cell carcinoma. Nat. Rev. Dis. Primers 2020, 6, 92. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Eichberger, J.; Schulz, D.; Pscheidl, K.; Fiedler, M.; Reichert, T.E.; Bauer, R.J.; Ettl, T. PD-L1 Influences Cell Spreading, Migration and Invasion in Head and Neck Cancer Cells. Int. J. Mol. Sci. 2020, 21, 8089. [Google Scholar] [CrossRef] [PubMed]
- Perri, F.; Longo, F.; Caponigro, F.; Sandomenico, F.; Guida, A.; Della Vittoria Scarpati, G.; Ottaiano, A.; Muto, P.; Ionna, F. Management of HPV-Related Squamous Cell Carcinoma of the Head and Neck: Pitfalls and Caveat. Cancers 2020, 12, 975. [Google Scholar] [CrossRef] [Green Version]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell. Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Aiello, N.M.; Brabletz, T.; Kang, Y.; Nieto, M.A.; Weinberg, R.A.; Stanger, B.Z. Upholding a role for EMT in pancreatic cancer metastasis. Nature 2017, 547, E7–E8. [Google Scholar] [CrossRef]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef]
- Ye, X.; Brabletz, T.; Kang, Y.; Longmore, G.D.; Nieto, M.A.; Stanger, B.Z.; Yang, J.; Weinberg, R.A. Upholding a role for EMT in breast cancer metastasis. Nature 2017, 547, E1–E3. [Google Scholar] [CrossRef]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef] [Green Version]
- Baumeister, P.; Zhou, J.; Canis, M.; Gires, O. Epithelial-to-Mesenchymal Transition-Derived Heterogeneity in Head and Neck Squamous Cell Carcinomas. Cancers 2021, 13, 5355. [Google Scholar] [CrossRef]
- Gonzalez-Gonzalez, R.; Ortiz-Sarabia, G.; Molina-Frechero, N.; Salas-Pacheco, J.M.; Salas-Pacheco, S.M.; Lavalle-Carrasco, J.; Lopez-Verdin, S.; Tremillo-Maldonado, O.; Bologna-Molina, R. Epithelial-Mesenchymal Transition Associated with Head and Neck Squamous Cell Carcinomas: A Review. Cancers 2021, 13, 3027. [Google Scholar] [CrossRef] [PubMed]
- Meyer, T.J.; Stoth, M.; Moratin, H.; Ickrath, P.; Herrmann, M.; Kleinsasser, N.; Hagen, R.; Hackenberg, S.; Scherzad, A. Cultivation of Head and Neck Squamous Cell Carcinoma Cells with Wound Fluid Leads to Cisplatin Resistance via Epithelial-Mesenchymal Transition Induction. Int. J. Mol. Sci. 2021, 22, 4474. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leemans, C.R.; Snijders, P.J.F.; Brakenhoff, R.H. The molecular landscape of head and neck cancer. Nat. Rev. Cancer 2018, 18, 269–282. [Google Scholar] [CrossRef]
- Puram, S.V.; Tirosh, I.; Parikh, A.S.; Patel, A.P.; Yizhak, K.; Gillespie, S.; Rodman, C.; Luo, C.L.; Mroz, E.A.; Emerick, K.S.; et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 2017, 171, 1611–1624.e24. [Google Scholar] [CrossRef] [Green Version]
- Ren, X.; Wang, J.; Lin, X.; Wang, X. E-cadherin expression and prognosis of head and neck squamous cell carcinoma: Evidence from 19 published investigations. Onco Targets Ther. 2016, 9, 2447–2453. [Google Scholar] [CrossRef] [Green Version]
- Su, L.; Morgan, P.R.; Lane, E.B. Keratin 14 and 19 expression in normal, dysplastic and malignant oral epithelia. A study using in situ hybridization and immunohistochemistry. J. Oral. Pathol. Med. 1996, 25, 293–301. [Google Scholar] [CrossRef]
