
New Avenues of Heme Synthesis Regulation



Abstract
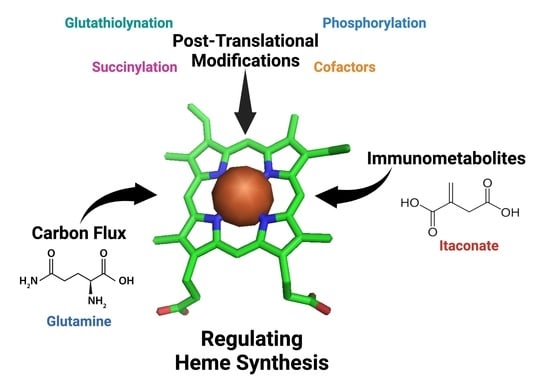
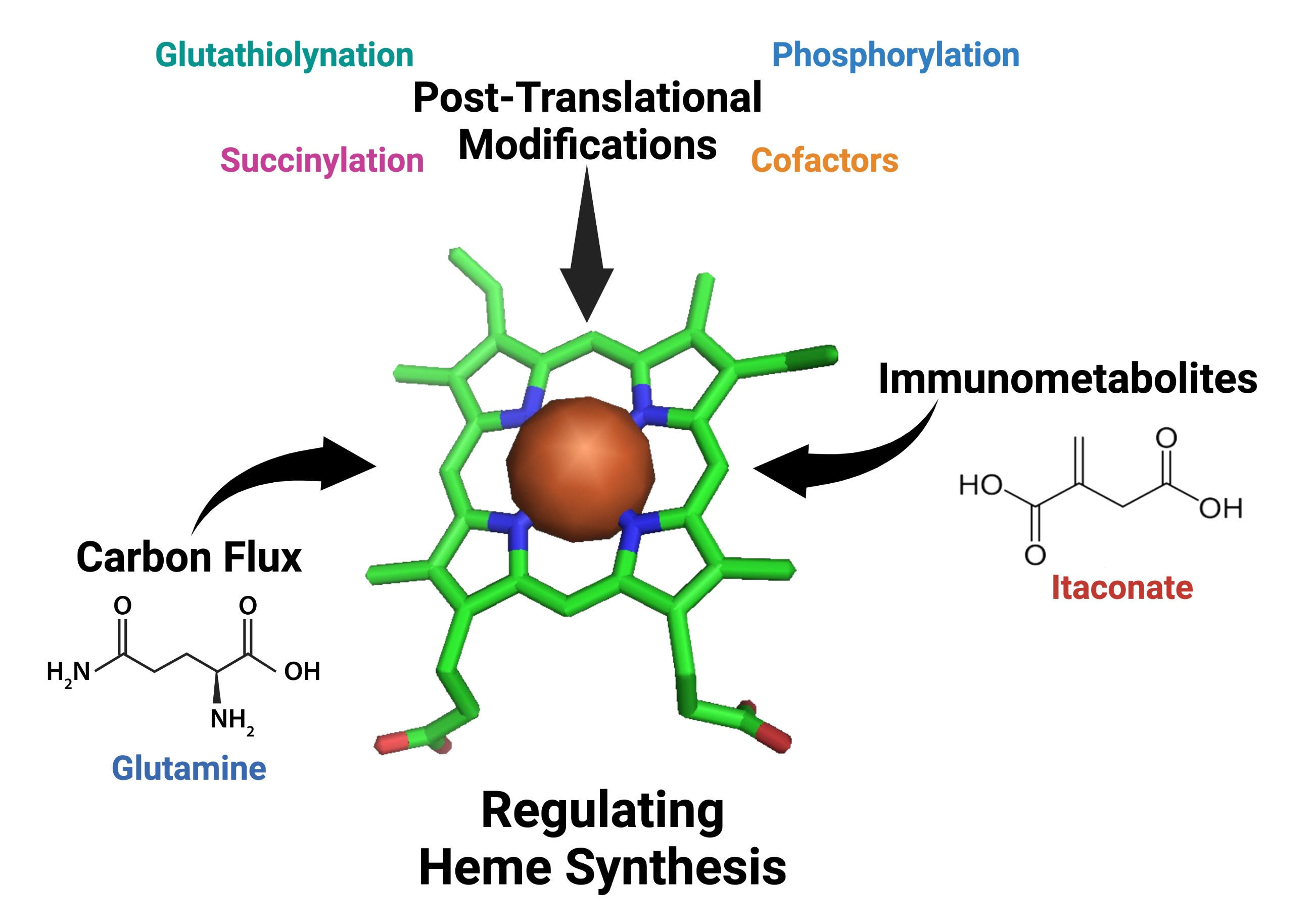
:
{kind=link}
{kind=link}
{kind=link}
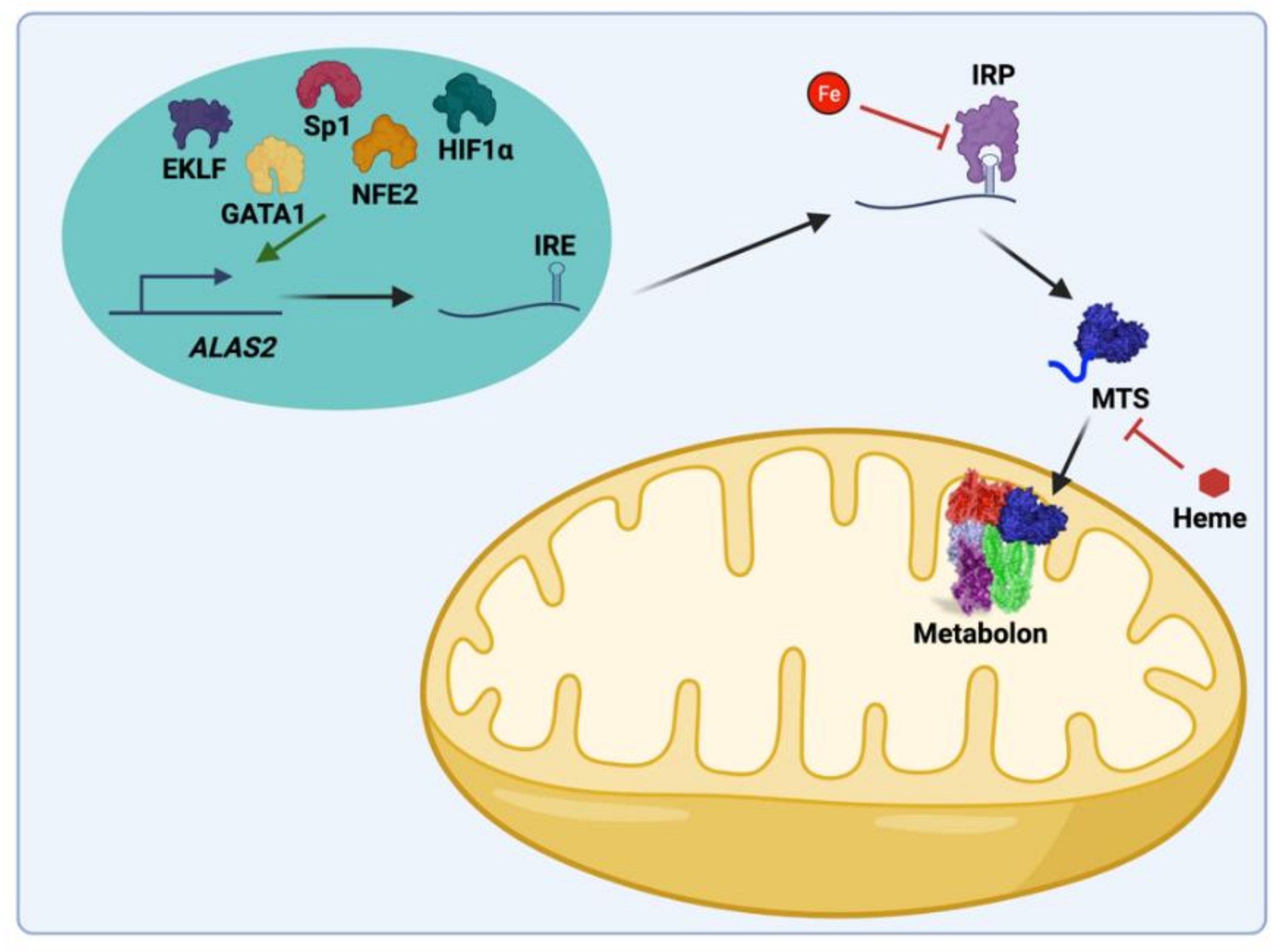{kind=link}
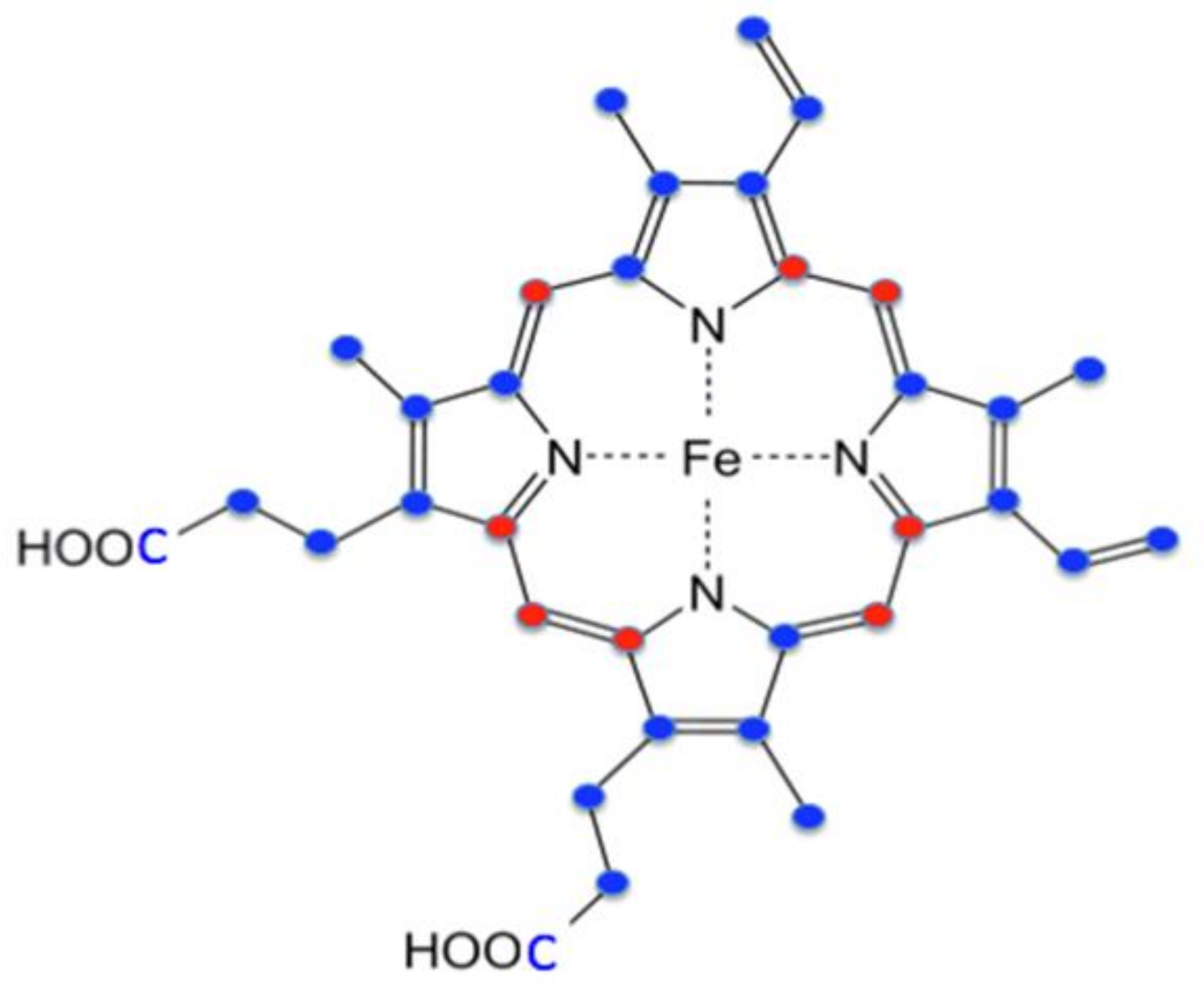{kind=link}
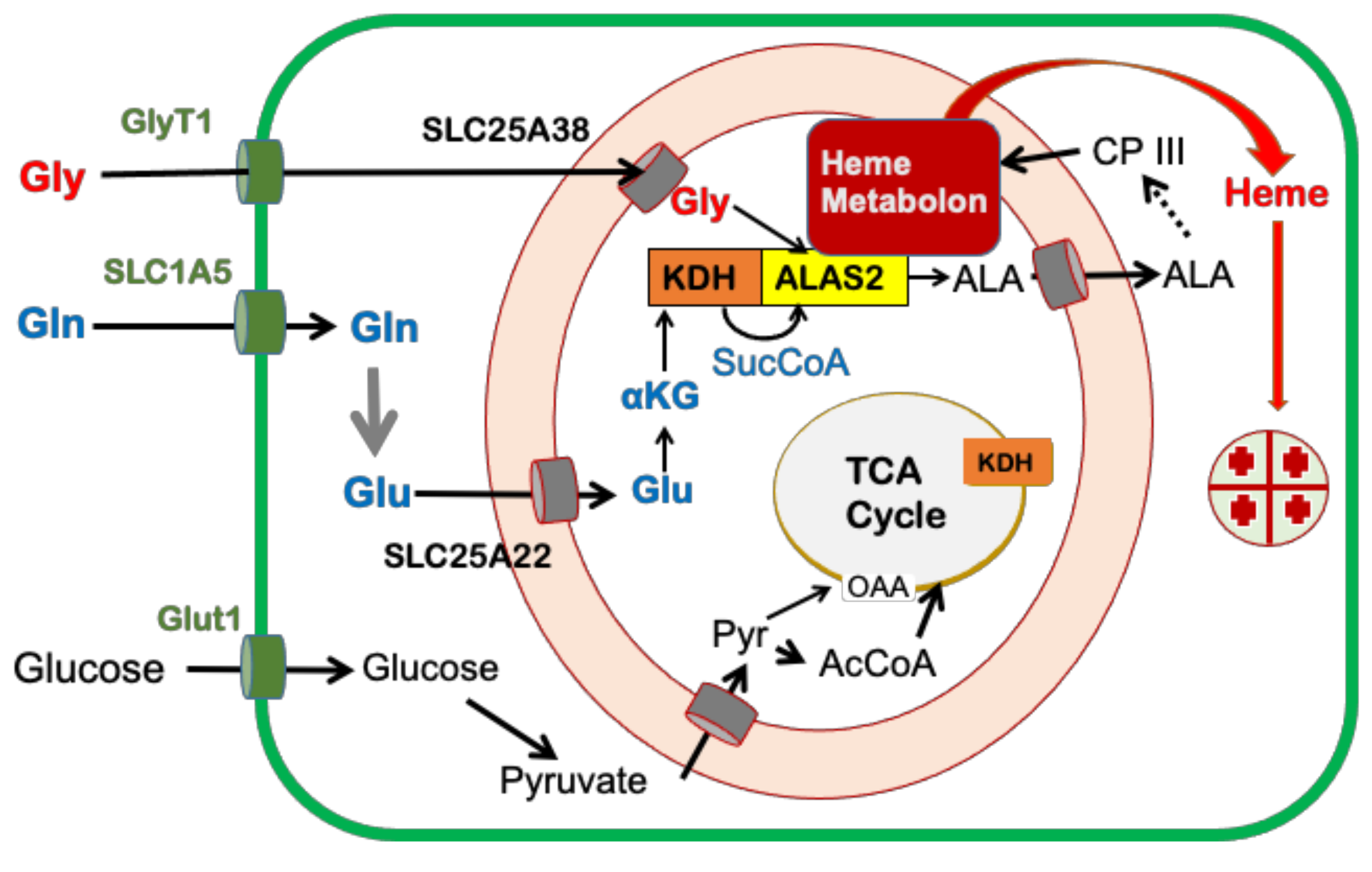{kind=link}
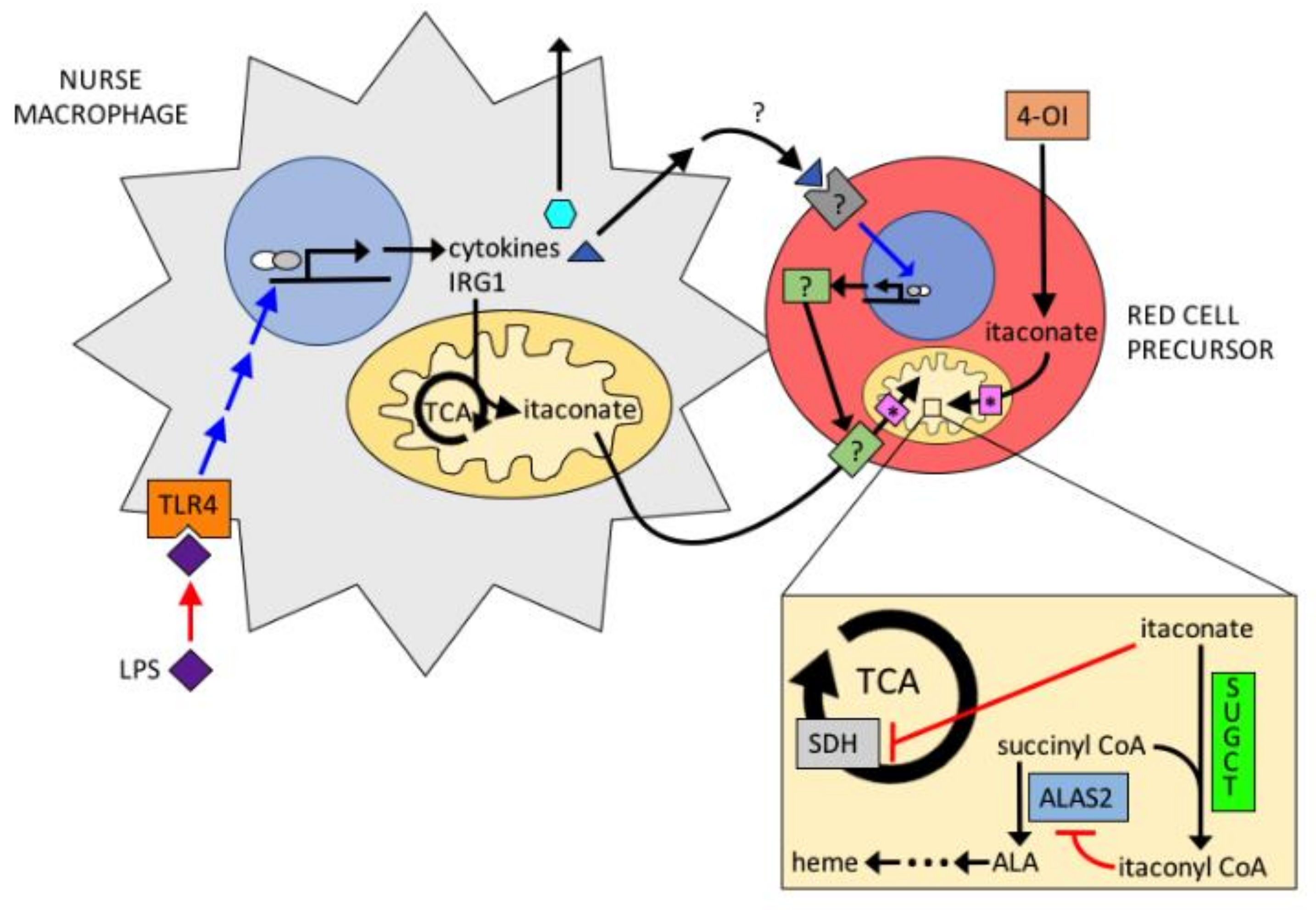{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
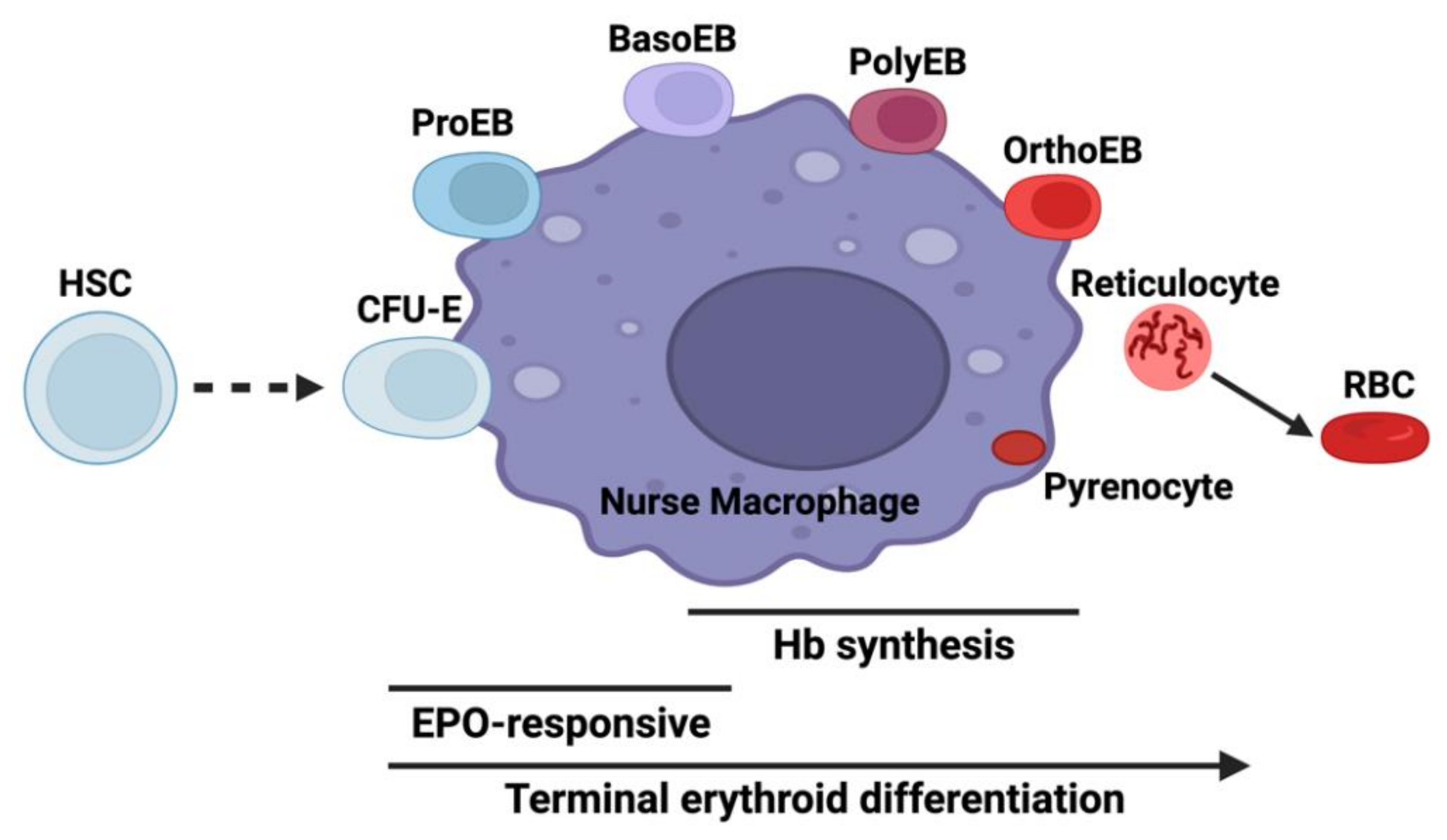
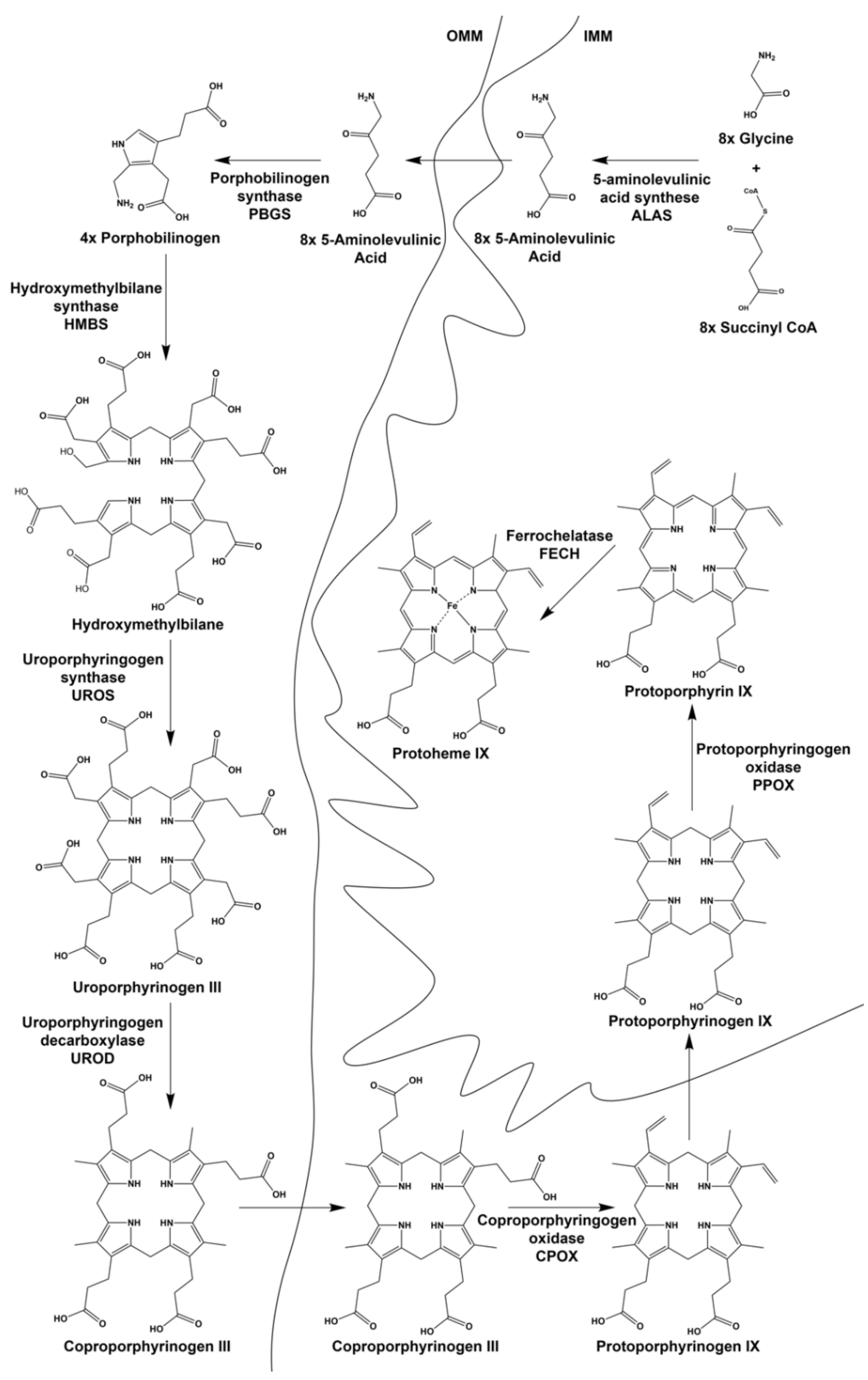
2. Provision of Glycine and Succinyl-CoA for 5-Aminolevulinic Acid (ALA) Synthesis during Erythropoiesis
3. Role of the Immunometabolite Itaconate on Erythropoiesis
4. PTMs of Heme Synthesis Enzymes
4.1. Cofactor Assembly
4.2. Phosphorylation
4.3. Glutathionylation
4.4. Succinylation
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Song, S.; Starunov, V.; Bailly, X.; Ruta, C.; Kerner, P.; Cornelissen, A.J.M.; Balavoine, G. Globins in the marine annelid Platynereis dumerilii shed new light on hemoglobin evolution in bilaterians. BMC Evol. Biol. 2020, 20, 165. [Google Scholar] [CrossRef] [PubMed]
- Palis, J. Primitive and definitive erythropoiesis in mammals. Front. Physiol. 2014, 5, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orkin, S.H.; Zon, L.I. Hematopoiesis: An evolving paradigm for stem cell biology. Cell 2008, 132, 631–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hattangadi, S.M.; Wong, P.; Zhang, L.; Flygare, J.; Lodish, H.F. From stem cell to red cell: Regulation of erythropoiesis at multiple levels by multiple proteins, RNAs, and chromatin modifications. Blood 2011, 118, 6258–6268. [Google Scholar] [CrossRef] [Green Version]
- Bessis, M. Erythroblastic island, functional unity of bone marrow. Rev. Hematol. 1958, 13, 8–11. [Google Scholar]
- Manwani, D.; Bieker, J.J. The erythroblastic island. Curr. Top. Dev. Biol. 2008, 82, 23–53. [Google Scholar] [CrossRef] [Green Version]
- An, X.; Schulz, V.P.; Li, J.; Wu, K.; Liu, J.; Xue, F.; Hu, J.; Mohandas, N.; Gallagher, P.G. Global transcriptome analyses of human and murine terminal erythroid differentiation. Blood 2014, 123, 3466–3477. [Google Scholar] [CrossRef] [Green Version]
- Bishop, D.F.; Henderson, A.S.; Astrin, K.H. Human delta-aminolevulinate synthase: Assignment of the housekeeping gene to 3p21 and the erythroid-specific gene to the X chromosome. Genomics 1990, 7, 207–214. [Google Scholar] [CrossRef]
- Cox, T.C.; Bawden, M.J.; Abraham, N.G.; Bottomley, S.S.; May, B.K.; Baker, E.; Chen, L.Z.; Sutherland, G.R. Erythroid 5-aminolevulinate synthase is located on the X chromosome. Am. J. Hum. Genet. 1990, 46, 107–111. [Google Scholar]
- Sutherland, G.R.; Baker, E.; Callen, D.F.; Hyland, V.J.; May, B.K.; Bawden, M.J.; Healy, H.M.; Borthwick, I.A. 5-Aminolevulinate synthase is at 3p21 and thus not the primary defect in X-linked sideroblastic anemia. Am. J. Hum. Genet. 1988, 43, 331–335. [Google Scholar]
- Medlock, A.E.; Dailey, H.A. Regulation of Mammalian Heme Biosynthesis. In Tetrapyrroles: Birth, Lift and Death; Landes Bioscience Springer Science+Business Media: New York, NY, USA, 2009; pp. 116–127. [Google Scholar]
- Bhasker, C.R.; Burgiel, G.; Neupert, B.; Emery-Goodman, A.; Kuhn, L.C.; May, B.K. The putative iron-responsive element in the human erythroid 5-aminolevulinate synthase mRNA mediates translational control. J. Biol. Chem. 1993, 268, 12699–12705. [Google Scholar] [CrossRef]
