
Mechanistic Interrogation of Cell Transformation In Vitro: The Transformics Assay as an Exemplar of Oncotransformation

 , and
, and
Abstract
:1. Introduction
2. Results
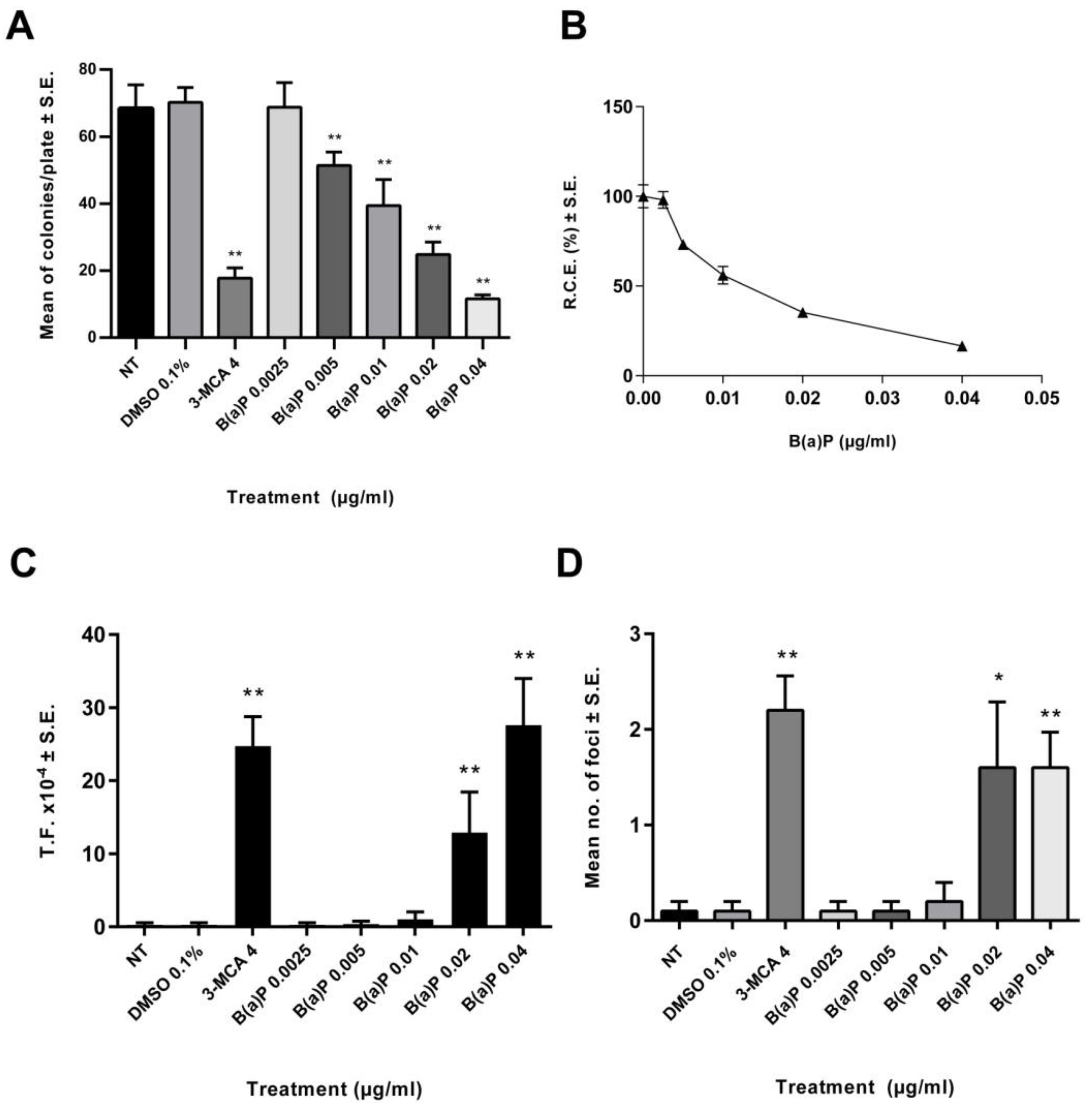
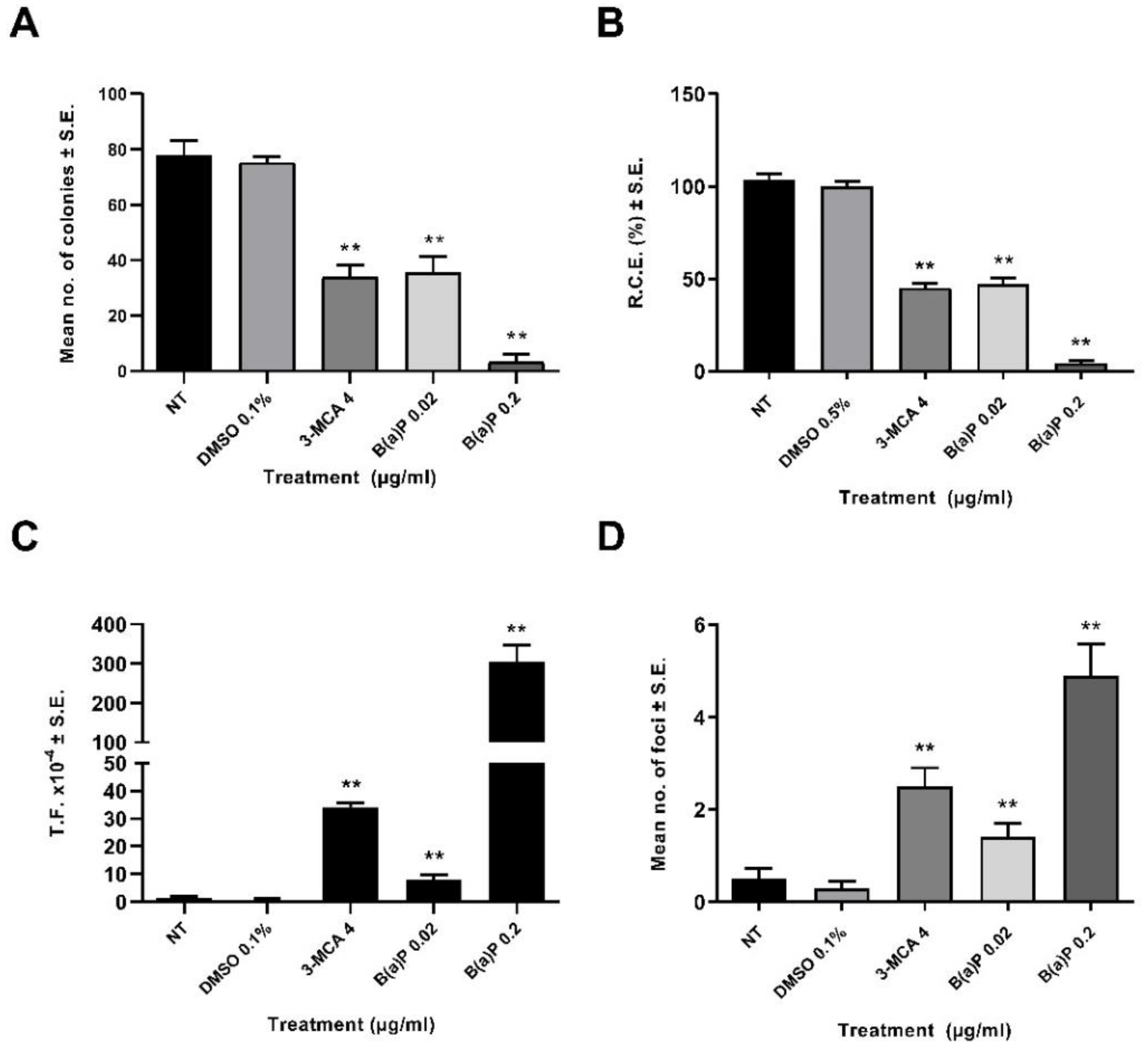
2.1. CTA
2.2. Statistical Analysis of Microarray Experiment
2.3. Biological Interpretation of Microarray Experiments
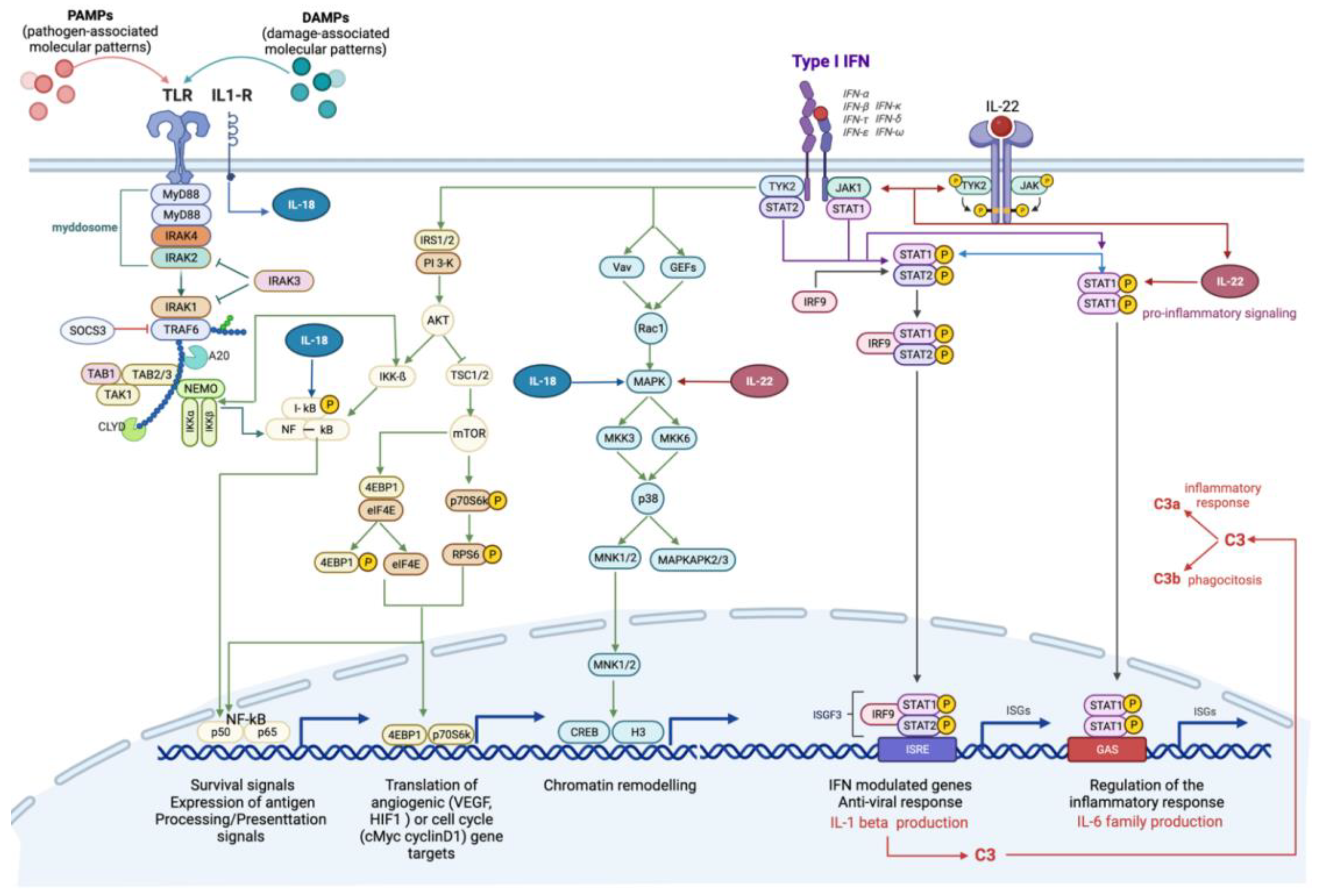
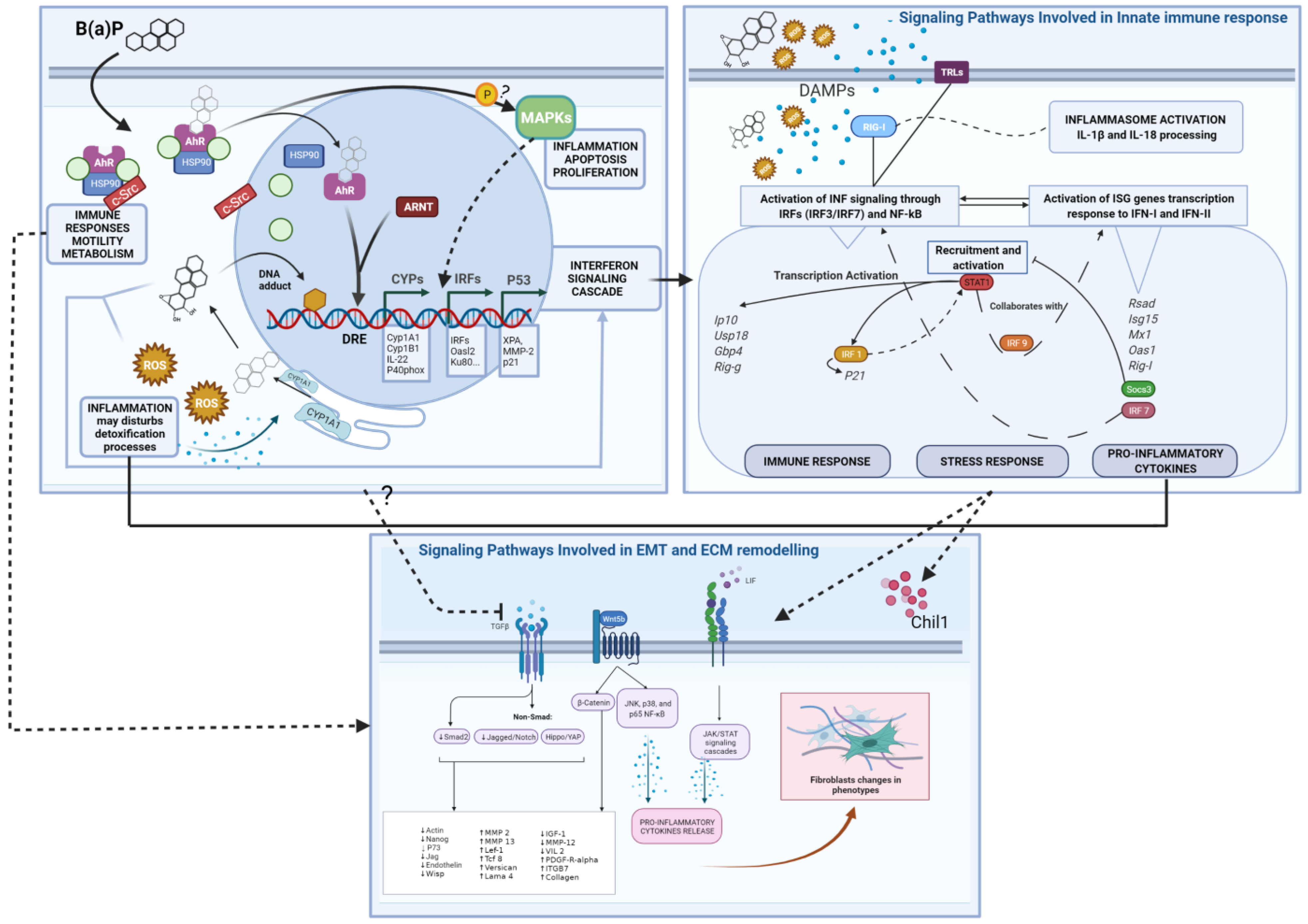
2.3.1. Pathway and Processes Related to Immune Response
2.3.2. Pathways and Processes Related to EMC Modification and EMT/MET
2.3.3. Genes Involved in the Metabolism of Benzo (a) Pyrene
3. Discussion
4. Materials and Methods
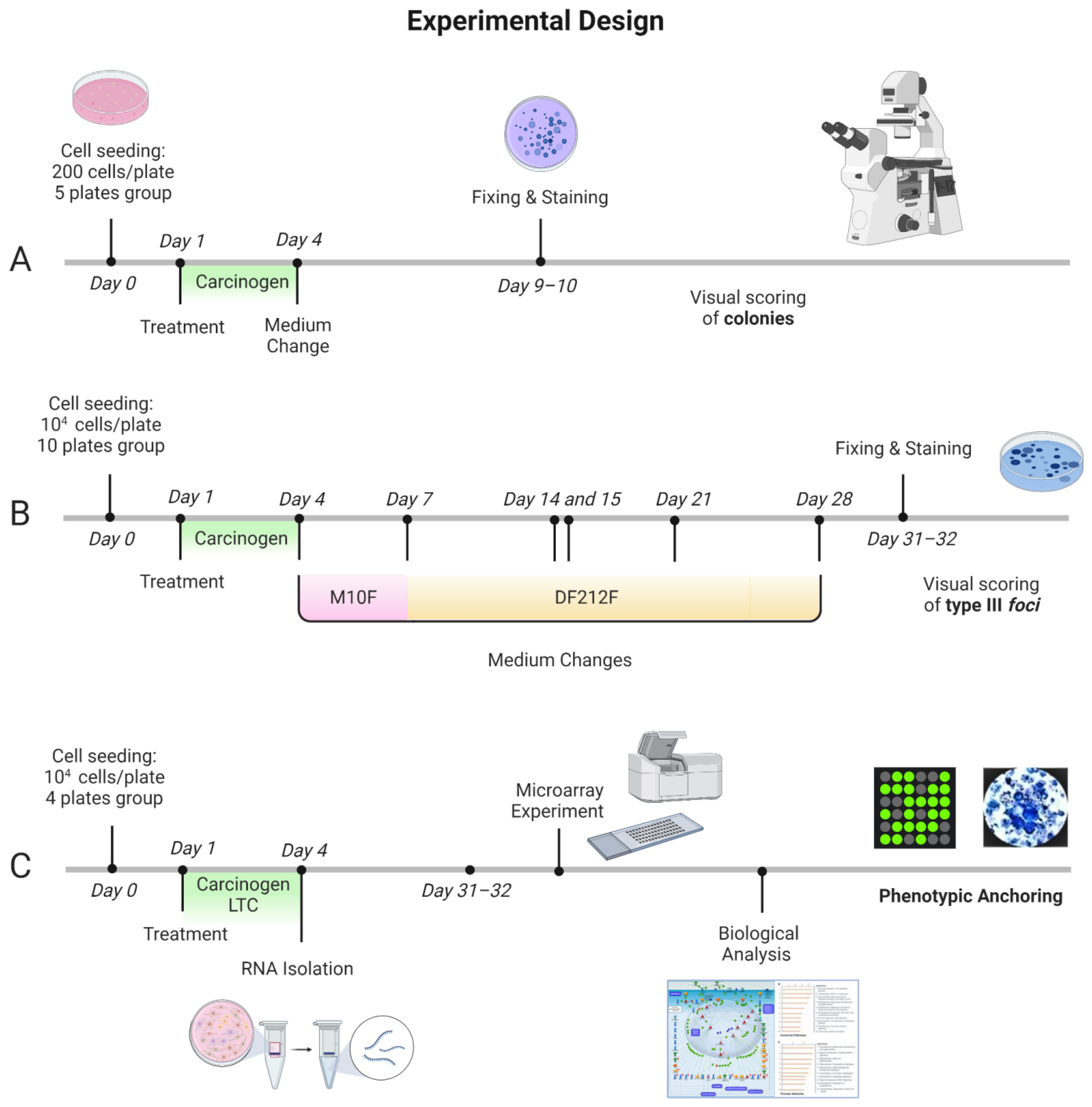
4.1. Experimental Design
4.2. Cells
4.3. Chemicals and Solutions
4.4. CTA and Cytotoxicity Assessment
4.5. Total RNA Extraction
4.6. Total RNA Labeling and Hybridization
4.7. Statistical Analysis of Microarray Data
4.8. Tools of Biological Interpretation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Disclaimer
References
- Cohen, S.M.; Arnold, L.L. Chemical carcinogenesis. Toxicol. Sci. Off. J. Soc. Toxicol. 2011, 120 (Suppl. S1), S76–S92. [Google Scholar] [CrossRef] [PubMed]
- Colacci, A.; Vaccari, M. Children’s and Adult Involuntary and Occupational Exposures and Cancer. In Translational Toxicology and Therapeutics: Windows of Developmental Susceptibility in Reproduction and Cancer; Waters, M.D., Hughes, C.L., Eds.; Wiley: Hoboken, NJ, USA, 2017. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, M.T.; Guyton, K.Z.; Gibbons, C.F.; Fritz, J.M.; Portier, C.J.; Rusyn, I.; DeMarini, D.M.; Caldwell, J.C.; Kavlock, R.J.; Lambert, P.F.; et al. Key Characteristics of Carcinogens as a Basis for Organizing Data on Mechanisms of Carcinogenesis. Environ. Health Perspect. 2016, 124, 713–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guyton, K.Z.; Rieswijk, L.; Wang, A.; Chiu, W.A.; Smith, M.T. Key Characteristics Approach to Carcinogenic Hazard Identification. Chem. Res. Toxicol. 2018, 31, 1290–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helm, J.S.; Rudel, R.A. Adverse outcome pathways for ionizing radiation and breast cancer involve direct and indirect DNA damage, oxidative stress, inflammation, genomic instability, and interaction with hormonal regulation of the breast. Arch. Toxicol. 2020, 94, 1511–1549. [Google Scholar] [CrossRef]
- Felter, S.P.; Bhat, V.S.; Botham, P.A.; Bussard, D.A.; Casey, W.; Hayes, A.W.; Hilton, G.M.; Magurany, K.A.; Sauer, U.G.; Ohanian, E.V. Assessing chemical carcinogenicity: Hazard identification, classification, and risk assessment. Insight from a Toxicology Forum state-of-the-science workshop. Crit. Rev. Toxicol. 2021, 51, 653–694. [Google Scholar] [CrossRef]
- Smith, C.J.; Perfetti, T.A.; Berry, S.C.; Brash, D.E.; Bus, J.; Calabrese, E.; Clemens, R.A.; Fowle, J.R.J., 3rd; Greim, H.; MacGregor, J.T.; et al. Bruce Nathan Ames-Paradigm shifts inside the cancer research revolution. Mutat. Res. Rev. Mutat. Res. 2021, 787, 108363. [Google Scholar] [CrossRef]
- Corvi, R.; Madia, F.; Guyton, K.Z.; Kasper, P.; Rudel, R.; Colacci, A.; Kleinjans, J.; Jennings, P. Moving forward in carcinogenicity assessment: Report of an EURL ECVAM/ESTIV workshop. Toxicol. Vitr. Int. J. Publ. Assoc. BIBRA 2017, 45 Pt 3, 278–286. [Google Scholar] [CrossRef]
- Jacobs, M.N.; Colacci, A.; Corvi, R.; Vaccari, M.; Aguila, M.C.; Corvaro, M.; Delrue, N.; Desaulniers, D.; Ertych, N.; Jacobs, A.; et al. Chemical carcinogen safety testing: OECD expert group international consensus on the development of an integrated approach for the testing and assessment of chemical non-genotoxic carcinogens. Arch. Toxicol. 2020, 94, 2899–2923. [Google Scholar] [CrossRef]
- Hwang, S.H.; Yeom, H.; Han, B.I.; Ham, B.J.; Lee, Y.M.; Han, M.R.; Lee, M. Predicting Carcinogenic Mechanisms of Non-Genotoxic Carcinogens via Combined Analysis of Global DNA Methylation and In Vitro Cell Transformation. Int. J. Mol. Sci. 2020, 21, 5387. [Google Scholar] [CrossRef]
