
Gene Expression over Time during Cell Transformation Due to Non-Genotoxic Carcinogen Treatment of Bhas 42 Cells


Abstract
:1. Introduction
2. Results
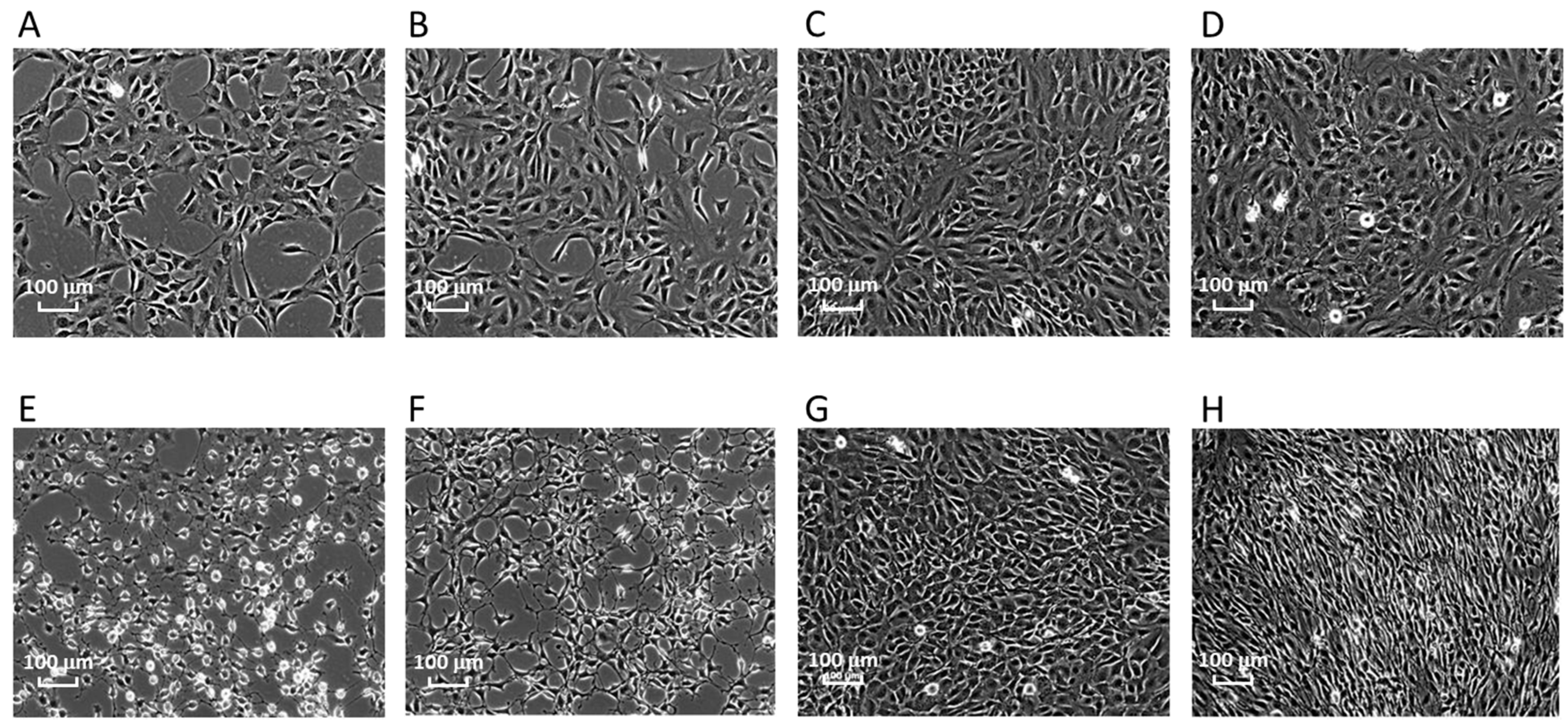
2.1. Morphological Changes and Focus Formation by Bhas 42 Cells at the Tumor-Promotion Stage of the Cell-Transformation Assay
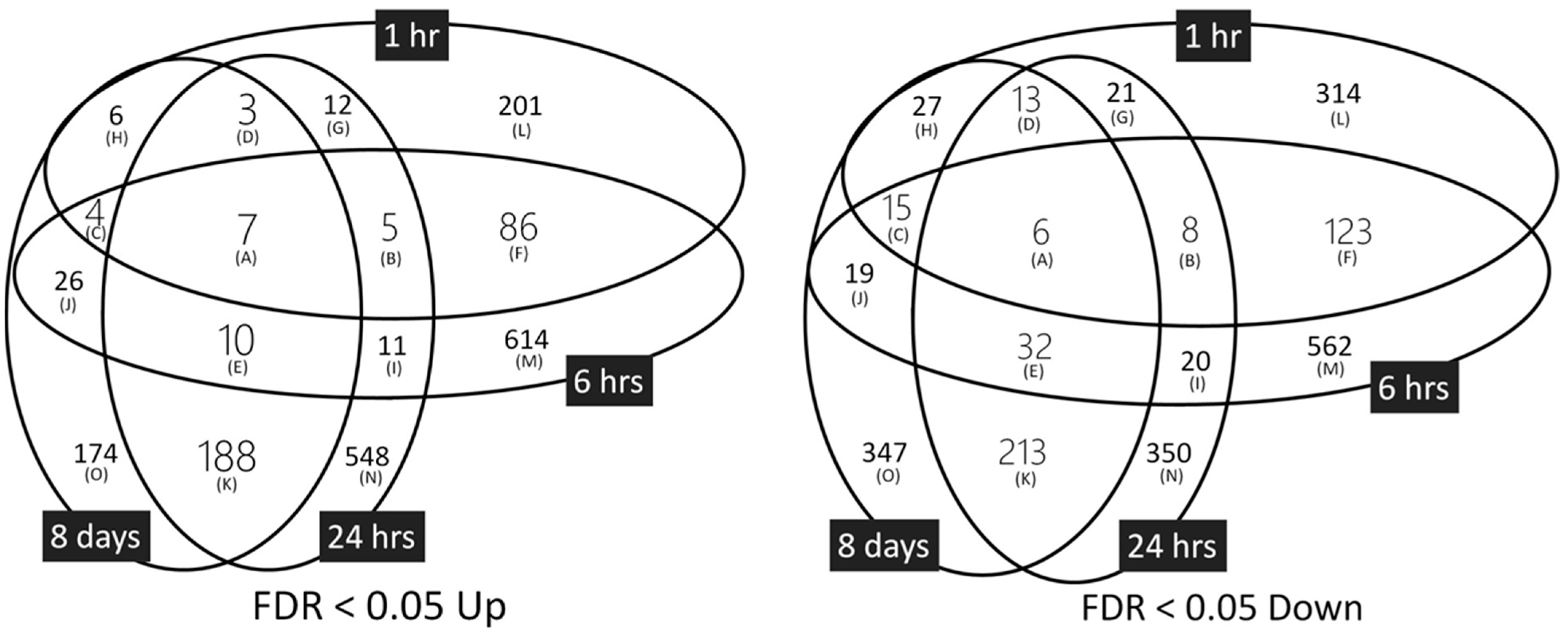
2.2. Quantification of DNA Microarray Data and Detection of Differentially Expressed Genes
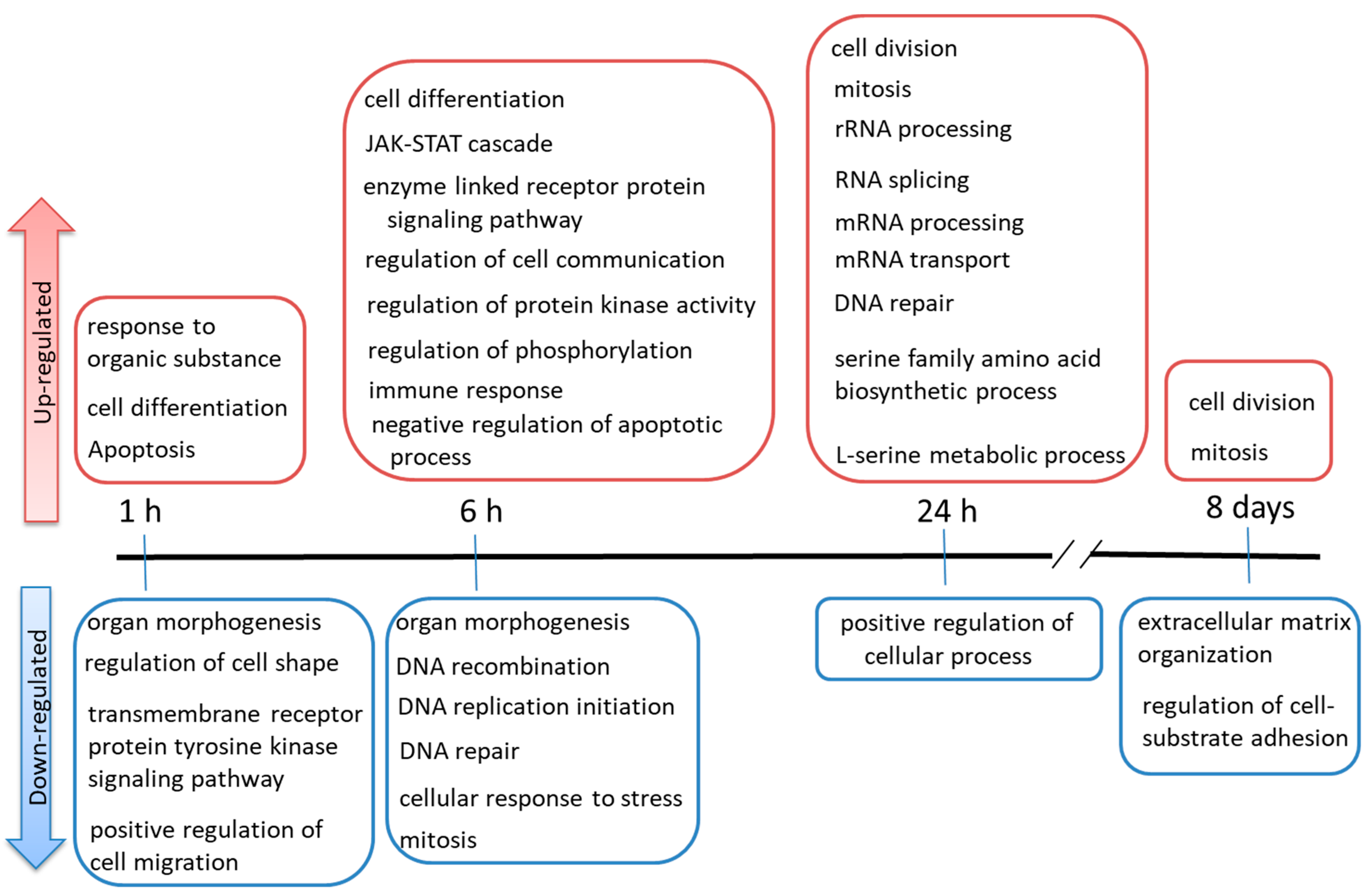
2.3. Gene Ontology (GO) Terms
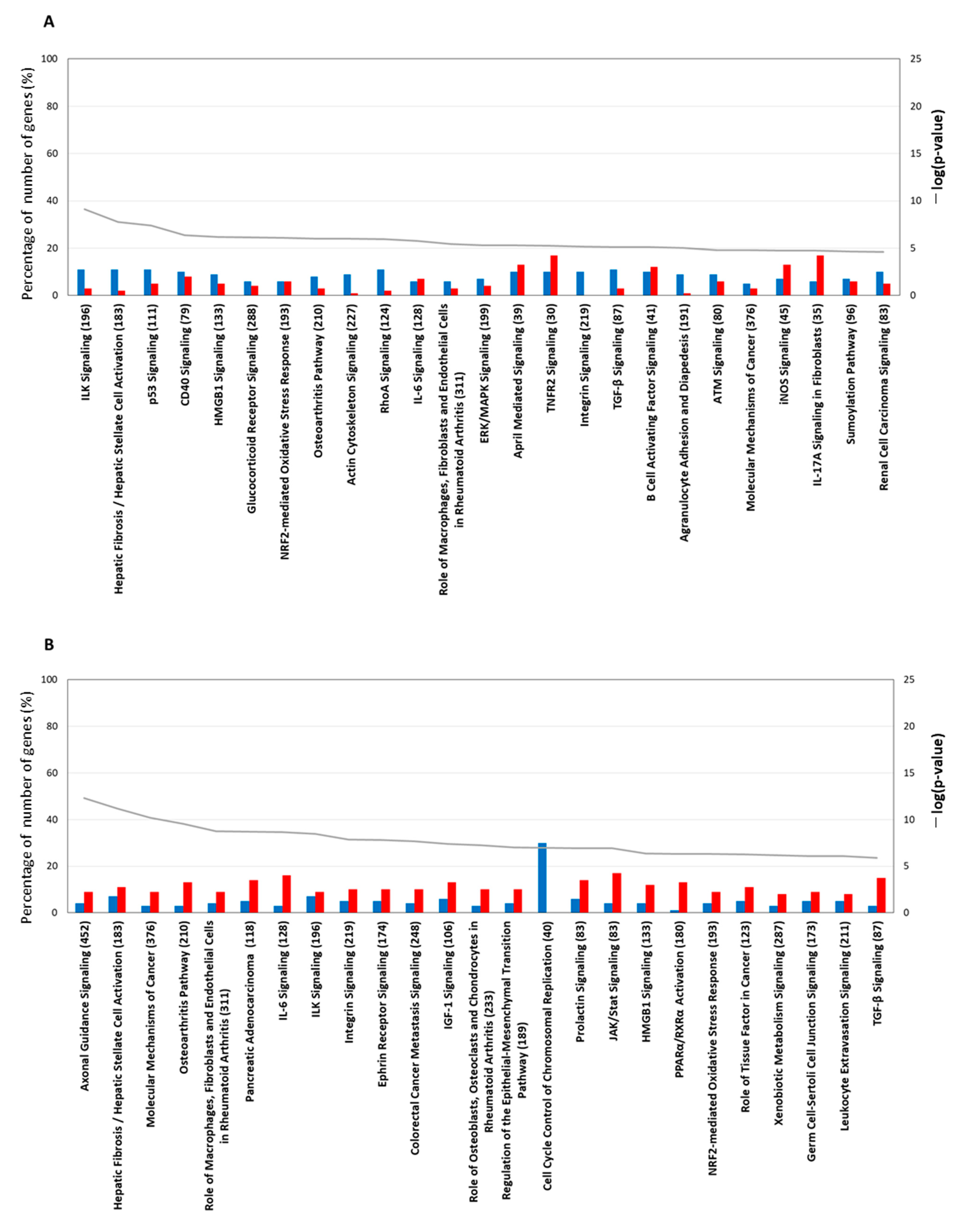
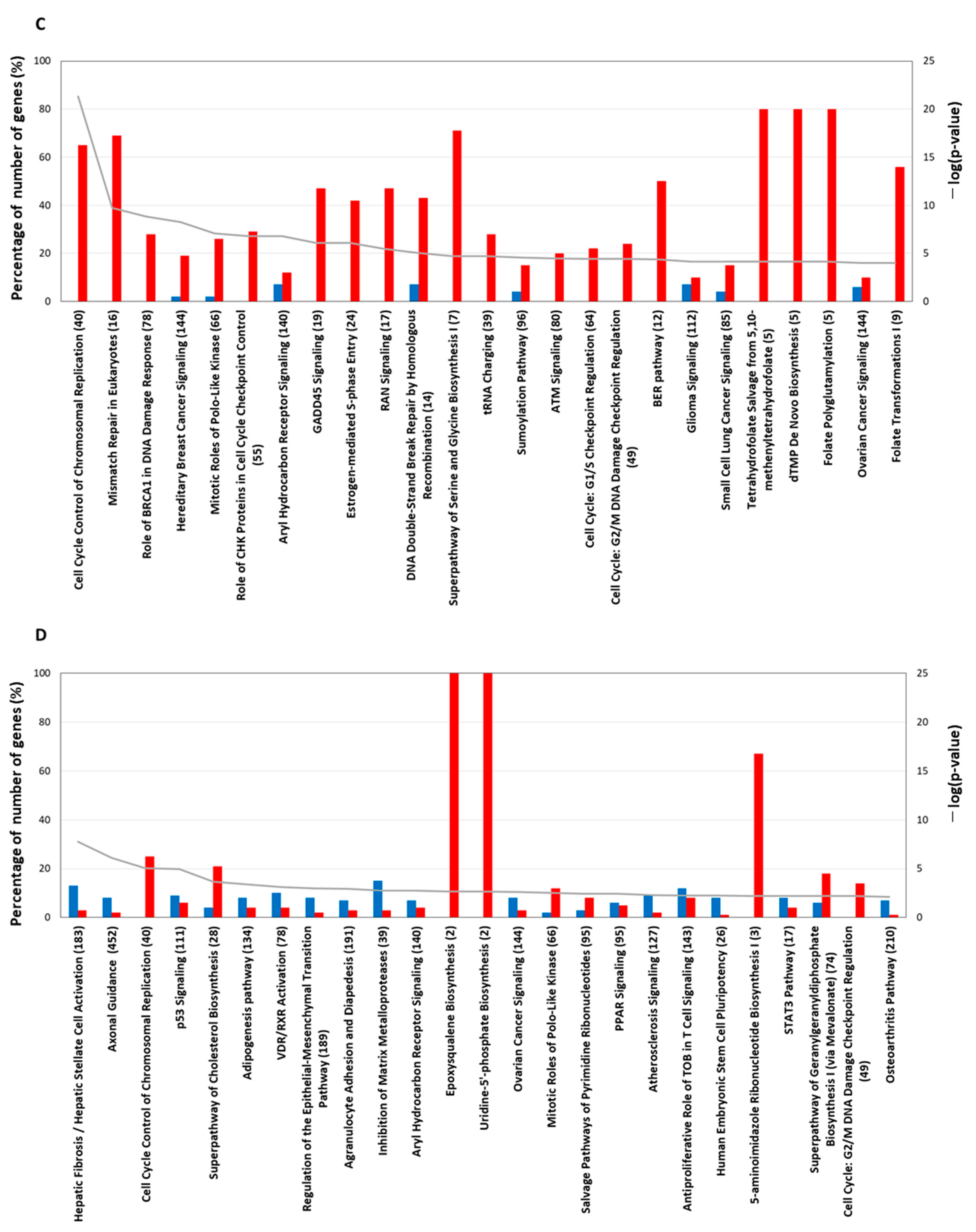
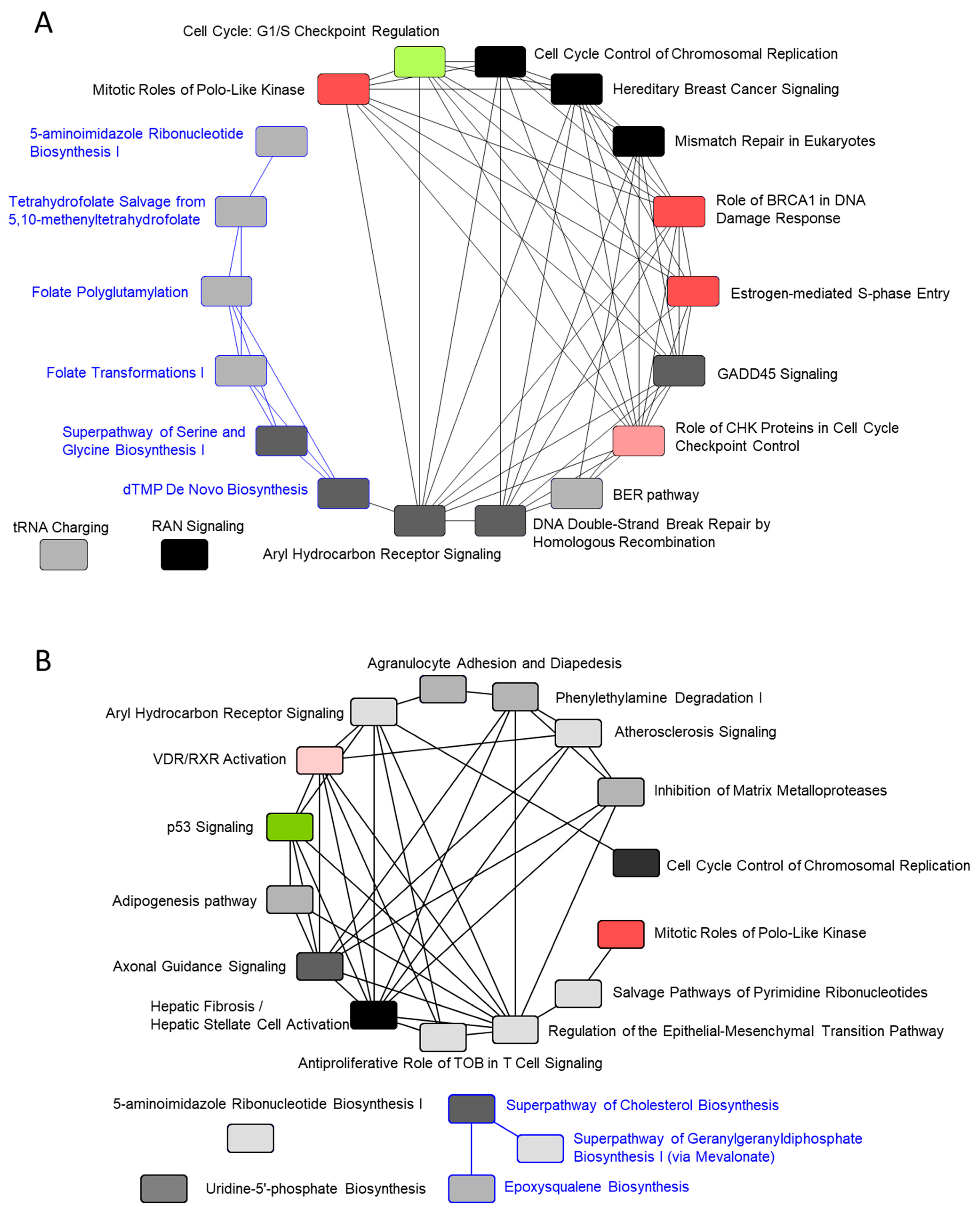
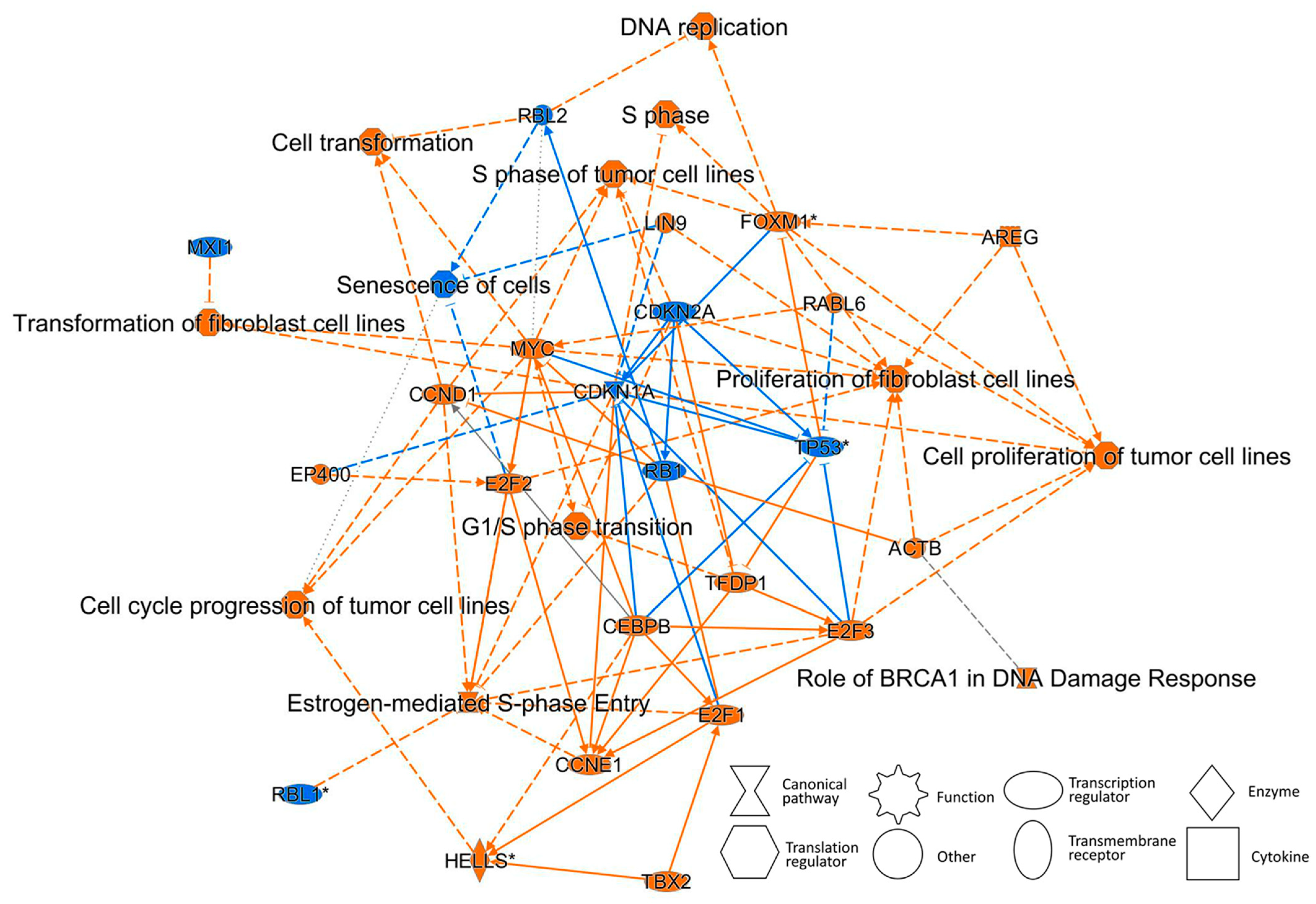
2.4. Pathway Analysis Using IPA Software
2.4.1. Gene Expression Characteristics for 1 H TPA Treatment
2.4.2. Gene-Expression Characteristics of 6 H TPA Treatment
2.4.3. Gene-Expression Characteristics of 24 H TPA Treatment
2.4.4. Gene-Expression Characteristics of 8-Day TPA Treatment
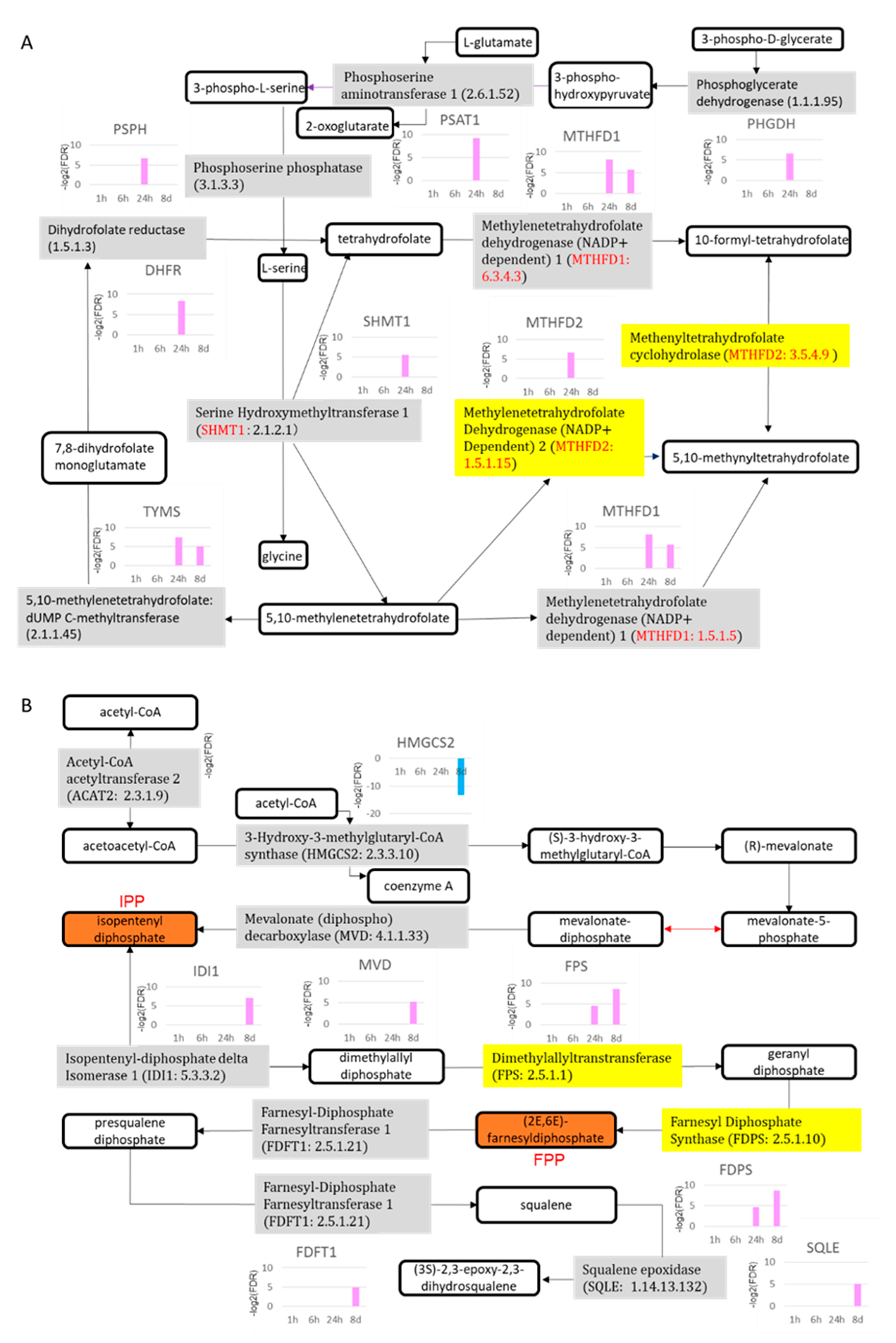
2.4.5. DNA Methylation
2.4.6. γ-Glutamyl Cycle and Cytochrome P450 Family
2.5. Collation of the Pathways of Cell Transformation and the Hallmarks of Cancer
2.5.1. Hallmarks of Cancer with 1 H TPA Treatment
2.5.2. Hallmarks of Cancer with 6 H TPA Treatment
2.5.3. Hallmarks of Cancer with 24 H TPA Treatment
2.5.4. Hallmarks of Cancer with 8-Day TPA Treatment
3. Discussion
4. Conclusions
5. Methods
5.1. Cell Culture