- Melefors, O.; Goossen, B.; Johansson, H.E.; Stripecke, R.; Gray, N.K.; Hentze, M.W. Translational control of 5-aminolevulinate synthase mRNA by iron-responsive elements in erythroid cells. J. Biol. Chem. 1993, 268, 5974–5978. [Google Scholar] [CrossRef]
- Schranzhofer, M.; Schifrer, M.; Cabrera, J.A.; Kopp, S.; Chiba, P.; Beug, H.; Mullner, E.W. Remodeling the regulation of iron metabolism during erythroid differentiation to ensure efficient heme biosynthesis. Blood 2006, 107, 4159–4167. [Google Scholar] [CrossRef] [PubMed]
- Doyle, F.; Tenenbaum, S.A. Trans-regulation of RNA-binding protein motifs by microRNA. Front. Genet. 2014, 5, 79. [Google Scholar] [CrossRef] [Green Version]
- Abu-Farha, M.; Niles, J.; Willmore, W.G. Erythroid-specific 5-aminolevulinate synthase protein is stabilized by low oxygen and proteasomal inhibition. Biochem. Cell Biol. 2005, 83, 620–630. [Google Scholar] [CrossRef]
- Dailey, T.A.; Woodruff, J.H.; Dailey, H.A. Examination of mitochondrial protein targeting of haem synthetic enzymes: In vivo identification of three functional haem-responsive motifs in 5-aminolaevulinate synthase. Biochem. J. 2005, 386, 381–386. [Google Scholar] [CrossRef] [Green Version]
- Lathrop, J.T.; Timko, M.P. Regulation by heme of mitochondrial protein transport through a conserved amino acid motif. Science 1993, 259, 522–525. [Google Scholar] [CrossRef]
- Yamauchi, K.; Hayashi, N.; Kikuchi, G. Translocation of delta-aminolevulinate synthase from the cytosol to the mitochondria and its regulation by hemin in the rat liver. J. Biol. Chem. 1980, 255, 1746–1751. [Google Scholar] [CrossRef]
- Dailey, H.A.; Meissner, P.N. Erythroid heme biosynthesis and its disorders. Cold Spring Harb. Perspect. Med. 2013, 3, a011676. [Google Scholar] [CrossRef] [Green Version]
- Conder, L.H.; Woodard, S.I.; Dailey, H.A. Multiple mechanisms for the regulation of haem synthesis during erythroid cell differentiation. Possible role for coproporphyrinogen oxidase. Biochem. J. 1991, 275 Pt 2, 321–326. [Google Scholar] [CrossRef] [Green Version]
- Yin, X.; Dailey, H.A. Erythroid 5-aminolevulinate synthase is required for erythroid differentiation in mouse embryonic stem cells. Blood Cells Mol. Dis. 1998, 24, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Paw, B.H. Cellular and mitochondrial iron homeostasis in vertebrates. Biochim. Biophys. Acta 2012, 1823, 1459–1467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Murray, J.P.; Prykhozhij, S.V.; Dufay, J.N.; Steele, S.L.; Gaston, D.; Nasrallah, G.K.; Coombs, A.J.; Liwski, R.S.; Fernandez, C.V.; Berman, J.N.; et al. Glycine and Folate Ameliorate Models of Congenital Sideroblastic Anemia. PLoS Genet. 2016, 12, e1005783. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Santos, D.; Schranzhofer, M.; Bergeron, R.; Sheftel, A.D.; Ponka, P. Extracellular glycine is necessary for optimal hemoglobinization of erythroid cells. Haematologica 2017, 102, 1314–1323. [Google Scholar] [CrossRef] [PubMed]
- Burch, J.S.; Marcero, J.R.; Maschek, J.A.; Cox, J.E.; Jackson, L.K.; Medlock, A.E.; Phillips, J.D.; Dailey, H.A., Jr. Glutamine via alpha-ketoglutarate dehydrogenase provides succinyl-CoA for heme synthesis during erythropoiesis. Blood 2018, 132, 987–998. [Google Scholar] [CrossRef] [Green Version]