- Berenjeno, I.M.; Núñez, F.; Bustelo, X.R. Transcriptomal profiling of the cellular transformation induced by Rho subfamily GTPases. Oncogene 2007, 26, 4295–4305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohrbeck, A.; Salinas, G.; Maaser, K.; Linge, J.; Salovaara, S.; Corvi, R.; Borlak, J. Toxicogenomics applied to in vitro carcinogenicity testing with Balb/c 3T3 cells revealed a gene signature predictive of chemical carcinogens. Toxicol. Sci. Off. J. Soc. Toxicol. 2010, 118, 31–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahu, B.; Pihlajamaa, P.; Zhang, K.; Palin, K.; Ahonen, S.; Cervera, A.; Ristimäki, A.; Aaltonen, L.A.; Hautaniemi, S.; Taipale, J. Human cell transformation by combined lineage conversion and oncogene expression. Oncogene 2021, 40, 5533–5547. [Google Scholar] [CrossRef]
- Landkocz, Y.; Poupin, P.; Atienzar, F.; Vasseur, P. Transcriptomic effects of di-(2-ethylhexyl)-phthalate in Syrian hamster embryo cells: An important role of early cytoskeleton disturbances in carcinogenesis? BMC Genom. 2011, 12, 524. [Google Scholar] [CrossRef] [Green Version]
- Perdichizzi, S.; Mascolo, M.G.; Rotondo, F.; Vaccari, M.; Grilli, S.; Colacci, A. Transcriptomics and Cell Transformation Assay: An Integrated Approach to Evaluate the Effects of Low Dose Ionizing Radiation. Int. J. Oncol. Radiother. 2021, 2, 12–19. [Google Scholar] [CrossRef]
- Ohmori, K.; Kamei, A.; Watanabe, Y.; Abe, K. Gene Expression over Time during Cell Transformation Due to Non-Genotoxic Carcinogen Treatment of Bhas 42 Cells. Int. J. Mol. Sci. 2022, 23, 3216. [Google Scholar] [CrossRef]
- Ding, Z.; Xiang, X.; Li, J.; Wu, S. Molecular Mechanism of Malignant Transformation of Balb/c-3T3 Cells Induced by Long-Term Exposure to 1800 MHz Radiofrequency Electromagnetic Radiation (RF-EMR). Bioengineering 2022, 9, 43. [Google Scholar] [CrossRef]
- Jacobs, M.N.; Colacci, A.; Louekari, K.; Luijten, M.; Hakkert, B.C.; Paparella, M.; Vasseur, P. International regulatory needs for development of an IATA for non-genotoxic carcinogenic chemical substances. ALTEX 2016, 33, 359–392. [Google Scholar] [CrossRef] [Green Version]
- Adler, S.; Basketter, D.; Creton, S.; Pelkonen, O.; van Benthem, J.; Zuang, V.; Andersen, K.E.; Angers-Loustau, A.; Aptula, A.; Bal-Price, A.; et al. Alternative (non-animal) methods for cosmetics testing: Current status and future prospects-2010. Arch. Toxicol. 2011, 85, 367–485. [Google Scholar] [CrossRef]
- Goodson, W.H.; Lowe, L.; Carpenter, D.O.; Gilbertson, M.; Manaf Ali, A.; Lopez de Cerain Salsamendi, A.; Lasfar, A.; Carnero, A.; Azqueta, A.; Amedei, A.; et al. Assessing the carcinogenic potential of low-dose exposures to chemical mixtures in the environment: The challenge ahead. Carcinogenesis 2015, 36 (Suppl. S1), S254–S296. [Google Scholar] [CrossRef] [Green Version]
- Madia, F.; Pillo, G.; Worth, A.; Corvi, R.; Prieto, P. Integration of data across toxicity endpoints for improved safety assessment of chemicals: The example of carcinogenicity assessment. Arch. Toxicol. 2021, 95, 1971–1993. [Google Scholar] [CrossRef] [PubMed]
- Mascolo, M.G.; Perdichizzi, S.; Vaccari, M.; Rotondo, F.; Zanzi, C.; Grilli, S.; Paparella, M.; Jacobs, M.N.; Colacci, A. The Transformics Assay: First steps for the development of an integrated approach to investigate the malignant cell transformation in vitro. Carcinogenesis 2018, 39, 955–967. [Google Scholar] [CrossRef] [PubMed]
- Malik, D.-e.-s.; David, R.M.; Gooderham, N.J. Mechanistic evidence that benzo[a]pyrene promotes an inflammatory microenvironment that drives the metastatic potential of human mammary cells. Arch. Toxicol. 2018, 92, 3223–3239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.; Zhang, S.; An, X.; Tan, W.; Pang, D.; Ouyang, H. Toxicological effects of benzo[a]pyrene on DNA methylation of whole genome in ICR mice. Cell. Mol. Biol. 2015, 61, 115–119. [Google Scholar]