5.2. Cell-Transformation Assay at the Stationary Phase Using Bhas 42 Cells (Bhas 42 CTA Promotion Test)
5.3. Statistical Analysis and Criteria of Judgment
5.4. Isolation of Total RNA
5.5. DNA-Microarray Assay
5.6. DNA-Microarray Data Analysis
5.7. GO Analysis
5.8. Functional Analysis and Pathway Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Bhas 42 CTA | Bhas 42 cell-transformation assay |
| OECD | Organization for Economic Cooperation and Development |
| NGTxC | Non-genotoxic carcinogen |
| TPA | 12-O-Tetradecanoylphorbol-13-acetate |
| IATA | Integrated approaches for testing and assessment |
| DMSO | Dimethyl sulfoxide |
| FDR | False-discovery rate |
| GO | Gene Ontology |
| DAVID | Database for Annotation, Visualization, and Integrated Discovery |
| MIE | Molecular initiating event |
| FBS | Fetal bovine serum |
References
- Morita, T.; Hamada, S.; Masumura, K.; Wakata, A.; Maniwa, J.; Takasawa, H.; Yasunaga, K.; Hashizumef, T.; Honma, M. Evaluation of the sensitivity and specificity of in vivo erythrocyte micronucleus and transgenic rodent gene mutation tests to detect rodent carcinogens. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2016, 802, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Combes, R.D. Detection of non-genotoxic carcinogens: Major barriers to replacement of the rodent assays. In Proceedings of the 2nd World Congress on Alternatives and Animal Use in the Life Sciences, Utrecht, The Netherlands, 20–24 October 1996; Van Zutphen, L.F.M., Balls, M., Eds.; Elsevier: Amsterdam, The Netherlands, 1997; pp. 627–634. [Google Scholar]