- Medlock, A.E.; Shiferaw, M.T.; Marcero, J.R.; Vashisht, A.A.; Wohlschlegel, J.A.; Phillips, J.D.; Dailey, H.A. Identification of the Mitochondrial Heme Metabolism Complex. PLoS ONE 2015, 10, e0135896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piel, R.B., 3rd; Dailey, H.A., Jr.; Medlock, A.E. The mitochondrial heme metabolon: Insights into the complex(ity) of heme synthesis and distribution. Mol. Genet. Metab. 2019, 128, 198–203. [Google Scholar] [CrossRef]
- Piel, R.B., 3rd; Shiferaw, M.T.; Vashisht, A.A.; Marcero, J.R.; Praissman, J.L.; Phillips, J.D.; Wohlschlegel, J.A.; Medlock, A.E. A Novel Role for Progesterone Receptor Membrane Component 1 (PGRMC1): A Partner and Regulator of Ferrochelatase. Biochemistry 2016, 55, 5204–5217. [Google Scholar] [CrossRef] [Green Version]
- Carrico, C.; Meyer, J.G.; He, W.; Gibson, B.W.; Verdin, E. The Mitochondrial Acylome Emerges: Proteomics, Regulation by Sirtuins, and Metabolic and Disease Implications. Cell Metab. 2018, 27, 497–512. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, S.J.; James, D.E.; Mann, M. Protein Phosphorylation: A Major Switch Mechanism for Metabolic Regulation. Trends Endocrinol. Metab. 2015, 26, 676–687. [Google Scholar] [CrossRef]
- Zhang, J.; Ye, Z.W.; Singh, S.; Townsend, D.M.; Tew, K.D. An evolving understanding of the S-glutathionylation cycle in pathways of redox regulation. Free Radic. Biol. Med. 2018, 120, 204–216. [Google Scholar] [CrossRef] [PubMed]
- Homedan, C.; Laafi, J.; Schmitt, C.; Gueguen, N.; Lefebvre, T.; Karim, Z.; Desquiret-Dumas, V.; Wetterwald, C.; Deybach, J.C.; Gouya, L.; et al. Acute intermittent porphyria causes hepatic mitochondrial energetic failure in a mouse model. Int. J. Biochem. Cell Biol. 2014, 51, 93–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lunetti, P.; Damiano, F.; De Benedetto, G.; Siculella, L.; Pennetta, A.; Muto, L.; Paradies, E.; Marobbio, C.M.; Dolce, V.; Capobianco, L. Characterization of Human and Yeast Mitochondrial Glycine Carriers with Implications for Heme Biosynthesis and Anemia. J. Biol. Chem. 2016, 291, 19746–19759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schranzhofer, M.; Bergeron, R.; Ponka, P. Glycine Transporter 1 Plays a Crucial Role in Hemoglobinization. Blood 2011, 118, 345. [Google Scholar] [CrossRef]
- Heeney, M.M.; Berhe, S.; Campagna, D.R.; Oved, J.H.; Kurre, P.; Shaw, P.J.; Teo, J.; Shanap, M.A.; Hassab, H.M.; Glader, B.E.; et al. SLC25A38 congenital sideroblastic anemia: Phenotypes and genotypes of 31 individuals from 24 families, including 11 novel mutations, and a review of the literature. Hum. Mutat. 2021, 42, 1367–1383. [Google Scholar] [CrossRef]
- Winter, M.; Funk, J.; Korner, A.; Alberati, D.; Christen, F.; Schmitt, G.; Altmann, B.; Pospischil, A.; Singer, T. Effects of GlyT1 inhibition on erythropoiesis and iron homeostasis in rats. Exp. Hematol. 2016, 44, 964–974.e964. [Google Scholar] [CrossRef]
- Furuyama, K.; Sassa, S. Interaction between succinyl CoA synthetase and the heme-biosynthetic enzyme ALAS-E is disrupted in sideroblastic anemia. J. Clin. Investig. 2000, 105, 757–764. [Google Scholar] [CrossRef] [Green Version]
- Labbe, R.F.; Kurumada, T.; Onisawa, J. The role of succinyl-CoA synthetase in the control of heme biosynthesis. Biochim. Biophys. Acta 1965, 111, 403–415. [Google Scholar] [CrossRef]