- Murray, I.A.; Patterson, A.D.; Perdew, G.H. Aryl hydrocarbon receptor ligands in cancer: Friend and foe. Nat. Rev. Cancer 2014, 14, 801–814. [Google Scholar] [CrossRef]
- Corvi, R.; Aardema, M.J.; Gribaldo, L.; Hayashi, M.; Hoffmann, S.; Schechtman, L.; Vanparys, P. ECVAM prevalidation study on in vitro cell transformation assays: General outline and conclusions of the study. Mutat. Res. 2012, 744, 12–19. [Google Scholar] [CrossRef]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef] [Green Version]
- Czerkies, M.; Korwek, Z.; Prus, W.; Kochańczyk, M.; Jaruszewicz-Błońska, J.; Tudelska, K.; Błoński, S.; Kimmel, M.; Brasier, A.R.; Lipniacki, T. Cell fate in antiviral response arises in the crosstalk of IRF, NF-κB and JAK/STAT pathways. Nat. Commun. 2018, 9, 493. [Google Scholar] [CrossRef]
- Cohen, N.; Shani, O.; Raz, Y.; Sharon, Y.; Hoffman, D.; Abramovitz, L.; Erez, N. Fibroblasts drive an immunosuppressive and growth-promoting microenvironment in breast cancer via secretion of Chitinase 3-like 1. Oncogene 2017, 36, 4457–4468. [Google Scholar] [CrossRef] [Green Version]
- Salomon, J.; Piotrowska, A.; Matusiak, Ł.; Dzięgiel, P.; Szepietowski, J.C. Chitinase-3-like Protein 1 (YKL-40) Expression in Squamous Cell Skin Cancer. Anticancer Res. 2018, 38, 4753–4758. [Google Scholar] [CrossRef] [Green Version]
- Bissey, P.A.; Law, J.H.; Bruce, J.P.; Shi, W.; Renoult, A.; Chua, M.L.K.; Yip, K.W.; Liu, F.-F. Dysregulation of the MiR-449b target TGFBI alters the TGFβ pathway to induce cisplatin resistance in nasopharyngeal carcinoma. Oncogenesis 2018, 7, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.-J.; Li, Y.; Tan, Y.; Xiao, M.; Zhang, J.; Radtke, F.; Velazquez, O.C. Inhibition of fibroblast growth by Notch1 signaling is mediated by induction of Wnt11-dependent WISP-1. PLoS ONE 2012, 7, e38811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, H.; Cai, L.; Moller, M.; Issac, B.; Zhang, L.; Owyong, M.; Moscowitz, A.E.; Vazquez-Padron, R.; Radtke, F.; Liu, Z.-J. Notch1-WISP-1 axis determines the regulatory role of mesenchymal stem cell-derived stromal fibroblasts in melanoma metastasis. Oncotarget 2016, 7, 79262–79273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albrengues, J.; Bertero, T.; Grasset, E.; Bonan, S.; Maiel, M.; Bourget, I.; Philippe, C.; Herraiz Serrano, C.; Benamar, S.; Croce, O.; et al. Epigenetic switch drives the conversion of fibroblasts into proinvasive cancer-associated fibroblasts. Nat. Commun. 2015, 6, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biffi, G.; Oni, T.E.; Spielman, B.; Hao, Y.; Elyada, E.; Park, Y.; Preall, J.; Tuveson, D.A. IL1-Induced JAK/STAT signaling is antagonized by TGFβ to shape CAF heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discov. 2019, 9, 282–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, G.J.; Azuma, A.; Miura, Y.; Orimo, A. Activated fibroblast program orchestrates tumor initiation and progression; molecular mechanisms and the associated therapeutic strategies. Int. J. Mol. Sci. 2019, 20, 2256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prochaska, H.J.; De Long, M.J.; Talalay, P. On the mechanisms of induction of cancer-protective enzymes: A unifying proposal. Proc. Natl. Acad. Sci. USA 1985, 82, 8232–8236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moffat, I.; Chepelev, N.L.; Labib, S.; Bourdon-Lacombe, J.; Kuo, B.; Buick, J.K.; Lemieux, F.; Williams, A.; Halappanavar, S.; Malik, A.I.; et al. Comparison of toxicogenomics and traditional approaches to inform mode of action and points of departure in human health risk assessment of benzo[a]pyrene in drinking water. Crit. Rev. Toxicol. 2015, 45, 1–43. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, M.N. Nuclear receptors and dietary ligands. In Nutrients and Cell Signaling, 1st ed.; Zemplini, J., Dakshinamurt, K., Eds.; CRC Taylor & Francis: New York, NY, USA, 2005; pp. 35–90. [Google Scholar]
- Pohjanvirta, R. The AH Receptor in Biology and Toxicology; John Wiley & Sons: Hoboken, NJ, USA, 2011; pp. 469–500. [Google Scholar]