- Ohmori, K.; Sasaki, K.; Asada, S.; Tanaka, N.; Umeda, M. An assay method for the prediction of tumor promoting potential of chemicals by the use of Bhas 42 cells. Mutat. Res. 2004, 557, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, K.; Mizusawa, H.; Ishidate, M. Isolation and characterization of ras-transfected BALB/3T3 clone showing morphological transformation by 12-O-tetradecanoyl-phorbol-13-acetate. Jpn. J. Cancer Res. 1988, 79, 921–930. [Google Scholar] [CrossRef]
- Ohmori, K.; Umeda, M.; Tanaka, N.; Takagi, T.; Yoshimura, I.; Sasaki, K.; Asada, S.; Sakai, A.; Araki, H.; Asakura, M.; et al. An inter-laboratory collaborative study by the Non-Genotoxic Carcinogen Study Group in Japan, on a cell transformation assay for tumour promoters using Bhas 42 cells. Altern. Lab. Anim. 2005, 33, 619–639. [Google Scholar] [CrossRef] [PubMed]
- Asada, S.; Sasaki, K.; Tanaka, N.; Takeda, K.; Hayashi, M.; Umeda, M. Detection of initiating as well as promoting activity of chemicals by a novel cell transformation assay using v-Ha-ras-transfected BALB/c 3T3 cells (Bhas 42 cells). Mutat. Res. 2005, 588, 7–21. [Google Scholar] [CrossRef]
- Sakai, A.; Sasaki, K.; Muramatsu, D.; Arai, S.; Endou, N.; Kuroda, S.; Hayashi, K.; Lim, Y.M.; Yamazaki, S.; Umeda, M.; et al. A Bhas 42 cell transformation assay on 98 chemicals: The characteristics and performance for the prediction of chemical carcinogenicity. Mutat. Res. 2010, 702, 100–122. [Google Scholar] [CrossRef]