- Gibson, K.D.; Laver, W.G.; Neuberger, A. Initial stages in the biosynthesis of porphyrins. 2. The formation of delta-aminolaevulic acid from glycine and succinyl-coenzyme A by particles from chicken erythrocytes. Biochem. J. 1958, 70, 71–81. [Google Scholar] [CrossRef]
- Laver, W.G.; Neuberger, A.; Udenfriend, S. Initial stages in the biosynthesis of porphyrins. I. The formation of delta-am-inolaevulic acid by particles obtained from chicken erythrocytes. Biochem. J. 1958, 70, 4–14. [Google Scholar] [CrossRef] [Green Version]
- Kacso, G.; Ravasz, D.; Doczi, J.; Nemeth, B.; Madgar, O.; Saada, A.; Ilin, P.; Miller, C.; Ostergaard, E.; Iordanov, I.; et al. Two transgenic mouse models for beta-subunit components of succinate-CoA ligase yielding pleiotropic metabolic alterations. Biochem. J. 2016, 473, 3463–3485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bishop, D.F.; Tchaikovskii, V.; Nazarenko, I.; Desnick, R.J. Molecular expression and characterization of erythroid-specific 5-aminolevulinate synthase gain-of-function mutations causing X-linked protoporphyria. Mol. Med. 2013, 19, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Neuberger, A. Aspects of the metabolism of glycine and of porphyrins. Biochem. J. 1961, 78, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dailey, H.A.; Finnegan, M.G.; Johnson, M.K. Human ferrochelatase is an iron-sulfur protein. Biochemistry 1994, 33, 403–407. [Google Scholar] [CrossRef]
- Owen, O.E.; Kalhan, S.C.; Hanson, R.W. The key role of anaplerosis and cataplerosis for citric acid cycle function. J. Biol. Chem. 2002, 277, 30409–30412. [Google Scholar] [CrossRef] [Green Version]
- Greenfest-Allen, E.; Malik, J.; Palis, J.; Stoeckert, C.J., Jr. Stat and interferon genes identified by network analysis differentially regulate primitive and definitive erythropoiesis. BMC Syst. Biol. 2013, 7, 38. [Google Scholar] [CrossRef] [Green Version]
- Kingsley, P.D.; Greenfest-Allen, E.; Frame, J.M.; Bushnell, T.P.; Malik, J.; McGrath, K.E.; Stoeckert, C.J.; Palis, J. Ontogeny of erythroid gene expression. Blood 2013, 121, e5–e13. [Google Scholar] [CrossRef] [Green Version]
- Lyubarev, A.E.; Kurganov, B.I. Supramolecular organization of tricarboxylic acid cycle enzymes. Biosystems 1989, 22, 91–102. [Google Scholar] [CrossRef]
- Oburoglu, L.; Tardito, S.; Fritz, V.; de Barros, S.C.; Merida, P.; Craveiro, M.; Mamede, J.; Cretenet, G.; Mongellaz, C.; An, X.; et al. Glucose and glutamine metabolism regulate human hematopoietic stem cell lineage specification. Cell Stem. Cell 2014, 15, 169–184. [Google Scholar] [CrossRef] [Green Version]
- Katt, W.P.; Cerione, R.A. Glutaminase regulation in cancer cells: A druggable chain of events. Drug Discov. Today 2014, 19, 450–457. [Google Scholar] [CrossRef] [Green Version]
- Diaz, G.A.; Banikazemi, M.; Oishi, K.; Desnick, R.J.; Gelb, B.D. Mutations in a new gene encoding a thiamine transporter cause thiamine-responsive megaloblastic anaemia syndrome. Nat. Genet. 1999, 22, 309–312. [Google Scholar] [CrossRef] [PubMed]
- Fleming, J.C.; Tartaglini, E.; Steinkamp, M.P.; Schorderet, D.F.; Cohen, N.; Neufeld, E.J. The gene mutated in thiamine-responsive anaemia with diabetes and deafness (TRMA) encodes a functional thiamine transporter. Nat. Genet. 1999, 22, 305–308. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.L.; Littlewood, T.J. The investigation and treatment of secondary anaemia. Blood Rev. 2012, 26, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.Z.; Devalaraja, S.; Haldar, M. The Heme Connection: Linking Erythrocytes and Macrophage Biology. Front. Immunol. 2017, 8, 33. [Google Scholar] [CrossRef] [Green Version]