- Wada, T.; Sunaga, H.; Ohkawara, R.; Shimba, S. Aryl hydrocarbon receptor modulates NADPH oxidase activity via direct transcriptional regulation of p40phox expressions. Mol. Pharm. 2013, 83, 1133–1140. [Google Scholar] [CrossRef]
- Ecvam. Balb/c 3T3 Cell Transformation Assay Prevalidation Study Report; Ecvam: Ispra, Italy, 2010. [Google Scholar]
- Villeneuve, D.L.; Landesmann, B.; Allavena, P.; Ashley, N.; Bal-Price, A.; Corsini, E.; Halappanavar, S.; Hussell, T.; Laskin, D.; Lawrence, T.; et al. Representing the process of inflammation as Key Events in Adverse Outcome Pathways. Toxicol. Sci. 2018, 163, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mescoli, A.; Maffei, G.; Pillo, G.; Bortone, G.; Marchesi, S.; Morandi, E.; Ranzi, A.; Rotondo, F.; Serra, S.; Vaccari, M.; et al. The Secretive Liaison of Particulate Matter and SARS-CoV-2. A Hypothesis and Theory Investigation. Front. Genet. 2020, 11, 579964. [Google Scholar] [CrossRef]
- Anders, H.-J.; Schaefer, L. Beyond tissue injury-damage-associated molecular patterns, toll-like receptors, and inflammasomes also drive regeneration and fibrosis. J. Am. Soc. Nephrol. 2014, 25, 1387–1400. [Google Scholar] [CrossRef] [PubMed]
- Legler, J.; Zalko, D.; Jourdan, F.; Jacobs, M.; Fromenty, B.; Balaguer, P.; Bourguet, W.; Munic Kos, V.; Nadal, A.; Beausoleil, C.; et al. The GOLIATH Project: Towards an Internationally Harmonised Approach for Testing Metabolism Disrupting Compounds. Int. J. Mol. Sci. 2020, 21, 3480. [Google Scholar] [CrossRef] [PubMed]
- Puga, A.; Ma, C.; Marlowe, J.L. The aryl hydrocarbon receptor cross-talks with multiple signal transduction pathways. Biochem. Pharm. 2009, 77, 713–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, B.; Yang, J.; Zhao, Y.; Ivanciuc, T.; Sun, H.; Garofalo, R.P.; Brasier, A.R. BRD4 couples NF-κB/RelA with airway inflammation and the IRF-RIG-I amplification loop in respiratory syncytial virus infection. J. Virol. 2017, 91, e00007-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vázquez-Gómez, G.; Rocha-Zavaleta, L.; Rodríguez-Sosa, M.; Petrosyan, P.; Rubio-Lightbourn, J. Benzo(a)pyrene activates an AhR/Src/ERK axis that contributes to CYP1A1 induction and stable DNA adducts formation in lung cells. Toxicol. Lett. 2018, 289, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Nastasi, C.; Mannarino, L.; D’Incalci, M. DNA Damage Response and Immune Defense. Int. J. Mol. Sci. 2020, 21, 7504. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Hao, G. The role of angiotensin-converting enzyme 2 in coronaviruses/influenza viruses and cardiovascular disease. Cardiovasc. Res. 2020, 116, 1932–1936. [Google Scholar] [CrossRef] [PubMed]
- Weber-Gerlach, M.; Weber, F. Standing on three legs: Antiviral activities of RIG-I against influenza viruses. Curr. Opin. Immunol. 2016, 42, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Huang, G.; Haizhu, S.; Chen, Y.; Chen, L. Cancer Associated Fibroblasts: An essential role in the tumor microenvironment. Oncol. Lett. 2017, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reya, T.; Clevers, H. Wnt signalling in stem cells and cancer. Nature 2005, 434, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Neuzillet, C.; Tijeras-Raballand, A.; Cohen, R.; Cros, J.; Faivre, S.; Raymond, E.; de Gramont, A. Targeting the TGFβ pathway for cancer therapy. Pharmacol. Ther. 2015, 147, 22–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syed, V. TGF-β Signaling in Cancer. J. Cell. Biochem. 2016, 117, 1279–1287. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Cao, Y.; Luo, Y.; Hu, H.; Ling, H. Signalling mechanism(s) of epithelial–mesenchymal transition and cancer stem cells in tumour therapeutic resistance. Clin. Chim. Acta J. 2018, 483, 156–163. [Google Scholar] [CrossRef] [PubMed]