- EU Reference Laboratory for Alternatives to Animal Testing. EURL ECVAM Recommendation on the Cell Transformation Assay Based on the Bhas 42 Cell Line; Ispra, Italy, 2013. Available online: https://data.europa.eu/doi/10.2788/42908 (accessed on 21 February 2015).
- Organization for Economic Co-operation and Development (OECD). Bhas 42 Cell Transformation Assay Validation Study Report; Series on Testing and Assessment No. 208; OECD Environment Directorate, Environment, Health and Safety Division: Paris, France, 2014. Available online: https://www.oecd.org/env/ehs/testing/Text_Bhas_Validation_Study_Report.pdf (accessed on 21 February 2015).
- Organization for Economic Co-operation and Development (OECD). Guidance Document on the In Vitro Bhas 42 Cell Transformation Assay; Series on Testing & Assessment No. 231; OECD Environment Directorate, Environment, Health and Safety Division: Paris, France, 2016. Available online: https://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?cote=ENV/JM/MONO(2016)1&doclanguage=en (accessed on 21 February 2015).
- Organization for Economic Co-operation and Development (OECD). Environment Directorate Joint Meeting of the Chemicals Committee and the Working Party on Chemicals, Pesticides and Biotechnology. Guidance Document for the Use of Adverse Outcome Pathways in Developing Integrated Approaches to Testing and Assessment (IATA); Series on Testing & Assessment No. 260; OECD