- Gao, L.; Zhang, W.; Zhong, W.Q.; Liu, Z.J.; Li, H.M.; Yu, Z.L.; Zhao, Y.F. Tumor associated macrophages induce epithelial to mesenchymal transition via the EGFR/ERK1/2 pathway in head and neck squamous cell carcinoma. Oncol. Rep. 2018, 40, 2558–2572. [Google Scholar] [CrossRef]
- Zheng, Y.; Wang, Z.; Xiong, X.; Zhong, Y.; Zhang, W.; Dong, Y.; Li, J.; Zhu, Z.; Zhang, W.; Wu, H.; et al. Membrane-tethered Notch1 exhibits oncogenic property via activation of EGFR-PI3K-AKT pathway in oral squamous cell carcinoma. J. Cell. Physiol. 2019, 234, 5940–5952. [Google Scholar] [CrossRef]
- Zhang, Z.; Dong, Z.; Lauxen, I.S.; Filho, M.S.; Nör, J.E. Endothelial cell-secreted EGF induces epithelial to mesenchymal transition and endows head and neck cancer cells with stem-like phenotype. Cancer Res. 2014, 74, 2869–2881. [Google Scholar] [CrossRef] [Green Version]
- Xu, Q.; Zhang, Q.; Ishida, Y.; Hajjar, S.; Tang, X.; Shi, H.; Dang, C.V.; Le, A.D. EGF induces epithelial-mesenchymal transition and cancer stem-like cell properties in human oral cancer cells via promoting Warburg effect. Oncotarget 2017, 8, 9557–9571. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Peng, Y.; Fan, S.; Li, Y.; Xiao, Z.X.; Li, C. A double dealing tale of p63: An oncogene or a tumor suppressor. Cell. Mol. Life Sci. 2018, 75, 965–973. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.; Tan, Y.; Wang, L.; Cai, J.; Li, X.; Zeng, Z.; Xiong, W.; Li, G.; Li, X.; Tan, P.; et al. TP63 links chromatin remodeling and enhancer reprogramming to epidermal differentiation and squamous cell carcinoma development. Cell. Mol. Life Sci. 2020, 77, 4325–4346. [Google Scholar] [CrossRef] [PubMed]
- Lo Muzio, L.; Santarelli, A.; Caltabiano, R.; Rubini, C.; Pieramici, T.; Trevisiol, L.; Carinci, F.; Leonardi, R.; De Lillo, A.; Lanzafame, S.; et al. p63 overexpression associates with poor prognosis in head and neck squamous cell carcinoma. Hum. Pathol. 2005, 36, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Lo Muzio, L.; Campisi, G.; Farina, A.; Rubini, C.; Pastore, L.; Giannone, N.; Colella, G.; Leonardi, R.; Carinci, F. Effect of p63 expression on survival in oral squamous cell carcinoma. Cancer Investig. 2007, 25, 464–469. [Google Scholar] [CrossRef] [PubMed]
- Ramer, N.; Wu, H.; Sabo, E.; Ramer, Y.; Emanuel, P.; Orta, L.; Burstein, D.E. Prognostic value of quantitative p63 immunostaining in adenoid cystic carcinoma of salivary gland assessed by computerized image analysis. Cancer 2010, 116, 77–83. [Google Scholar] [CrossRef]
- Saghravanian, N.; Anvari, K.; Ghazi, N.; Memar, B.; Shahsavari, M.; Aghaee, M.A. Expression of p63 and CD44 in oral squamous cell carcinoma and correlation with clinicopathological parameters. Arch. Oral. Biol. 2017, 82, 160–165. [Google Scholar] [CrossRef]
- Takahashi, Y.; Noguchi, T.; Takeno, S.; Kimura, Y.; Okubo, M.; Kawahara, K. Reduced expression of p63 has prognostic implications for patients with esophageal squamous cell carcinoma. Oncol. Rep. 2006, 15, 323–328. [Google Scholar] [CrossRef] [Green Version]