- Soares, M.P.; Hamza, I. Macrophages and Iron Metabolism. Immunity 2016, 44, 492–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brendt, P.; Rehfeld, I.; Kamphausen, A.; Kreissig, C.; Peters, J. Lipopolysaccharide interference in erythropoiesis in mice. Anaesthesia 2012, 67, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Cordes, T.; Michelucci, A.; Hiller, K. Itaconic Acid: The Surprising Role of an Industrial Compound as a Mammalian Antimicrobial Metabolite. Annu. Rev. Nutr. 2015, 35, 451–473. [Google Scholar] [CrossRef]
- Cordes, T.; Wallace, M.; Michelucci, A.; Divakaruni, A.S.; Sapcariu, S.C.; Sousa, C.; Koseki, H.; Cabrales, P.; Murphy, A.N.; Hiller, K.; et al. Immunoresponsive Gene 1 and Itaconate Inhibit Succinate Dehydrogenase to Modulate Intracellular Succinate Levels. J. Biol. Chem. 2016, 291, 14274–14284. [Google Scholar] [CrossRef] [Green Version]
- Lampropoulou, V.; Sergushichev, A.; Bambouskova, M.; Nair, S.; Vincent, E.E.; Loginicheva, E.; Cervantes-Barragan, L.; Ma, X.; Huang, S.C.; Griss, T.; et al. Itaconate Links Inhibition of Succinate Dehydrogenase with Macrophage Metabolic Remodeling and Regulation of Inflammation. Cell Metab. 2016, 24, 158–166. [Google Scholar] [CrossRef] [Green Version]
- Luan, H.H.; Medzhitov, R. Food Fight: Role of Itaconate and Other Metabolites in Antimicrobial Defense. Cell Metab. 2016, 24, 379–387. [Google Scholar] [CrossRef] [Green Version]
- Michelucci, A.; Cordes, T.; Ghelfi, J.; Pailot, A.; Reiling, N.; Goldmann, O.; Binz, T.; Wegner, A.; Tallam, A.; Rausell, A.; et al. Immune-responsive gene 1 protein links metabolism to immunity by catalyzing itaconic acid production. Proc. Natl. Acad. Sci. USA 2013, 110, 7820–7825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemeth, B.; Doczi, J.; Csete, D.; Kacso, G.; Ravasz, D.; Adams, D.; Kiss, G.; Nagy, A.M.; Horvath, G.; Tretter, L.; et al. Abolition of mitochondrial substrate-level phosphorylation by itaconic acid produced by LPS-induced Irg1 expression in cells of murine macrophage lineage. FASEB J. 2016, 30, 286–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meiser, J.; Kraemer, L.; Jaeger, C.; Madry, H.; Link, A.; Lepper, P.M.; Hiller, K.; Schneider, J.G. Itaconic acid indicates cellular but not systemic immune system activation. Oncotarget 2018, 9, 32098–32107. [Google Scholar] [CrossRef] [PubMed]
- Marcero, J.R.; Cox, J.E.; Bergonia, H.A.; Medlock, A.E.; Phillips, J.D.; Dailey, H.A. The immunometabolite itaconate inhibits heme synthesis and remodels cellular metabolism in erythroid precursors. Blood Adv. 2021, 5, 4831–4841. [Google Scholar] [CrossRef]
- Hruz, T.; Laule, O.; Szabo, G.; Wessendorp, F.; Bleuler, S.; Oertle, L.; Widmayer, P.; Gruissem, W.; Zimmermann, P. Genevestigator v3: A reference expression database for the meta-analysis of transcriptomes. Adv. Bioinform. 2008, 2008, 420747. [Google Scholar] [CrossRef]
- Sassa, S.; Wolpe, S.; Cerami, A. Inhibition of erythroid differentiation of mouse erythroleukemia cells by a macrophage product(s). Blood Cells 1987, 13, 161–169. [Google Scholar]