Environment Directorate, Environment, Health and Safety Division: Paris, France, 2016. Available online: https://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?cote=env/jm/mono(2016)67&doclanguage=en (accessed on 21 February 2015).
- Jacobs, M.N.; Colacci, A.; Louekari, K.; Luijten, M.; Hakkert, B.C.; Paparella, M.; Vasseur, P. International regulatory needs for development of an IATA for non-genotoxic carcinogenic chemical substances. ALTEX 2016, 33, 359–392. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, M.N.; Colacci, A.; Corvi, R.; Vaccari, M.; Aguila, M.C.; Corvaro, M.; Delrue, N.; Desaulniers, D.; Ertych, N.; Jacobs, A.; et al. Chemical carcinogen safety testing: OECD expert group international consensus on the development of an integrated approach for the testing and assessment of chemical non-genotoxic carcinogens. Arch. Toxicol. 2020, 94, 2899–2923. [Google Scholar] [CrossRef] [PubMed]
- Iversen, O.H. TPA (12-O-tetradecanoyl-phorbol- 13-acetate) as a carcinogen for mouse skin. A positive dose-response relationship. Virchows Arch. Cell Pathol. 1985, 49, 129–135. [Google Scholar] [CrossRef]
- Soper, C.J.; Evance, F.J. Investigations into the mode of action of the cocarcinogen 12-O.Tetradecanoyl-phorbol-13-acetate using auxotrophic bacteria. Cancer Res. 1977, 37, 2487–2491. Available online: https://aacrjournals.org/cancerres/article/37/8_Part_1/2487/482262/Investigations-into-the-Mode-of-Action-of-the (accessed on 21 February 2015).