- Urist, M.J.; Di Como, C.J.; Lu, M.L.; Charytonowicz, E.; Verbel, D.; Crum, C.P.; Ince, T.A.; McKeon, F.D.; Cordon-Cardo, C. Loss of p63 expression is associated with tumor progression in bladder cancer. Am. J. Pathol. 2002, 161, 1199–1206. [Google Scholar] [CrossRef] [Green Version]
- Koga, F.; Kawakami, S.; Fujii, Y.; Saito, K.; Ohtsuka, Y.; Iwai, A.; Ando, N.; Takizawa, T.; Kageyama, Y.; Kihara, K. Impaired p63 expression associates with poor prognosis and uroplakin III expression in invasive urothelial carcinoma of the bladder. Clin. Cancer Res. 2003, 9, 5501–5507. [Google Scholar]
- Moses, M.A.; George, A.L.; Sakakibara, N.; Mahmood, K.; Ponnamperuma, R.M.; King, K.E.; Weinberg, W.C. Molecular Mechanisms of p63-Mediated Squamous Cancer Pathogenesis. Int. J. Mol. Sci. 2019, 20, 3590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, K.E.; Reddi, D.M.; Ponnamperuma, R.M.; Gerdes, M.; Weinberg, W.C. Dysregulated ΔNp63α negatively regulates the maspin promoter in keratinocytes via blocking endogenous p73 binding. Mol. Carcinog. 2014, 53, 698–710. [Google Scholar] [CrossRef] [PubMed]
- Kouwenhoven, E.N.; van Heeringen, S.J.; Tena, J.J.; Oti, M.; Dutilh, B.E.; Alonso, M.E.; de la Calle-Mustienes, E.; Smeenk, L.; Rinne, T.; Parsaulian, L.; et al. Genome-wide profiling of p63 DNA-binding sites identifies an element that regulates gene expression during limb development in the 7q21 SHFM1 locus. PLoS Genet. 2010, 6, e1001065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oti, M.; Kouwenhoven, E.N.; Zhou, H. Genome-wide p63-regulated gene expression in differentiating epidermal keratinocytes. Genom. Data 2015, 5, 159–163. [Google Scholar] [CrossRef] [Green Version]
- Carroll, D.K.; Carroll, J.S.; Leong, C.O.; Cheng, F.; Brown, M.; Mills, A.A.; Brugge, J.S.; Ellisen, L.W. p63 regulates an adhesion programme and cell survival in epithelial cells. Nat. Cell. Biol. 2006, 8, 551–561. [Google Scholar] [CrossRef]
- Carroll, D.K.; Brugge, J.S.; Attardi, L.D. p63, cell adhesion and survival. Cell Cycle 2007, 6, 255–261. [Google Scholar] [CrossRef] [Green Version]
- Barbieri, C.E.; Tang, L.J.; Brown, K.A.; Pietenpol, J.A. Loss of p63 leads to increased cell migration and up-regulation of genes involved in invasion and metastasis. Cancer Res. 2006, 66, 7589–7597. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Xiao, Z.X. Regulation of p63 protein stability via ubiquitin-proteasome pathway. Biomed Res. Int. 2014, 2014, 175721. [Google Scholar] [CrossRef]
- Malumbres, M. Cyclin-dependent kinases. Genome Biol. 2014, 15, 122. [Google Scholar] [CrossRef] [Green Version]
- Candas, D.; Fan, M.; Nantajit, D.; Vaughan, A.T.; Murley, J.S.; Woloschak, G.E.; Grdina, D.J.; Li, J.J. CyclinB1/Cdk1 phosphorylates mitochondrial antioxidant MnSOD in cell adaptive response to radiation stress. J. Mol. Cell. Biol. 2013, 5, 166–175. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Xie, B.; Fan, M.; Candas-Green, D.; Jiang, J.X.; Wei, R.; Wang, Y.; Chen, H.W.; Hu, Y.; Li, J.J. Low-Level Saturated Fatty Acid Palmitate Benefits Liver Cells by Boosting Mitochondrial Metabolism via CDK1-SIRT3-CPT2 Cascade. Dev. Cell 2020, 52, 196–209.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asghar, U.; Witkiewicz, A.K.; Turner, N.C.; Knudsen, E.S. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat. Rev. Drug Discov. 2015, 14, 130–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, B.; Wang, S.; Jiang, N.; Li, J.J. Cyclin B1/CDK1-regulated mitochondrial bioenergetics in cell cycle progression and tumor resistance. Cancer Lett. 2019, 443, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Santamaría, D.; Barrière, C.; Cerqueira, A.; Hunt, S.; Tardy, C.; Newton, K.; Cáceres, J.F.; Dubus, P.; Malumbres, M.; Barbacid, M. Cdk1 is sufficient to drive the mammalian cell cycle. Nature 2007, 448, 811–815. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.X.; Wang, X.Q.; Chok, S.H.; Man, K.; Tsang, S.H.Y.; Chan, A.C.Y.; Ma, K.W.; Xia, W.; Cheung, T.T. Blocking CDK1/PDK1/β-Catenin signaling by CDK1 inhibitor RO3306 increased the efficacy of sorafenib treatment by targeting cancer stem cells in a preclinical model of hepatocellular carcinoma. Theranostics 2018, 8, 3737–3750. [Google Scholar] [CrossRef]
- Li, M.; He, F.; Zhang, Z.; Xiang, Z.; Hu, D. CDK1 serves as a potential prognostic biomarker and target for lung cancer. J. Int. Med. Res. 2020, 48, 300060519897508. [Google Scholar] [CrossRef] [Green Version]
- Ravindran Menon, D.; Luo, Y.; Arcaroli, J.J.; Liu, S.; KrishnanKutty, L.N.; Osborne, D.G.; Li, Y.; Samson, J.M.; Bagby, S.; Tan, A.C.; et al. CDK1 Interacts with Sox2 and Promotes Tumor Initiation in Human Melanoma. Cancer Res. 2018, 78, 6561–6574. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Kawakami, H.; Liu, W.; Zeng, X.; Strebhardt, K.; Tao, K.; Huang, S.; Sinicrope, F.A. Targeting CDK1 and MEK/ERK Overcomes Apoptotic Resistance in BRAF-Mutant Human Colorectal Cancer. Mol. Cancer Res. 2018, 16, 378–389. [Google Scholar] [CrossRef] [Green Version]
- Izadi, S.; Nikkhoo, A.; Hojjat-Farsangi, M.; Namdar, A.; Azizi, G.; Mohammadi, H.; Yousefi, M.; Jadidi-Niaragh, F. CDK1 in Breast Cancer: Implications for Theranostic Potential. Anticancer. Agents Med. Chem. 2020, 20, 758–767. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, F.H.; Chen, Q.E.; Wang, Y.Y.; Wang, Y.L.; He, J.C.; Zhou, J. The clinical significance of CDK1 expression in oral squamous cell carcinoma. Med. Oral. Patol. Oral. Cir. Bucal. 2015, 20, e7–e12. [Google Scholar] [CrossRef]
- Chang, J.T.; Wang, H.M.; Chang, K.W.; Chen, W.H.; Wen, M.C.; Hsu, Y.M.; Yung, B.Y.; Chen, I.H.; Liao, C.T.; Hsieh, L.L.; et al. Identification of differentially expressed genes in oral squamous cell carcinoma (OSCC): Overexpression of NPM, CDK1 and NDRG1 and underexpression of CHES1. Int. J. Cancer 2005, 114, 942–949. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Pang, Y.; Cao, H.; Liu, X.; Tu, L.; Shen, Y.; Jia, X.; Lee, J.C.; Wang, Y. Integrated Screens Identify CDK1 as a Therapeutic Target in Advanced Gastrointestinal Stromal Tumors. Cancer Res. 2021, 81, 2481–2494. [Google Scholar] [CrossRef]
- Fan, X.; He, W.; Hu, K.; Chen, H.; Chen, L.; Fan, S.; Li, C. Pin1 and JNK1 cooperatively modulate TAp63γ. FEBS Open Bio. 2021, 11, 890–897. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Chen, Y.; Jiang, Y.; Hu, K.; Li, C. Prefoldin subunit MM1 promotes cell migration via facilitating filopodia formation. Biochem. Biophys. Res. Commun. 2020, 533, 613–619. [Google Scholar] [CrossRef]