- van Dijk, E.M.; Menzen, M.H.; Spanjer, A.I.R.; Middag, L.D.C.; Brandsma, C.-A.A.; Gosens, R. Noncanonical WNT-5B signaling induces inflammatory responses in human lung fibroblasts. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2016, 310, L1166–L1176. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Duran, A.; Carvajal-Gonzalez, J.M.; Mulero-Navarro, S.; Santiago-Josefat, B.; Puga, A.; Fernandez-Salguero, P.M. Fitting a xenobiotic receptor into cell homeostasis: How the dioxin receptor interacts with TGFβ signaling. Biochem. Pharm. 2009, 77, 700–712. [Google Scholar] [CrossRef]
- Haarmann-Stemmann, T.; Bothe, H.; Abel, J. Growth factors, cytokines and their receptors as downstream targets of arylhydrocarbon receptor (AhR) signaling pathways. Biochem. Pharm. 2009, 77, 508–520. [Google Scholar] [CrossRef] [PubMed]
- Nakano, N.; Sakata, N.; Katsu, Y.; Nochise, D.; Sato, E.; Takahashi, Y.; Yamaguchi, S.; Haga, Y.; Ikeno, S.; Motizuki, M.; et al. Dissociation of the AhR/ARNT complex by TGF-β/Smad signaling represses CYP1A1 gene expression and inhibits benze[a]pyrene-mediated cytotoxicity. J. Biol. Chem. 2020, 295, 9033–9051. [Google Scholar] [CrossRef] [PubMed]
- Giannone, J.V.; Li, W.; Probst, M.; Okey, A.B. Prolonged depletion of AH receptor without alteration of receptor mRNA levels after treatment of cells in culture with 2,3,7,8-tetrachlorodibenzo-p-dioxin. Biochem. Pharm. 1998, 55, 489–497. [Google Scholar] [CrossRef]
- Tsai, C.H.; Li, C.H.; Cheng, Y.W.; Lee, C.C.; Liao, P.L.; Lin, C.H.; Huang, S.H.; Kang, J.J. The inhibition of lung cancer cell migration by AhR-regulated autophagy. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Šmerdová, L.; Neča, J.; Svobodová, J.; Topinka, J.; Schmuczerová, J.; Kozubík, A.; Machala, M.; Vondráček, J. Inflammatory mediators accelerate metabolism of benzo(a)pyrene in rat alveolar type II cells: The role of enhanced cytochrome P450 1B1 expression. Toxicology 2013, 314, 30–38. [Google Scholar] [CrossRef]
- Shi, Q.; Godschalk, R.W.L.; Van Schooten, F.J. Inflammation and the chemical carcinogen benzo(a)pyrene: Partners in crime. Mutat. Res. 2017, 774, 12–24. [Google Scholar] [CrossRef]
- Shi, Q.; Fijten, R.R.; Spina, D.; Riffo Vasquez, Y.; Arlt, V.M.; Godschalk, R.W.; Van Schooten, F.J. Altered gene expression profiles in the lungs of benzo(a)pyrene-exposed mice in the presence of lipopolysaccharide-induced pulmonary inflammation. Toxicol. Appl. Pharmacol. 2017, 336, 8–19. [Google Scholar] [CrossRef] [Green Version]
- Fang, J.-L.; Beland, F.A.; Doerge, D.R.; Wiener, D.; Guillemette, C.; Marques, M.M.; Lazarus, P. Characterization of benzo(a)pyrene-trans-7,8-dihydrodiol glucuronidation by human tissue microsomes and overexpressed UDP-glucuronosyltransferase enzymes. Cancer Res. 2002, 53, 1529–1533. [Google Scholar]
- Bock, K.W.; Köhle, C. UDP-glucuronosyltransferase 1A6: Structural, functional, and regulatory aspects. Methods Enzymol. 2005, 400, 57–75. [Google Scholar]
- Nguyen, N.T.; Hanieh, H.; Nakahama, T.; Kishimoto, T. The roles of aryl hydrocarbon receptor in immune responses. Int. Immunol. 2013, 25, 335–343. [Google Scholar] [CrossRef] [Green Version]
- Chepelev, N.L.; Moffat, I.D.; Bowers, W.J.; Yauk, C.L. Neurotoxicity may be an overlooked consequence of benzo[a]pyrene exposure that is relevant to human health risk assessment. Mutat. Res. Rev. Mutat. Res. 2015, 764, 64–89. [Google Scholar] [CrossRef]
- Sasaki, K.; Bohnenberger, S.; Hayashi, K.; Kunkelmann, T.; Muramatsu, D.; Phrakonkham, P.; Poth, A.; Sakai, A.; Salovaara, S.; Tanaka, N.; et al. Recommended protocol for the BALB/c 3T3 Cell Transformation Assay. Mutat. Res.-Genet. Toxicol. Environ. Mutagenesis 2012, 744, 30–35. [Google Scholar] [CrossRef]
- Tanaka, N.; Bohnenberger, S.; Kunkelmann, T.; Munaro, B.; Ponti, J.; Poth, A.; Sabbioni, E.; Sakai, A.; Salovaara, S.; Sasaki, K.; et al. Prevalidation study of the BALB/c 3T3 cell transformation assay for assessment of carcinogenic potential of chemicals. Mutat. Res. 2012, 744, 20–29. [Google Scholar] [CrossRef]
- OECD. Detailed Review Paper on Cell Transformation Assay for Detection of Chemicals Carcinogens; OECD: Paris, France, 2007.