- Lamph, W.W.; Wamsley, S.C.P.; Verma, I.M. Induction of proto-oncogene JUN/AP-1 by serum and TPA. Nature 1988, 334, 629–631. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.Y.; Mercer, S.E.; Ewton, D.Z.; Yan, Z.; Jin, K.; Friedman, E. The stress-activated protein kinases p38 alpha and JNK1 stabilize p21(Cip1) by phosphorylation. J. Biol. Chem. 2002, 277, 29792–29802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bank, S.; Andersen, P.S.; Burisch, J.; Pedersen, N.; Roug, S.; Galsgaard, J.; Turino, S.Y.; Brodersen, J.B.; Rashid, S.; Rasmussen, B.K. Associations between functional polymorphisms in the NFκB signaling pathway and response to anti-TNF treatment in Danish patients with inflammatory bowel disease. Pharmacogenom. J. 2014, 14, 526–534. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 2016, 7, 11624. [Google Scholar] [CrossRef] [Green Version]
- Kikuchi, K.; Niikura, Y.; Kitagawa, K.; Kikuchi, A. Dishevelled, a Wnt signalling component, is involved in mitotic progression in cooperation with Plk1. EMBO J. 2010, 29, 3470–3483. [Google Scholar] [CrossRef] [Green Version]
- Boudhraa, Z.; Carmona, E.; Provencher, D.; Mes-Masson, A.M. Ran GTPase: A key player in tumor progression and metastasis. Front. Cell Dev. Biol. 2020, 8, 345. [Google Scholar] [CrossRef]
- Deng, L.; Lu, Y.; Zhao, X.; Sun, Y.; Shi, Y.; Fan, H.; Liu, C.; Zhou, J.; Nie, Y.; Wu, K.; et al. Ran GTPase protein promotes human pancreatic cancer proliferation by deregulating the expression of Survivin and cell cycle proteins. Biochem. Biophys. Res. Commun. 2013, 440, 322–329. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Han, Y.; Suarez Saiz, F.; Minden, M.D. A tumor suppressor and oncogene: The WT1 story. Leukemia 2007, 21, 868–876. [Google Scholar] [CrossRef] [PubMed]
- Van Driessche, A.; Berneman, Z.N.; Van Tendeloo, V.F. Active specific immunotherapy targeting the Wilms’ tumor protein 1 (WT1) for patients with hematological malignancies and solid tumors: Lessons from early clinical trials. Oncologist 2012, 17, 250–259. [Google Scholar] [CrossRef] [Green Version]
- Jaenisch, R.; Bird, A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat. Genet. 2003, 33, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Sharapova, T.; Talaty, N.; Buck, W.R.; Fossey, S.; Liguori, M.J.; Van Vleet, T.R. Reduced hepatic global hydroxymethylation in mice treated with non-genotoxic carcinogens is transiently reversible with a methyl supplemented diet. Toxicol. Appl. Pharmacol. 2021, 415, 115439. [Google Scholar] [CrossRef]
- Thomson, J.P.; Moggs, J.G.; Wolf, C.R.; Meehan, R.R. Epigenetic profiles as defined signatures of xenobiotic exposure. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2014, 764–765, 3–9. [Google Scholar] [CrossRef]
- Amano, T.; Eishi, Y.; Yamada, T.; Uchida, K.; Minegishi, K.; Tamura, T.; Kobayashi, D.; Hiroshi, K.; Suzuki, T.; Board, P.G. Widespread expression of γ-glutamyl cyclotransferase suggests it is not a general tumor marker. J. Histochem. Cytochem. 2012, 60, 76–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bansal, A.; Celeste Simon, M. Glutathione metabolism in cancer progression and treatment resistance. J. Cell Biol. 2018, 217, 2291–2298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, M.T.; Guyton, K.Z.; Gibbons, C.F.; Fritz, J.M.; Portier, C.J.; Rusyn, I.; DeMarini, D.M.; Caldwell, J.C.; Kavlock, R.J.; Lambert, P.F.; et al. Key characteristics of carcinogens as a basis for organizing data on mechanisms of carcinogenesis. Environ. Health Perspect. 2016, 124, 713–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.H.; Park, Y.Y.; Kim, S.W.; Lee, J.S.; Wang, D.; DuBois, R.N. ANGPTL4 induction by prostaglandin E2 under hypoxic conditions promotes colorectal cancer progression. Cancer Res. 2011, 71, 7010–7020. [Google Scholar] [CrossRef] [Green Version]
- Zhu, P.; Tan, M.J.; Huang, R.L.; Tan, C.K.; Chong, H.C.; Pal, M.; Lam, C.R.I.; Boukamp, P.; Pan, J.Y.; Tan, S.H. Angiopoietin-like 4 protein elevates the prosurvival intracellular O2−:H2O2 ratio and confers anoikis resistance to tumors. Cancer Cell 2011, 19, 401–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lund, T.; Holtlund, J.; Laland, S.G. On the phosphorylation of low molecular mass HMG (high mobility group) proteins in Ehrlich ascites cells. FEBS Lett. 1985, 180, 275–279. [Google Scholar] [CrossRef] [Green Version]
- Schoenmakers, E.F.; Wanschura, S.; Mols, R.; Bullerdiek, J.; Van den Berghe, H.; Van de Ven, W.J. Recurrent rearrangements in the high mobility group protein gene, HMGI-C, in benign mesenchymal tumours. Nat. Genet. 1995, 10, 436–444. [Google Scholar] [CrossRef]
- Hess, J.L. Chromosomal translocations in benign tumors: The HMGI proteins. Am. J. Clin. Pathol. 1998, 109, 251–261. [Google Scholar] [CrossRef] [Green Version]
- Sarhadi, V.K.; Wikman, H.; Salmenkivi, K.; Kuosma, E.; Sioris, T.; Salo, J.; Karjalainen, A.; Knuutila, S.; Anttila, S. Increased expression of high mobility group A proteins in lung cancer. J. Pathol. 2006, 209, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Fusco, A.; Fedele, M. Roles of HMGA proteins in cancer. Nat. Rev. Cancer 2007, 7, 899–910. [Google Scholar] [CrossRef] [PubMed]
- Cello, F.D.; Hillion, J.; Hristov, A.; Wood, L.J.; Mukherjee, M.; Schuldenfrei, A.; Kowalski, J.; Bhattacharya, R.; Ashfaq, R.; Resar, L.M.S. HMGA2 participates in transformation in human lung cancer. Mol. Cancer Res. 2008, 6, 743–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Resar, L.M. The high mobility group A1 gene: Transforming inflammatory signals into cancer? Cancer Res. 2010, 70, 436–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, S.N.; Resar, L.M. High mobility group A1 and cancer: Potential biomarker and therapeutic target. Histol. Histopathol. 2012, 27, 567–579. [Google Scholar] [CrossRef] [PubMed]
- Morishita, A.; Zaidi, M.Z.; Mitoro, A.; Sankarasharma, D.; Szabolcs, M.; Okada, Y.; D’Armiento, J.; Chada, K. HMGA2 is a driver of tumor metastasis. Cancer Res. 2013, 73, 4289–4299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Resar, L.; Chia, L.; Xian, L. Lessons from the Crypt: HMGA1—Amping up Wnt for stem cells and tumor progression. Cancer Res. 2018, 78, 1890–1897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unachukwu, U.; Kiran Chada, K.; D’Armiento, J. High mobility group AT-hook 2 (HMGA2) oncogenicity in mesenchymal and epithelial neoplasia. Int. J. Mol. Sci. 2020, 21, 3151. [Google Scholar] [CrossRef]
- Xian, L.; Georgess, D.; Huso, T.; Cope, L.; Belton, A.; Chang, Y.T.; Kuang, W.; Gu, Q.; Zhang, X.; Senger, S.; et al. Hmga1amplifies Wnt Signaling and expands the intestinal stem cell compartment and Paneth cell niche. Nat. Comm. 2017, 8, 15008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mansoori, B.; Mohammadi, A.; Ditzel, H.J.; Duijf, P.H.G.; Khaze, V.; Gjerstorff, M.F.; Baradaran, B. HMGA2 as a critical regulator in cancer development. Genes 2021, 12, 269. [Google Scholar] [CrossRef] [PubMed]
- Stangeland, B.; Mughal, A.A.; Grieg, Z.; Sandberg, C.J.; Joel, M.; Nygård, S.; Meling, T.; Murrell, W.; Vik Mo, E.O.; Langmoen, I.A. Combined expressional analysis, bioinformatics and targeted proteomics identify new potential therapeutic targets in glioblastoma stem cells. Oncotarget 2015, 6, 26192–26195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derry, J.M.; Kerns, J.A.; Francke, U. RBM3, a novel human gene in Xp11.23 with a putative RNA-binding domain. Hum. Mol. Genet. 1995, 4, 2307–2311. [Google Scholar] [CrossRef] [PubMed]