- Osada, M.; Nagakawa, Y.; Park, H.L.; Yamashita, K.; Wu, G.; Kim, M.S.; Fomenkov, A.; Trink, B.; Sidransky, D. p63-specific activation of the BPAG-1e promoter. J. Investig. Dermatol. 2005, 125, 52–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeisberg, M.; Neilson, E.G. Biomarkers for epithelial-mesenchymal transitions. J. Clin. Investig. 2009, 119, 1429–1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, A.K.; Yuen, P.W.; Liu, Y.; Wang, X.H.; Cheung, A.L.; Wong, Y.C.; Tsao, S.W. Downregulation of hemidesmosomal proteins in nasopharyngeal carcinoma cells. Cancer Lett. 2001, 163, 117–123. [Google Scholar] [CrossRef]
- Yoon, S.O.; Shin, S.; Mercurio, A.M. Hypoxia stimulates carcinoma invasion by stabilizing microtubules and promoting the Rab11 trafficking of the alpha6beta4 integrin. Cancer Res. 2005, 65, 2761–2769. [Google Scholar] [CrossRef] [Green Version]
- Hussein, A.A.; Forouzanfar, T.; Bloemena, E.; de Visscher, J.; Brakenhoff, R.H.; Leemans, C.R.; Helder, M.N. A review of the most promising biomarkers for early diagnosis and prognosis prediction of tongue squamous cell carcinoma. Br. J. Cancer 2018, 119, 724–736. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Li, Z. MicroRNA expression and its implications for diagnosis and therapy of tongue squamous cell carcinoma. J. Cell. Mol. Med. 2016, 20, 10–16. [Google Scholar] [CrossRef]
- Hsieh, M.H.; Choe, J.H.; Gadhvi, J.; Kim, Y.J.; Arguez, M.A.; Palmer, M.; Gerold, H.; Nowak, C.; Do, H.; Mazambani, S.; et al. p63 and SOX2 Dictate Glucose Reliance and Metabolic Vulnerabilities in Squamous Cell Carcinomas. Cell Rep. 2019, 28, 1860–1878.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramsey, M.R.; Wilson, C.; Ory, B.; Rothenberg, S.M.; Faquin, W.; Mills, A.A.; Ellisen, L.W. FGFR2 signaling underlies p63 oncogenic function in squamous cell carcinoma. J. Clin. Investig. 2013, 123, 3525–3538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Sen, T.; Nagpal, J.; Upadhyay, S.; Trink, B.; Ratovitski, E.; Sidransky, D. ATM kinase is a master switch for the Delta Np63 alpha phosphorylation/degradation in human head and neck squamous cell carcinoma cells upon DNA damage. Cell Cycle 2008, 7, 2846–2855. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Shi, H.; Qi, J.; Liu, D.; Yang, Z.; Li, C. JNK1 inhibits transcriptional and pro-apoptotic activity of TAp63γ. FEBS Lett. 2015, 589, 3686–3690. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, A.; Chang, X.; Sen, T.; Ravi, R.; Bedi, A.; Sidransky, D. Regulation of p53 family member isoform DeltaNp63alpha by the nuclear factor-kappaB targeting kinase IkappaB kinase beta. Cancer Res. 2010, 70, 1419–1429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, S.R.; Wu, H.; Wang, B.; Abuetabh, Y.; Sergi, C.; Leng, R.P. The Regulation of Tumor Suppressor p63 by the Ubiquitin-Proteasome System. Int. J. Mol. Sci. 2016, 17, 2041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuppi, M.; Kehrloesser, S.; Coutandin, D.W.; Rossi, V.; Luh, L.M.; Strubel, A.; Hötte, K.; Hoffmeister, M.; Schäfer, B.; De Oliveira, T.; et al. Oocyte DNA damage quality control requires consecutive interplay of CHK2 and CK1 to activate p63. Nat. Struct Mol. Biol. 2018, 25, 261–269. [Google Scholar] [CrossRef]