- Adler, J.; Wang, S.F.; Lardy, H.A. The metabolism of itaconic acid by liver mitochondria. J. Biol. Chem. 1957, 229, 865–879. [Google Scholar] [CrossRef]
- Wang, S.F.; Adler, J.; Lardy, H.A. The pathway of itaconate metabolism by liver mitochondria. J. Biol. Chem. 1961, 236, 26–30. [Google Scholar] [CrossRef]
- Mills, E.L.; Ryan, D.G.; Prag, H.A.; Dikovskaya, D.; Menon, D.; Zaslona, Z.; Jedrychowski, M.P.; Costa, A.S.H.; Higgins, M.; Hams, E.; et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 2018, 556, 113–117. [Google Scholar] [CrossRef]
- Nemeth, E.; Valore, E.V.; Territo, M.; Schiller, G.; Lichtenstein, A.; Ganz, T. Hepcidin, a putative mediator of anemia of inflammation, is a type II acute-phase protein. Blood 2003, 101, 2461–2463. [Google Scholar] [CrossRef]
- Mailloux, R.J.; Willmore, W.G. S-glutathionylation reactions in mitochondrial function and disease. Front. Cell Dev. Biol. 2014, 2, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, J.C.; He, W.; Verdin, E. Mitochondrial protein acylation and intermediary metabolism: Regulation by sirtuins and implications for metabolic disease. J. Biol. Chem. 2012, 287, 42436–42443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rardin, M.J.; He, W.; Nishida, Y.; Newman, J.C.; Carrico, C.; Danielson, S.R.; Guo, A.; Gut, P.; Sahu, A.K.; Li, B.; et al. SIRT5 regulates the mitochondrial lysine succinylome and metabolic networks. Cell Metab. 2013, 18, 920–933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.; Wang, F.; Sun, R.; Chen, X.; Zhang, M.; Xu, Q.; Wang, Y.; Wang, S.; Xiong, Y.; Guan, K.L.; et al. SIRT5 promotes IDH2 desuccinylation and G6PD deglutarylation to enhance cellular antioxidant defense. EMBO Rep. 2016, 17, 811–822. [Google Scholar] [CrossRef]
- McDonagh, B.; Pedrajas, J.R.; Padilla, C.A.; Barcena, J.A. Thiol redox sensitivity of two key enzymes of heme biosynthesis and pentose phosphate pathways: Uroporphyrinogen decarboxylase and transketolase. Oxid. Med. Cell Longev. 2013, 2013, 932472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, J.; Wittig, J.G.; Ghamari, A.; Maeda, M.; Dailey, T.A.; Bergonia, H.; Kafina, M.D.; Coughlin, E.E.; Minogue, C.E.; Hebert, A.S.; et al. Erythropoietin signaling regulates heme biosynthesis. eLife 2017, 6, 24767. [Google Scholar] [CrossRef] [Green Version]
- Shah, D.I.; Takahashi-Makise, N.; Cooney, J.D.; Li, L.; Schultz, I.J.; Pierce, E.L.; Narla, A.; Seguin, A.; Hattangadi, S.M.; Medlock, A.E.; et al. Mitochondrial Atpif1 regulates haem synthesis in developing erythroblasts. Nature 2012, 491, 608–612. [Google Scholar] [CrossRef] [Green Version]
- Jordan, P.M.; Warren, M.J. Evidence for a dipyrromethane cofactor at the catalytic site of E. coli porphobilinogen deaminase. FEBS Lett. 1987, 225, 87–92. [Google Scholar] [CrossRef] [Green Version]
- Shoolingin-Jordan, P.M.; Al-Dbass, A.; McNeill, L.A.; Sarwar, M.; Butler, D. Human porphobilinogen deaminase mutations in the investigation of the mechanism of dipyrromethane cofactor assembly and tetrapyrrole formation. Biochem. Soc. Trans. 2003, 31, 731–735. [Google Scholar] [CrossRef] [Green Version]
- Bailey, H.J.; Bezerra, G.A.; Marcero, J.R.; Padhi, S.; Foster, W.R.; Rembeza, E.; Roy, A.; Bishop, D.F.; Desnick, R.J.; Bulusu, G.; et al. Human aminolevulinate synthase structure reveals a eukaryotic-specific autoinhibitory loop regulating substrate binding and product release. Nat. Commun. 2020, 11, 2813. [Google Scholar] [CrossRef]