- Danno, S.; Nishiyama, H.; Higashitsuji, H.; Yokoi, H.; Xue, J.H.; Itoh, K.; Matsuda, T.; Fujita, J. Increased transcript level of RBM3, a member of the glycine-rich RNA-binding protein family, in human cells in response to cold stress. Biochem. Biophys. Res. Commun. 1997, 236, 804–807. [Google Scholar] [CrossRef]
- Dresios, J.; Aschrafi, A.; Owens, G.C.; Peter, W.; Vanderklish, P.W.; Edelman, G.M.; Mauro, V.P. Cold stress-induced protein Rbm3 binds 60S ribosomal subunits, alters microRNA levels, and enhances global protein synthesis. Proc. Natl. Acad. Sci. USA 2005, 102, 1865–1870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sureban, S.M.; Ramalingam, S.; Natarajan, G.; May, R.; Subramaniam, D.; Bishnupuri, K.S.; Morrison, A.R.; Dieckgraefe, B.K.; Brackett, D.J.; Postier, R.G.; et al. Translation regulatory factor RBM3 is a proto-oncogene that prevents mitotic catastrophe. Oncogene 2008, 27, 4544–4556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouchard, D.; Morisset, D.; Bourbonnais, Y.; Tremblay, G.M. Proteins with whey-acidic-protein motifs and cancer. Lancet Oncol. 2006, 7, 167–174. [Google Scholar] [CrossRef]
- Devoogdt, N.; Revets, H.; Kindt, A.; Liu, Y.Q.; Baetselier, P.D.; Ghassabeh, G.H. The tumor-promoting effect of TNF-alpha involves the induction of secretory leukocyte protease inhibitor. J. Immunol. 2006, 177, 8046–8052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weldon, S.; McGarry, N.; Taggart, C.C.; McElvaney, N.G. The role of secretory leucoprotease inhibitor in the resolution of inflammatory responses. Biochem. Soc. Trans. 2007, 35, 273–276. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Simmen, R.C.M.; Michel, F.J.; Zhao, G.; Vale-Cruz, D.; Simmen, A.S. Secretory leukocyte protease inhibitor mediates proliferation of human endometrial epithelial cells by positive and negative regulation of growth-associated genes. J. Biol. Chem. 2002, 277, 29999–30009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Famulski, J.K.; Vos, L.; Sun, X.; Chan, G. Stable hZW10 kinetochore residency, mediated by hZwint-1 interaction, is essential for the mitotic checkpoint. J. Cell Biol. 2008, 180, 507–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ying, H.; Simmen, R.C.M.; Michel, F.J.; Zhao, G.; Vale-Cruz, D.; Simmen, F.A. Overexpression of Zwint predicts poor prognosis and promotes the proliferation of hepatocellular carcinoma by regulating cell-cycle-related proteins. Onco. Targets Ther. 2018, 11, 689–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilsson, R.; Jain, M.; Madhusudhan, N.; Sheppard, N.G.; Strittmatter, L.; Kampf, C.; Huang, J.; Asplund, A.; Mootha, V.K. Metabolic enzyme expression highlights a key role for MTHFD2 and the mitochondrial folate pathway in cancer. Nat. Commun. 2014, 5, 3128. [Google Scholar] [CrossRef] [Green Version]
- Yang, G.; Chang, B.; Yang, F.; Guo, X.; Cai, K.Q.; Xiao, X.S.; Wang, H.; Sen, S.; Hung, M.C.; Mills, G.B.; et al. Aurora kinase A promotes ovarian tumorigenesis through dysregulation of the cell cycle and suppression of BRCA2. Clin. Cancer Res. 2010, 16, 3171–3181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castro, P.D.; Aguirre-Portolés, C.; Fernández-Miranda, G.; Cañamero, M.; Cowley, D.O.; Dyke, T.V.; Malumbres, M. Requirements for Aurora-A in tissue regeneration and tumor development in adult mammals. Cancer Res. 2013, 73, 6804–6815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullen, P.J.; Yu, R.; Longo, J.; Archer, M.C.; Penn, L.Z. The interplay between cell signalling and the mevalonate pathway in cancer. Nat. Rev. Cancer 2016, 16, 718–731. [Google Scholar] [CrossRef] [PubMed]
- Abate, M.; Laezza, C.; Pisanti, S.; Torelli, G.; Seneca, V.; Catapano, G.; Montella, F.; Ranieri, R.; Notarnicola, M.; Gazzerro, P.; et al. Deregulated expression and activity of farnesyl diphosphate synthase (FDPS) in glioblastoma. Sci. Rep. 2017, 7, 14123. [Google Scholar] [CrossRef] [Green Version]
- Kato, K.; Cox, A.D.; Hisaka, M.M.; Graham, S.M.; Buss, J.E.; Der, C.J. Isoprenoid addition to Ras protein is the critical modification for its membrane association and transforming activity. Proc. Natl. Acad. Sci. USA 1992, 89, 6403–6407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bos, J.L. Ras oncogenes in human cancer: A review. Cancer Res. 1989, 49, 4682–4689. Available online: https://aacrjournals.org/cancerres/article/49/17/4682/494105/ras-Oncogenes-in-Human-Cancer-A-Review1 (accessed on 21 February 2015). [PubMed]
- Philips, M.R. Ras hitchhikes on PDE6δ. Nat. Cell Biol. 2012, 14, 128–129. [Google Scholar] [CrossRef] [Green Version]
- Chandra, A.; Grecco, H.E.; Pisupati, V.; Perera, D.; Cassidy, L.; Skoulidis, F.; Ismail, S.A.; Hedberg, C.; Hanzal-Bayer, M.; Venkitaraman, A.R.; et al. The GDI-like solubilizing factor PDEδ sustains the spatial organization and signalling of Ras family proteins. Nat. Cell Biol. 2011, 14, 148–158. [Google Scholar] [CrossRef]
- Iwig, J.S.; Kuriyan, J. Fixing a hole where the Ras gets in. Cell 2013, 153, 1191–1193. [Google Scholar] [CrossRef] [Green Version]
- Hanzal-Bayer, M.; Renault, L.; Roversi, P.; Wittinghofer, A.; Hillig, R.C. The complex of Arl2-GTP and PDE delta: From structure to function. EMBO J. 2002, 21, 2095–2106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nancy, V.; Callebaut, I.; El Marjou, A.; de Gunzburg, J. The delta subunit of retinal rod cGMP phosphodiesterase regulates the membrane association of Ras and Rap GTPases. J. Biol. Chem. 2002, 277, 15076–15084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siperstein, M.D. Regulation of cholesterol synthesis in normal and malignant tissue. Curr. Top. Cell Regul. 1970, 2, 65–100. [Google Scholar] [CrossRef]
- Haeffner, E.W.; Hoffmann, C.J.; Stoehr, M.; Scherf, H. Cholesterol-induced growth stimulation, cell aggregation, and membrane properties of ascites tumor cells in culture. Cancer Res. 1984, 44, 2668–2676. [Google Scholar]
- Brusselmans, K.; Timmermans, L.; Van de Sande, T.; Van Veldhoven, P.P.; Guan, G.; Shechter, I.; Claessens, F.; Verhoeven, G.; Swinnen, J.V. Squalene synthase, a determinant of Raft-associated cholesterol and modulator of cancer cell proliferation. J. Biol. Chem. 2007, 282, 18777–18785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poburski, D.; Thierbach, R. Improvement of the BALB/c-3T3 cell transformation assay: A tool for investigating cancer mechanisms and therapies. Sci. Rep. 2016, 6, 32966. [Google Scholar] [CrossRef] [PubMed]
- Mascolo, M.G.; Perdichizzi, S.; Vaccari, M.; Rotondo, F.; Zanzi, C.; Grilli, S.; Paparella, M.; Jacobs, M.J.; Colacci, A. The transformics assay: First steps for the development of an integrated approach to investigate the malignant cell transformation in vitro. Carcinogenesis 2018, 39, 955–967. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.H.; Yeom, H.; Han, B.I.; Ham, B.J.; Lee, Y.M.; Han, M.R.; Lee, M. Predicting carcinogenic mechanisms of non-genotoxic carcinogens via combined analysis of global DNA methylation and in vitro cell transformation. Int. J. Mol. Sci. 2020, 15, 5387. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, A.; Mueller, O.; Stocker, S.; Salowsky, R.; Leiber, M.; Gassmann, M.; Lightfoot, S.; Menzel, W.; Granzow, M. Ragghe, The RIN: An RNA integrity number for assigning integrity values to RNA measurements. BMC Mol. Biol. 2006, 7, 3. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; McGee, M.; Liu, Q.; Scheuermann, R.H. A distribution free summarization method for Affymetrix GeneChip arrays. Bioinformatics 2007, 23, 321–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Foundation for Statistical Computing. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2006; Available online: https://cran-archive.r-project.org/bin/windows/base/old/2.7.1/ (accessed on 21 February 2015).