- Pitzius, S.; Osterburg, C.; Gebel, J.; Tascher, G.; Schäfer, B.; Zhou, H.; Münch, C.; Dötsch, V. TA*p63 and GTAp63 achieve tighter transcriptional regulation in quality control by converting an inhibitory element into an additional transactivation domain. Cell. Death Dis. 2019, 10, 686. [Google Scholar] [CrossRef] [Green Version]
- Demaria, M.; O’Leary, M.N.; Chang, J.; Shao, L.; Liu, S.; Alimirah, F.; Koenig, K.; Le, C.; Mitin, N.; Deal, A.M.; et al. Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer Discov. 2017, 7, 165–176. [Google Scholar] [CrossRef] [Green Version]
- Prasanna, P.G.; Citrin, D.E.; Hildesheim, J.; Ahmed, M.M.; Venkatachalam, S.; Riscuta, G.; Xi, D.; Zheng, G.; Deursen, J.V.; Goronzy, J.; et al. Therapy-Induced Senescence: Opportunities to Improve Anticancer Therapy. J. Natl. Cancer Inst. 2021, 113, 1285–1298. [Google Scholar] [CrossRef]
- Zhang, B.; Zhang, M.; Li, Q.; Yang, Y.; Shang, Z.; Luo, J. TPX2 mediates prostate cancer epithelial-mesenchymal transition through CDK1 regulated phosphorylation of ERK/GSK3β/SNAIL pathway. Biochem. Biophys. Res. Commun. 2021, 546, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Li, K.; Zhang, J.; Wang, L.; Sheng, L.; Yan, L. Inhibition of CDK1 Reverses the Resistance of 5-Fu in Colorectal Cancer. Cancer Manag. Res. 2020, 12, 11271–11283. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Hou, P.; Fan, D.; Dong, M.; Ma, M.; Li, H.; Yao, R.; Li, Y.; Wang, G.; Geng, P.; et al. The degradation of EZH2 mediated by lncRNA ANCR attenuated the invasion and metastasis of breast cancer. Cell Death Differ. 2017, 24, 59–71. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.; Wang, B.; Ma, Y.; Kuang, J.; Liang, J.; Yuan, Y. NUCKS1 Promotes Proliferation, Invasion and Migration of Non-Small Cell Lung Cancer by Upregulating CDK1 Expression. Cancer Manag. Res. 2020, 12, 13311–13323. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wu, Y.; Li, Q.; Liang, J.; He, Q.; Zhao, L.; Chen, J.; Cheng, M.; Huang, Z.; Ren, H.; et al. METTL3 Promotes Tumorigenesis and Metastasis through BMI1 m(6)A Methylation in Oral Squamous Cell Carcinoma. Mol. Ther. 2020, 28, 2177–2190. [Google Scholar] [CrossRef]
- Zhao, T.; He, Q.; Liu, Z.; Ding, X.; Zhou, X.; Wang, A. Angiotensin II type 2 receptor-interacting protein 3a suppresses proliferation, migration and invasion in tongue squamous cell carcinoma via the extracellular signal-regulated kinase-Snai2 pathway. Oncol. Lett. 2016, 11, 340–344. [Google Scholar] [CrossRef] [Green Version]
- Ji, M.; Wang, W.; Yan, W.; Chen, D.; Ding, X.; Wang, A. Dysregulation of AKT1, a miR-138 target gene, is involved in the migration and invasion of tongue squamous cell carcinoma. J. Oral. Pathol. Med. 2017, 46, 731–737. [Google Scholar] [CrossRef]
- Liu, Z.; He, Q.; Ding, X.; Zhao, T.; Zhao, L.; Wang, A. SOD2 is a C-myc target gene that promotes the migration and invasion of tongue squamous cell carcinoma involving cancer stem-like cells. Int. J. Biochem Cell. Biol. 2015, 60, 139–146. [Google Scholar] [CrossRef]