- Whiting, M.J.; Granick, S. Delta-Aminolevulinic acid synthase from chick embryo liver mitochondria. I. Purification and some properties. J. Biol. Chem. 1976, 251, 1340–1346. [Google Scholar] [CrossRef]
- Liu, G.; Sil, D.; Maio, N.; Tong, W.H.; Bollinger, J.M., Jr.; Krebs, C.; Rouault, T.A. Heme biosynthesis depends on previously unrecognized acquisition of iron-sulfur cofactors in human amino-levulinic acid dehydratase. Nat. Commun. 2020, 11, 6310. [Google Scholar] [CrossRef] [PubMed]
- Dailey, T.A.; Dailey, H.A. Human protoporphyrinogen oxidase: Expression, purification, and characterization of the cloned enzyme. Protein Sci. 1996, 5, 98–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakaino, M.; Ishigaki, M.; Ohgari, Y.; Kitajima, S.; Masaki, R.; Yamamoto, A.; Taketani, S. Dual mitochondrial localization and different roles of the reversible reaction of mammalian ferrochelatase. FEBS J. 2009, 276, 5559–5570. [Google Scholar] [CrossRef]
- Chung, J.; Chen, C.; Paw, B.H. Heme metabolism and erythropoiesis. Curr. Opin. Hematol. 2012, 19, 156–162. [Google Scholar] [CrossRef]
- Nilsson, R.; Schultz, I.J.; Pierce, E.L.; Soltis, K.A.; Naranuntarat, A.; Ward, D.M.; Baughman, J.M.; Paradkar, P.N.; Kingsley, P.D.; Culotta, V.C.; et al. Discovery of genes essential for heme biosynthesis through large-scale gene expression analysis. Cell Metab. 2009, 10, 119–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medlock, A.E.; Dailey, T.A.; Ross, T.A.; Dailey, H.A.; Lanzilotta, W.N. A pi-helix switch selective for porphyrin deprotonation and product release in human ferrochelatase. J. Mol. Biol. 2007, 373, 1006–1016. [Google Scholar] [CrossRef] [Green Version]
- Hoggins, M.; Dailey, H.A.; Hunter, C.N.; Reid, J.D. Direct measurement of metal ion chelation in the active site of human ferrochelatase. Biochemistry 2007, 46, 8121–8127. [Google Scholar] [CrossRef] [Green Version]
- Fra, A.; Yoboue, E.D.; Sitia, R. Cysteines as Redox Molecular Switches and Targets of Disease. Front. Mol. Neurosci. 2017, 10, 167. [Google Scholar] [CrossRef] [Green Version]
- Klomsiri, C.; Karplus, P.A.; Poole, L.B. Cysteine-based redox switches in enzymes. Antioxid Redox Signal. 2011, 14, 1065–1077. [Google Scholar] [CrossRef] [Green Version]
- Gallogly, M.M.; Mieyal, J.J. Mechanisms of reversible protein glutathionylation in redox signaling and oxidative stress. Curr. Opin. Pharmacol. 2007, 7, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Brennan, J.P.; Miller, J.I.; Fuller, W.; Wait, R.; Begum, S.; Dunn, M.J.; Eaton, P. The utility of N,N-biotinyl glutathione disulfide in the study of protein S-glutathiolation. Mol. Cell Proteomics 2006, 5, 215–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meissner, F.; Molawi, K.; Zychlinsky, A. Superoxide dismutase 1 regulates caspase-1 and endotoxic shock. Nat. Immunol. 2008, 9, 866–872. [Google Scholar] [CrossRef]
- Wu, C.K.; Dailey, H.A.; Rose, J.P.; Burden, A.; Sellers, V.M.; Wang, B.C. The 2.0 A structure of human ferrochelatase, the terminal enzyme of heme biosynthesis. Nat. Struct. Biol. 2001, 8, 156–160. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.; Liu, S.; Ando, H.; Ishii, R.; Tateno, S.; Kaneko, Y.; Yugami, M.; Sakamoto, S.; Yamaguchi, Y.; Nureki, O.; et al. Salicylic acid induces mitochondrial injury by inhibiting ferrochelatase heme biosynthesis activity. Mol. Pharmacol. 2013, 84, 824–833. [Google Scholar] [CrossRef] [Green Version]