- Gentleman, R.C.; Gentleman, R.C.; Carey, V.J.; Bates, D.M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; et al. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004, 5, R80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, R.; Shimodaira, H. Pvclust: An R package for assessing the uncertainty in hierarchical clustering. Bioinformatics 2006, 22, 1540–1542. [Google Scholar] [CrossRef]
- Breitling, R.; Armengaud, P.; Amtmann, A.; Herzyk, P. Rank products: A simple, yet powerful, new method to detect differentially regulated genes in replicated microarray experiments. FEBS Lett. 2004, 573, 83–92. [Google Scholar] [CrossRef]
- Dennis, G., Jr.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003, 4, R60. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Hosack, D.A.; Dennis, G., Jr.; Sherman, B.T.; Lane, H.C.; Lempicki, R.A. Identifying biological themes within lists of genes with EASE. Genome Biol. 2003, 4, R70. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. 1995, 57, 289–300. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hallmark | Pathway | 1 H | 6 H | 24 H | 8 Days |
|---|---|---|---|---|---|
| Deregulated Cellular Metabolism | Aryl Hydrocarbon Receptor Signaling | ↑CYP1A1, ↑CYP1B1 | |||
| Evading Anti-growth Signaling | HIPPO Signaling | ↓LATS1/2 | ↓LATS1/2, ↑YAP/TAZ, ↑TEAD | ||
| Gap Junction Signaling | ↓CONNEXIN | ||||
| Resisting Programmed Cell Death | Apoptosis Signaling | ↑BIRC6 | ↑CASP9, ↑,CASP3 | ↓CASP6,↓clAP, ↑PARP,↑BIRC6 | ↓GAS2, ↓clAP |
| Avoiding Immune Destruction | Interferon Signaling | ↑IFNgRb,↑JAK2 | ↑STAT2 | ||
| PD-1, PDL-1 cancer immunotherapy pathway | ↓ TGF-β | ↑CBLB,↑TGF-β ↑TNFR, ↑IFNγR2, ↑IL2R | ↓CBLB, ↓PTEN | ↓CDKN1B(P27kip),↓PDCD4,↓TGF-β | |
| Tumor-Promoting Inflammation | IL-1 Signaling | ↑IL1RAP, ↑TOLLIP | |||
| IL-2 Signaling | ↑IL2Rg, ↑STAT5 | ||||
| IL-6 Signaling | ↑IL-6R,↑JAK2, ↑STAT3 | ||||
| TNFR2 Signaling | ↑TNFR2 | ||||
| Tumor microenvironment | JAK/STAT signaling | ↑JAK2,↑STAT3 | |||
| Integrin signaling | ↑PDGFβ,↑MLK3, ↑PARVIN-β, ↑PXN,↑NEDD9, ↑FYN | ||||
| Tissue Invasion and Metastasis | Glioma Invasiveness Signaling | ↑CD44,↑MMP9 | ↑RAMM, ↑UPAR | ||
| Sustained Growth Signaling | Aryl Hydrocarbon Receptor Signaling | ↑AHR | |||
| Cell Cycle: G1/S Checkpoint Regulation | ↑CDK4/6, ↑CDC25A, ↑Cyclin D, ↑c-MYC, ↑Cyclin E,↑RB, ↑DP-1,↑E2F | ||||
| Cell Cycle: G2/M DNA Damage Checkpoint Regulation | ↑CDC2, ↑Cyclin B, ↑CKS1 | ||||
| VEGF Signaling | ↑VEGF,↑EIF, ↑RAS | ||||
| mTOR Signaling | ↑PPA2,↑elF4E, ↑elF4G,↑elF3, ↑40S,↑Ribosome | ||||
| Genetic Instability | DNA Methylation and Transcriptional Repression Signaling | ↑DNMT3A | ↑DNMT1A, ↑MBD3,↑Mi2 | ↑DNMT1A, ↑SAP30 | |
| Role of BRCA1 in DNA Damage Response | ↑BRCA1 | ||||
| Telomerase Signaling | ↑DKC1 | ||||
| Mismatch Repair in Eukaryotes | ↑Exo1,↑FEN1, ↑Polδ,↑PCNA, ↑MSH6,↑RPA | ||||
| Mitocondrial Dysfunction | ↑CYTC,↑GSR, ↑HtrA2, ↑ATP5G1 | ||||
| Role of CHK Proteins in Cell Cycle Checkpoint Control | ↑Chk1 | ↑Chk1 | |||
| Enabled Replication Immortality | Telomerase Signaling | ↑DKC1,↑HSP90, ↑p23 | |||
| Inducing New Blood Flow | VEGF Signaling | ↑VEGF,↑EIF, ↑RAS | |||
| mTOR Signaling | ↑PPA2,↑elF4E, ↑elF4G,↑elF3, ↑40S,↑Ribosome | ||||
| Deregulated Cellular Metabolism | Folate Biosynthesis | ↑DHFR,↑GART, ↑MTHFD1, ↑MTHFD1L, ↑MTHFD2, ↑PHGDH, ↑PSAT1,↑PSPH,↑SHMT1, ↑SHMT2,↑TYMS | ↑GART, ↑MTHFD1, ↑SHMT2,↑TYMS | ||
| Cholesterol Biosynthesis | ↑ACAT2,↑FDPS, ↑FPS,↑LBR | ↑DHCR24, ↑FDFT1,↑FDPS, ↑FPS,↑HMGCS2,↑IDI1,↑MVD, ↑SQLE |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ohmori, K.; Kamei, A.; Watanabe, Y.; Abe, K. Gene Expression over Time during Cell Transformation Due to Non-Genotoxic Carcinogen Treatment of Bhas 42 Cells. Int. J. Mol. Sci. 2022, 23, 3216. https://doi.org/10.3390/ijms23063216
Ohmori K, Kamei A, Watanabe Y, Abe K. Gene Expression over Time during Cell Transformation Due to Non-Genotoxic Carcinogen Treatment of Bhas 42 Cells. International Journal of Molecular Sciences. 2022; 23(6):3216. https://doi.org/10.3390/ijms23063216
Chicago/Turabian StyleOhmori, Kiyomi, Asuka Kamei, Yuki Watanabe, and Keiko Abe. 2022. "Gene Expression over Time during Cell Transformation Due to Non-Genotoxic Carcinogen Treatment of Bhas 42 Cells" International Journal of Molecular Sciences 23, no. 6: 3216. https://doi.org/10.3390/ijms23063216
APA StyleOhmori, K., Kamei, A., Watanabe, Y., & Abe, K. (2022). Gene Expression over Time during Cell Transformation Due to Non-Genotoxic Carcinogen Treatment of Bhas 42 Cells. International Journal of Molecular Sciences, 23(6), 3216. https://doi.org/10.3390/ijms23063216