- Harrill, J.A.; Viant, M.R.; Yauk, C.L.; Sachana, M.; Gant, T.W.; Auerbach, S.S.; Beger, R.D.; Bouhifd, M.; O’Brien, J.; Burgoon, L.; et al. Progress towards an OECD reporting framework for transcriptomics and metabolomics in regulatory toxicology. Regul. Toxicol. Pharmacol. RTP 2021, 125, 105020. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enrichment for Pathway Maps | ||||||
|---|---|---|---|---|---|---|
| Pathway Maps | p Value | FDR | Gene Ratio | Up-Regulated Network Objects from Active Data | Down-Regulated Network Objects | |
| 1 | Immune response_IFN-alpha/beta signaling via JAK/STAT | 1.419 × 10−11 | 9.964 × 10−9 | 15/64 | IRF1, Mx1, SOCS3, USP18, IRF7, p21, IRF9, IP10, GBP1, OAS1, ISG15, GBP4, RIG-G, RSAD2 | HIF1A |
| 2 | Immune response_IFN-alpha/beta signaling via MAPKs | 2.414 × 10−8 | 8.472 × 10−6 | 13/77 | Irgm2, IRF7, Oasl2, p21, IRF9, IP10, Oas1g, ISG15, PL scramblase 1, Ku80, RIG-G, RSAD2 | JNK (MAPK8-10) |
| 3 | Transcription_HIF-1 targets | 1.353 × 10−5 | 3.019 × 10−3 | 11/95 | WT1, Angiopoietin 2, MMP-2, p21 | HIF1A, Endothelin-1, PGK1, NIP3, GLUT1, Nucleophosmin, NANOG |
| 4 | Development_Regulation of epithelial-to-mesenchymal transition (EMT) | 1.720 × 10−5 | 3.019 × 10−3 | 9/64 | Lef-1, PDGF-R-alpha, TCF8, MMP-2, WNT | Jagged1, Endothelin-1, Frizzled, SMAD2 |
| 5 | Cell adhesion_ECM remodeling | 3.966 × 10−5 | 5.305 × 10−3 | 8/55 | Versican, MMP-2, LAMA4, MMP-13 | VIL2 (ezrin), Actin cytoskeletal, MMP-12, IGF-1 |
| 6 | Development_YAP/TAZ-mediated co-regulation of transcription | 4.534 × 10−5 | 5.305 × 10−3 | 8/56 | Lef-1, Bcl-XL, ID3, | HIF1A, Endothelin-1, NANOG, p73, SMAD2 |
| 7 | Glomerular injury in Lupus Nephritis | 5.815 × 10−5 | 5.831 × 10−3 | 10/92 | IRF1, Decorin, VCAM1, CX3CL1, RIG-I, IFI56, C3a, IP10 | HIF1A, JNK (MAPK8-10) |
| 8 | Immune response_IFN-alpha/beta signaling via PI3K and NF-kB pathways | 6.997 × 10−5 | 6.140 × 10−3 | 10/94 | IFIT1, Mx1, IRF7, p21, GBP1, ISG15, I-kB, RSAD2 | PI3K cat class IA, IRS-1 |
| 9 | Development_TGF-beta-dependent induction of EMT via SMADs | 1.474 × 10−4 | 1.150 × 10−2 | 6/35 | Lef-1, TCF8, MMP-2 | Jagged1, Endothelin-1, SMAD2 |
| 10 | Cell adhesion_Alpha-4 integrins in cell migration and adhesion | 1.734 × 10−4 | 1.187 × 10−2 | 6/36 | VCAM1, ITGB7 | ERM proteins, F-Actin cytoskeleton, Actin cytoskeletal, PI3K cat class IA |
| 11 | Signal transduction_Additional pathways of NF-kB activation (in the cytoplasm) | 2.006 × 10−4 | 1.187 × 10−2 | 7/52 | PKC-zeta, I-kB, NFKBIA, Adenylate cyclase, TPL2 (MAP3K8), alpha chain CSNK2A1) | PI3K cat class IA, Casein kinase II |
| 12 | Development_Growth factors in regulation of oligodendrocyte precursor cell survival | 2.030 × 10−4 | 1.187 × 10−2 | 6/37 | PDGF-R-alpha, Bcl-XL, NFKBIA | PI3K cat class IA, IRS-1, IGF-1 |
| 13 | Immune response_Classical complement pathway | 2.266 × 10−4 | 1.223 × 10−2 | 7/53 | C3c, C3a, C3b, C3dg, C3, iC3b, C1s | |
| 14 | Apoptosis and survival_APRIL and BAFF signaling | 2.739 × 10−4 | 1.373 × 10−2 | 6/39 | NF-AT2 (NFATC1), Bcl-XL, I-kB, RelB (NF-kB subunit) | JNK (MAPK8-10), TACI (TNFRSF13B) |
| 15 | Development_The role of GDNF ligand family/RET receptor in cell survival, growth and proliferation | 3.062 × 10−4 | 1.386 × 10−2 | 9/92 | GFRalpha2, Bcl-XL, NFKBIA, IL-22 | F-Actin cytoskeleton, PI3K cat class IA, JNK2 (MAPK9), HIF1A, IRS-1 |
| 16 | Development_Role of Activin A in cell differentiation and proliferation | 3.159 × 10−4 | 1.386 × 10−2 | 6/40 | Adenylate cyclase, p21, MAD, LHX3 | NANOG, SMAD2 |
| 17 | Immune response_TNF-R2 signaling pathways | 6.077 × 10−4 | 2.396 × 10−2 | 6/45 | TRAF1, Bcl-XL, I-kB, BMF | PI3K cat class IA, JNK (MAPK8-10) |
| 18 | Signal transduction_Additional pathways of NF-kB activation (in the nucleus) | 6.143 × 10−4 | 2.396 × 10−2 | 5/30 | PKC-zeta, STO, Adenylate cyclase, I-kB, NFKBIA | |
| 19 | Immune response_IL-1 signaling pathway | 6.724 × 10−4 | 2.405 × 10−2 | 8/82 | IRF1, PKC-zeta, IP10, TPL2 (MAP3K8), I-kB, MMP-13 | PI3K cat class IA, JNK (MAPK8-10) |
| 20 | Transcription_Androgen Receptor nuclear signaling | 6.853 × 10−4 | 2.405 × 10−2 | 6/46 | MMP-2, p21, WNT | VIL2 (ezrin), Frizzled, IGF-1 |
| 21 | Immune response_Histamine H1 receptor signaling in immune response | 7.703 × 10−4 | 2.458 × 10−2 | 6/47 | NF-AT2(NFATC1), VCAM1, I-kB, NFKBIA, MMP-13 | JNK(MAPK8-10) |
| 22 | Development_PIP3 signaling in cardiac myocytes | 7.703 × 10−4 | 2.458 × 10−2 | 6/47 | PKC-zeta, Bcl-XL | G-protein alpha-12 family, PI3K cat class IA, IRS-1, IGF-1 |
| 23 | Muscle contraction_Relaxin signaling pathway | 8.632 × 10−4 | 2.635 × 10−2 | 6/48 | PKC-zeta, MMP-2, I-kB, NFKBIA | PI3K cat class IA (p110-beta), Endothelin-1 |
| 24 | Ligand-independent activation of Androgen receptor in Prostate Cancer | 9.668 × 10−4 | 2.795 × 10−2 | 7/67 | Tcf (Lef), Bcl-XL | PP2A regulatory, Frizzled, PI3K cat class IA, IRS-1, IGF-1 |
| 25 | Immune response_Lectin induced complement pathway | 1.075 × 10−3 | 2.795 × 10−2 | 6/50 | C3c, C3a, C3b, C3dg, C3, iC3b | |
| 26 | G-protein signaling_RhoA regulation pathway | 1.111 × 10−3 | 2.795 × 10−2 | 5/34 | ARHGEF3 | VIL2 (ezrin), PRK1, G-protein alpha-12 family, IGF-1 |
| 27 | Androgen receptor activation and downstream signaling in Prostate cancer | 1.135 × 10−3 | 2.795 × 10−2 | 9/110 | ER81, Bcl-XL, p21, IL6RA | VIL2 (ezrin), Versican, PI3K cat class IA (p110-beta), IRS-1, IGF-1 |
| 28 | Signal transduction_PKA signaling | 1.195 × 10−3 | 2.795 × 10−2 | 6/51 | LBC, Adenylate cyclase, NFKBIA | PP2A regulatory, AKAP12, G-protein alpha-12 family |
| 29 | Development_IGF-1 receptor signaling | 1.195 × 10−3 | 2.795 × 10−2 | 6/51 | PKC-zeta, Bcl-XL, I-kB | PI3K cat class IA, IRS-1, IGF-1 |
| 30 | Some pathways of EMT in cancer cells | 1.195 × 10−3 | 2.795 × 10−2 | 6/51 | Lef-1, PDGF receptor, PDGF-R-alpha, I-kB | PI3K cat class IA, Endothelin-1 |
| 31 | Immune response_IL-9 signaling pathway | 1.449 × 10−3 | 3.115 × 10−2 | 5/36 | SOCS3, I-kB, IL-22 | PI3K cat class IA, IRS-1 |
| 32 | Apoptosis and survival_Role of PKR in stress-induced apoptosis | 1.464 × 10−3 | 3.115 × 10−2 | 6/53 | IRF1, p21, I-kB, NFKBIA | PACT, PP2A regulatory |