- Chen, J.J.; Zhang, W. High expression of WWP1 predicts poor prognosis and associates with tumor progression in human colorectal cancer. Am. J. Cancer Res. 2018, 8, 256–265. [Google Scholar]
- Han, A.; Li, J.; Li, Y.; Wang, Y.; Bergholz, J.; Zhang, Y.; Li, C.; Xiao Zh, X. p63α modulates c-Myc activity via direct interaction and regulation of MM1 protein stability. Oncotarget 2016, 7, 44277–44287. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Chen, L.; Jin, S.; Xiong, L.; Chen, H.; Hu, K.; Fan, X.; Fan, S.; Li, C. Ultraviolet B irradiation up-regulates MM1 and induces photoageing of the epidermis. Photodermatol. Photoimmunol. Photomed. 2021, 37, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Peng, B.L.; Li, W.J.; Ding, J.C.; He, Y.H.; Ran, T.; Xie, B.L.; Wang, Z.R.; Shen, H.F.; Xiao, R.Q.; Gao, W.W.; et al. A hypermethylation strategy utilized by enhancer-bound CARM1 to promote estrogen receptor α-dependent transcriptional activation and breast carcinogenesis. Theranostics 2020, 10, 3451–3473. [Google Scholar] [CrossRef] [PubMed]
- Guilhamon, P.; Eskandarpour, M.; Halai, D.; Wilson, G.A.; Feber, A.; Teschendorff, A.E.; Gomez, V.; Hergovich, A.; Tirabosco, R.; Fernanda Amary, M.; et al. Meta-analysis of IDH-mutant cancers identifies EBF1 as an interaction partner for TET2. Nat. Commun. 2013, 4, 2166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Chang, D.L.; Yang, Z.; Qi, J.; Liu, R.; He, H.; Li, D.; Xiao, Z.X. Pin1 modulates p63α protein stability in regulation of cell survival, proliferation and tumor formation. Cell. Death Dis. 2013, 4, e943. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, H.; Hu, K.; Xie, Y.; Qi, Y.; Li, W.; He, Y.; Fan, S.; Liu, W.; Li, C. CDK1 Promotes Epithelial–Mesenchymal Transition and Migration of Head and Neck Squamous Carcinoma Cells by Repressing ∆Np63α-Mediated Transcriptional Regulation. Int. J. Mol. Sci. 2022, 23, 7385. https://doi.org/10.3390/ijms23137385
Chen H, Hu K, Xie Y, Qi Y, Li W, He Y, Fan S, Liu W, Li C. CDK1 Promotes Epithelial–Mesenchymal Transition and Migration of Head and Neck Squamous Carcinoma Cells by Repressing ∆Np63α-Mediated Transcriptional Regulation. International Journal of Molecular Sciences. 2022; 23(13):7385. https://doi.org/10.3390/ijms23137385
Chicago/Turabian StyleChen, Huimin, Ke Hu, Ying Xie, Yucheng Qi, Wenjuan Li, Yaohui He, Shijie Fan, Wen Liu, and Chenghua Li. 2022. "CDK1 Promotes Epithelial–Mesenchymal Transition and Migration of Head and Neck Squamous Carcinoma Cells by Repressing ∆Np63α-Mediated Transcriptional Regulation" International Journal of Molecular Sciences 23, no. 13: 7385. https://doi.org/10.3390/ijms23137385
APA StyleChen, H., Hu, K., Xie, Y., Qi, Y., Li, W., He, Y., Fan, S., Liu, W., & Li, C. (2022). CDK1 Promotes Epithelial–Mesenchymal Transition and Migration of Head and Neck Squamous Carcinoma Cells by Repressing ∆Np63α-Mediated Transcriptional Regulation. International Journal of Molecular Sciences, 23(13), 7385. https://doi.org/10.3390/ijms23137385