- Medlock, A.E.; Najahi-Missaoui, W.; Ross, T.A.; Dailey, T.A.; Burch, J.; O’Brien, J.R.; Lanzilotta, W.N.; Dailey, H.A. Identification and characterization of solvent-filled channels in human ferrochelatase. Biochemistry 2012, 51, 5422–5433. [Google Scholar] [CrossRef] [Green Version]
- Burden, A.E.; Wu, C.; Dailey, T.A.; Busch, J.L.; Dhawan, I.K.; Rose, J.P.; Wang, B.; Dailey, H.A. Human ferrochelatase: Crystallization, characterization of the [2Fe-2S] cluster and determination that the enzyme is a homodimer. Biochim. Biophys. Acta 1999, 1435, 191–197. [Google Scholar] [CrossRef]
- Medlock, A.E.; Dailey, H.A. Human coproporphyrinogen oxidase is not a metalloprotein. J. Biol. Chem. 1996, 271, 32507–32510. [Google Scholar] [CrossRef] [Green Version]
- Najahi-Missaoui, W.; Dailey, H.A. Production and characterization of erythropoietic protoporphyric heterodimeric ferrochelatases. Blood 2005, 106, 1098–1104. [Google Scholar] [CrossRef]
- Park, J.; Chen, Y.; Tishkoff, D.X.; Peng, C.; Tan, M.; Dai, L.; Xie, Z.; Zhang, Y.; Zwaans, B.M.; Skinner, M.E.; et al. SIRT5-mediated lysine desuccinylation impacts diverse metabolic pathways. Mol. Cell 2013, 50, 919–930. [Google Scholar] [CrossRef] [Green Version]
- Weinert, B.T.; Scholz, C.; Wagner, S.A.; Iesmantavicius, V.; Su, D.; Daniel, J.A.; Choudhary, C. Lysine succinylation is a frequently occurring modification in prokaryotes and eukaryotes and extensively overlaps with acetylation. Cell Rep. 2013, 4, 842–851. [Google Scholar] [CrossRef] [Green Version]
- Du, J.; Zhou, Y.; Su, X.; Yu, J.J.; Khan, S.; Jiang, H.; Kim, J.; Woo, J.; Kim, J.H.; Choi, B.H.; et al. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science 2011, 334, 806–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, G.R.; Payne, R.M. Widespread and enzyme-independent Nepsilon-acetylation and Nepsilon-succinylation of proteins in the chemical conditions of the mitochondrial matrix. J. Biol. Chem. 2013, 288, 29036–29045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medlock, A.; Swartz, L.; Dailey, T.A.; Dailey, H.A.; Lanzilotta, W.N. Substrate interactions with human ferrochelatase. Proc. Natl. Acad. Sci. USA 2007, 104, 1789–1793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karim, Z.; Lyoumi, S.; Nicolas, G.; Deybach, J.C.; Gouya, L.; Puy, H. Porphyrias: A 2015 update. Clin. Res. Hepatol. Gastroenterol. 2015, 39, 412–425. [Google Scholar] [CrossRef] [PubMed]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Medlock, A.E.; Dailey, H.A. New Avenues of Heme Synthesis Regulation. Int. J. Mol. Sci. 2022, 23, 7467. https://doi.org/10.3390/ijms23137467
Medlock AE, Dailey HA. New Avenues of Heme Synthesis Regulation. International Journal of Molecular Sciences. 2022; 23(13):7467. https://doi.org/10.3390/ijms23137467
Chicago/Turabian StyleMedlock, Amy E., and Harry A. Dailey. 2022. "New Avenues of Heme Synthesis Regulation" International Journal of Molecular Sciences 23, no. 13: 7467. https://doi.org/10.3390/ijms23137467
APA StyleMedlock, A. E., & Dailey, H. A. (2022). New Avenues of Heme Synthesis Regulation. International Journal of Molecular Sciences, 23(13), 7467. https://doi.org/10.3390/ijms23137467