| 33 | Immune response_Alternative complement pathway | 1.464 × 10−3 | 3.115 × 10−2 | 6/53 | C3c, C3a, C3b, C3dg, C3, iC3b | |
| 34 | Immune response_IL-3 signaling via JAK/STAT, p38, JNK and NF-kB | 1.540 × 10−3 | 3.179 × 10−2 | 8/93 | SOCS3, Spi2a, Bcl-XL, I-kB | PI3K cat class IA, DHA2, MKP-1, TACI (TNFRSF13B) |
| 35 | Immune response_Platelet activating factor/PTAFR pathway signaling | 1.778 × 10−3 | 3.374 × 10−2 | 6/55 | NF-AT, NF-AT2 (NFATC1), Adenylate cyclase, NFKBIA | F-Actin cytoskeleton, PI3K cat class IA |
| 36 | Immune response_CD28 signaling | 1.778 × 10−3 | 3.374 × 10−2 | 6/55 | NF-AT, NF-AT2 (NFATC1), Bcl-XL, I-kB | PI3K cat class IA, JNK (MAPK8-10) |
| 37 | PGE2 pathways in cancer | 1.778 × 10−3 | 3.374 × 10−2 | 6/55 | Tcf (Lef), Lef-1, Adenylate cyclase, COX-1 (PTGS1) | HIF1A,PI3K cat class IA (p110-beta) |
| 38 | G-protein signaling_G-Protein alpha-12 signaling pathway | 1.856 × 10−3 | 3.429 × 10−2 | 5/38 | LBC | G-protein alpha-12 family, PI3K cat class IA, JNK (MAPK8-10), M-Ras |
| 39 | Transcription_P53 signaling pathway | 2.089 × 10−3 | 3.578 × 10−2 | 5/39 | XPA, MMP-2, p21 | JNK (MAPK8-10), MKP-1 |
| 40 | Development_Cytokine-mediated regulation of megakaryopoiesis | 2.141 × 10−3 | 3.578 × 10−2 | 6/57 | IRF1, Bcl-XL, p21, LIF, sIL6-RA | PI3K cat class IA |
| 41 | Immune response_Role of PKR in stress-induced antiviral cell response | 2.141 × 10−3 | 3.578 × 10−2 | 6/57 | IRF1, IRF7, I-kB, NFKBIA | PACT, JNK (MAPK8-10) |
| 42 | Immune response_TLR5, TLR7, TLR8 and TLR9 signaling pathways | 2.141 × 10−3 | 3.578 × 10−2 | 6/57 | IRF1, IRF7, I-kB, TPL2 (MAP3K8) | PI3K cat class IA, JNK (MAPK8-10) |
| 43 | Development_Prolactin receptor signaling | 2.341 × 10−3 | 3.737 × 10−2 | 6/58 | IRF1, SOCS3, Bcl-XL, OAS1 | PI3K cat class IA, IRS-1 |
| 44 | Apoptosis and survival_NGF activation of NF-kB | 2.342 × 10−3 | 3.737 × 10−2 | 5/40 | PKC-zeta, Bcl-XL, I-kB, NFKBIA | PI3K cat class IA |
| 45 | Main growth factor signaling cascades in multiple myeloma cells | 2.617 × 10−3 | 3.994 × 10−2 | 5/41 | I-kB | HIF1A, PI3K cat class IA, IRS-1, IGF-1 |
| 46 | Development_Regulation of lung epithelial progenitor cell differentiation | 2.617 × 10−3 | 3.994 × 10−2 | 5/41 | Tcf (Lef), WNT | Jagged1, FZD2, Frizzled, |
| 47 | Immune response_IL-18 signaling | 2.785 × 10−3 | 4.074 × 10−2 | 6/60 | VCAM1, IL-18, Bcl-XS, I-kB | PI3K cat class IA, JNK (MAPK8-10) |
| 48 | Immune response_LTBR1 signaling | 2.785 × 10−3 | 4.074 × 10−2 | 6/60 | PKC, I-kB Adenylate cyclase, | PI3K cat class IA, JNK (MAPK8-10), YES |
| 49 | Apoptosis and survival_Anti-apoptotic TNFs/NF-kB/Bcl-2 pathway | 2.915 × 10−3 | 4.176 × 10−2 | 5/42 | PKC-zeta, Bcl-XL, I-kB, RelB (NF-kB subunit) | TACI (TNFRSF13B) |
| 50 | Signal transduction_AKT signaling | 3.236 × 10−3 | 4.544E × 10−2 | 5/43 | Bcl-XL, p21, I-kB | PI3K cat class IA, IRS-1 |
| Enrichment for Process Networks | |||||
|---|---|---|---|---|---|
| Process Networks | p-Value | FDR | Gene Ratio | Network Objects from Active Data | |
| 1 | Immune response_Phagocytosis | 6.475 × 10−7 | 5.307 × 10−5 | 24/223 | MANR, ERM proteins, VIL2 (ezrin), Actin cytoskeletal, Actin, PI3K cat class IA, PI3K cat class IA (p110-beta), JNK (MAPK8-10), JNK2 (MAPK9), MRLC, C3b, C3dg, C3, iC3b, p40-phox, ENDO180, BLNK, HDL proteins, Casein kinase II, alpha chain (CSNK2A1), Casein kinase II, alpha chains, I-kB, NFKBIA, Casein kinase II, alpha’ chain (CSNK2A2), C/EBP |
| 2 | Signal transduction_WNT signaling | 6.760 × 10−7 | 5.307 × 10−5 | 21/177 | PP2A regulatory, NF-AT, NF-AT2 (NFATC1), Tcf (Lef), Lef-1, AKAP12, FZD2, Frizzled, JNK (MAPK8-10), JNK2 (MAPK9), MMP-2, WISP1, Adenylate cyclase, NANOG, p73, Casein kinase II, alpha chain (CSNK2A1), Casein kinase II, alpha chains, Casein kinase II, WNT5B, WNT, Casein kinase II, alpha’ chain (CSNK2A2) |
| 3 | Cell adhesion_Cell-matrix interactions | 3.832 × 10−5 | 2.006 × 10−3 | 20/211 | Perlecan, ADAM23, ECM1, Aggrecanase-2, Versican, Decorin, COL16A1, BETA-IG-H3, COL5A1, Collagen V, MMP-12, MMP-2, WISP1, Connexin 43, LAMA4, EMILIN-2, ADAM-TS1, ITGB7, TSG-6, MMP-13 |
| 4 | Immune response_BCR pathway | 7.205 × 10−5 | 2.494 × 10−3 | 15/137 | NF-AT, NF-AT2 (NFATC1), Actin cytoskeletal, Actin, PI3K cat class IA, PI3K cat class IA (p110-beta), JNK (MAPK8-10), JNK2 (MAPK9), C3dg, SHIP, Bcl-XL, BLNK, Casein kinase II, alpha chain (CSNK2A1), I-kB, NFKBIA |
| 5 | Inflammation_IFN-gamma signaling | 9.114 × 10−5 | 2.494 × 10−3 | 13/109 | PACT, IRF1, IFI16, IL-18, PI3K cat class IA, PI3K cat class IA (p110-beta), Bcl-XL, p21, IRF9, IP10, EIF2S3, I-kB, NFKBIA |
| 6 | Immune response_Phagosome in antigen presentation | 9.531 × 10−5 | 2.494 × 10−3 | 21/243 | ERM proteins, VIL2 (ezrin), Actin cytoskeletal, Actin, PI3K cat class IA, PI3K cat class IA (p110-beta), JNK (MAPK8-10), JNK2 (MAPK9), PSMB9, C3dg, C3, iC3b, SEC15L, PSME3, ENDO180, BLNK, Casein kinase II, alpha chain (CSNK2A1), Casein kinase II, alpha chains, I-kB, NFKBIA, Casein kinase II, alpha’ chain (CSNK2A2) |
| 7 | Inflammation_Interferon signaling | 3.945 × 10−4 | 8.848 × 10−3 | 12/110 | IRF1, SOCS3, IFI44, CCL8, IRF7, IFI56, MxA, Bcl-XL, IRF9, GBP1, GBP2, ISG15 |
| 8 | Cell adhesion_Cadherins | 5.422 × 10−4 | 1.064 × 10−2 | 16/182 | Tcf (Lef), PKC, Actin cytoskeletal, Actin, Frizzled, G-protein alpha-12, PI3K cat class IA, WISP1, MAST205, Casein kinase II, alpha chain (CSNK2A1), Casein kinase II, alpha chains, Casein kinase II, ACP1, WNT, Casein kinase II, alpha’ chain (CSNK2A2), YES |
| 9 | Development_Regulation of angiogenesis | 6.553 × 10−4 | 1.143 × 10−2 | 18/222 | HIF1A, PKC, PRK1, WT1, IL-18, TCF8, PI3K cat class IA, FXR, Endothelin-1, MMP-2, Cathepsin B, Connexin 43, Syndecan-3, Angiogenin, p21, SMAD2, I-kB, RelB (NF-kB subunit) |
| 10 | Inflammation_IL-6 signaling | 8.080 × 10−4 | 1.269 × 10−2 | 12/119 | SOCS3, PI3K cat class IA, PI3K cat class IA (p110-beta), SAA3, C3, p21, HDL proteins, I-kB, NFKBIA, C/EBP, IL-22, Alpha1-globin |
| 11 | Inflammation_MIF signaling | 1.079 × 10−3 | 1.500 × 10−2 | 13/140 | PLA2, PKC, VCAM1, JNK (MAPK8-10), JNK2 (MAPK9), Adenylate cyclase type VII, Adenylate cyclase, Casein kinase II, alpha chain (CSNK2A1), Casein kinase II, alpha chains, Casein kinase II, I-kB, NFKBIA, Casein kinase II, alpha’ chain (CSNK2A2) |
| 12 | Cell cycle_G1-S Growth factor regulation | 1.146 × 10−3 | 1.500 × 10−2 | 16/195 | PP2A regulatory, IRF1, Tcf (Lef), PKC, PDGF-R-alpha, PI3K cat class IA, PI3K cat class IA (p110-beta), JNK (MAPK8-10), JNK2 (MAPK9), p21, MAD, IRS-1, I-kB, NFKBIA, RelB (NF-kB subunit), IGF-1 |
| 13 | Cell adhesion_Cell junctions | 1.413 × 10−3 | 1.707 × 10−2 | 14/162 | ATP1B1, Tcf (Lef), PKC-zeta, PKC, Actin cytoskeletal, Actin, PI3K cat class IA, Endothelin-1, Connexin 43, Casein kinase II, alpha chain (CSNK2A1), Casein kinase II, Casein kinase II alpha chain (CSNK2A2), YES |
| 14 | Inflammation_Amphoterin signaling | 2.500 × 10−3 | 2.678 × 10−2 | 11/118 | VCAM1, Actin cytoskeletal, Actin, PI3K cat class IA, PI3K cat class IA (p110-beta), JNK (MAPK8-10), JNK2 (MAPK9), MRLC, I-kB, NFKBIA, MMP-13 |
| 15 | Signal Transduction_TGF-beta, GDF and Activin signaling | 2.559 × 10−3 | 2.678 × 10−2 | 13/154 | PP2A regulatory, HIF1A, Lef-1, PKC, PI3K cat class IA, JNK (MAPK8-10), IRF7, UNRIP, p21, SMAD2, IRS-1, IGF-1, TGF-beta receptor type III (betaglycan) |
| 16 | Inflammation_Histamine signaling | 2.852 × 10−3 | 2.752 × 10−2 | 16/213 | PLA2, NF-AT2 (NFATC1), VCAM1, Actin cytoskeletal, Actin, IL-18, JNK(MAPK8-10), JNK2 (MAPK9), MDP1, p40-phox, Adenylate cyclase type VII, Adenylate cyclase, IP10, COX-1 (PTGS1), I-kB, NFKBIA |
| 17 | Inflammation_IL-10 anti-inflammatory response | 3.016 × 10−3 | 2.752 × 10−2 | 9/87 | SOCS3, Nucleolysin TIAR, PI3K cat class IA, PI3K cat class IA (p110-beta), MMP-2, Bcl-XL, I-kB, NFKBIA, MMP-13 |
| 18 | Signal transduction_NOTCH signaling | 3.155 × 10−3 | 2.752 × 10−2 | 17/235 | Jagged1, HIF1A, PDGF receptor, PDGF-R-alpha, FZD2, Frizzled, PI3K cat class IA, JNK (MAPK8-10), JNK2 (MAPK9), GLUT1, p21, p73, SMAD2, WNT5B, WNT, I-kB, NFKBIA |
| 19 | Inflammation_Innate inflammatory response | 3.772 × 10−3 | 3.117 × 10−2 | 14/180 | PLA2, PKC-zeta, IL-18, JNK(MAPK8-10), JNK2 (MAPK9), IRF7, C3a, C3b, C3, PGRP-S, IP10, COX-1 (PTGS1), I-kB, NFKBIA |
| 20 | Reproduction_Male sex differentiation | 4.441 × 10−3 | 3.487 × 10−2 | 17/243 | PAFAH alpha (LIS1), PKC-zeta, PKC, PMEPA1, PDGF receptor, PDGF-R-alpha, WT1, BAPX1, AMH type II receptor, Adenylate cyclase type VII, p21, NANOG, STK39, Olfactory receptor, Casein kinase II, Casein kinase II, alpha’ chain (CSNK2A2), IGF-1 |
| 21 | Development_EMT_Regulation of epithelial-to-mesenchymal transition | 4.682 × 10−3 | 3.501 × 10−2 | 16/224 | Jagged1, HIF1A, NF-AT2 (NFATC1), Lef-1, Actin, PDGF receptor, PDGF-R-alpha, Frizzled, TCF8, PI3K cat class IA, JNK (MAPK8-10), Endothelin-1, MMP-2, SMAD2, WNT, I-kB |
| 22 | Immune response_TCR signaling | 7.223 × 10−3 | 4.933 × 10−2 | 13/174 | NF-AT, NF-AT2 (NFATC1), CD137(TNFRSF9), Actin cytoskeletal, Actin, PI3K cat class IA, JNK (MAPK8-10), JNK2 (MAPK9), TRAF1, Bcl-XL, TPL2 (MAP3K8), I-kB, NFKBIA |
| 23 | Inflammation_Inflammasome | 7.597 × 10−3 | 4.933 × 10−2 | 10/118 | PACT, RIG-I, IL-18, JNK (MAPK8-10), IRF7, TXNIP (VDUP1), EIF2S3, ISG15, I-kB, NFKBIA |
| 24 | Inflammation_IgE signaling | 7.782 × 10−3 | 4.933 × 10−2 | 11/137 | PLA2, NF-AT, NF-AT2 (NFATC1), PI3K cat class IA, PI3K cat class IA (p110-beta), JNK (MAPK8-10), JNK2 (MAPK9), MDP1, BLNK, I-kB, NFKBIA |
| 25 | Signal transduction_ESR1-nuclear pathway | 7.855 × 10−3 | 4.933 × 10−2 | 15/216 | SOCS3, WT1, PI3K cat class IA, MPG, SET, LBC, C3, Adenylate cyclase type VII, Adenylate cyclase, RAMP3, CYP1A1, p21, CYP1B1, IRS-1, IGF-1 |
| GO Process | p Value | FDR | Gene Ratio | |
|---|---|---|---|---|
| 1 | Response to stress | 3.713 × 10−22 | 2.771 × 10−18 | 233/4937 |
| 2 | Cellular response to cytokine stimulus | 3.510 × 10−19 | 1.310 × 10−15 | 98/1398 |
| 3 | Response to cytokine | 2.914 × 10−18 | 7.248 × 10−15 | 105/1611 |
| 4 | Response to external stimulus | 1.159 × 10−17 | 2.163 × 10−14 | 159/3089 |
| 5 | Cellular response to organic substance | 4.898 × 10−17 | 6.115 × 10−14 | 170/3457 |
| 6 | Response to organic substance | 4.917 × 10−17 | 6.115 × 10−14 | 207/4579 |
| 7 | Cellular response to chemical stimulus | 4.374 × 10−16 | 4.663 × 10−13 | 190/4141 |
| 8 | Negative regulation of biological process | 7.285 × 10−15 | 6.795 × 10−12 | 269/6845 |
| 9 | Response to interferon-beta | 5.701 × 10−14 | 4.727 × 10−11 | 17/57 |
| 10 | Response to virus | 1.456 × 10−13 | 1.087 × 10−10 | 41/403 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pillo, G.; Mascolo, M.G.; Zanzi, C.; Rotondo, F.; Serra, S.; Bortone, F.; Grilli, S.; Vaccari, M.; Jacobs, M.N.; Colacci, A. Mechanistic Interrogation of Cell Transformation In Vitro: The Transformics Assay as an Exemplar of Oncotransformation. Int. J. Mol. Sci. 2022, 23, 7603. https://doi.org/10.3390/ijms23147603
Pillo G, Mascolo MG, Zanzi C, Rotondo F, Serra S, Bortone F, Grilli S, Vaccari M, Jacobs MN, Colacci A. Mechanistic Interrogation of Cell Transformation In Vitro: The Transformics Assay as an Exemplar of Oncotransformation. International Journal of Molecular Sciences. 2022; 23(14):7603. https://doi.org/10.3390/ijms23147603
Chicago/Turabian StylePillo, Gelsomina, Maria Grazia Mascolo, Cristina Zanzi, Francesca Rotondo, Stefania Serra, Francesco Bortone, Sandro Grilli, Monica Vaccari, Miriam N. Jacobs, and Annamaria Colacci. 2022. "Mechanistic Interrogation of Cell Transformation In Vitro: The Transformics Assay as an Exemplar of Oncotransformation" International Journal of Molecular Sciences 23, no. 14: 7603. https://doi.org/10.3390/ijms23147603
APA StylePillo, G., Mascolo, M. G., Zanzi, C., Rotondo, F., Serra, S., Bortone, F., Grilli, S., Vaccari, M., Jacobs, M. N., & Colacci, A. (2022). Mechanistic Interrogation of Cell Transformation In Vitro: The Transformics Assay as an Exemplar of Oncotransformation. International Journal of Molecular Sciences, 23(14), 7603. https://doi.org/10.3390/ijms23147603