
Subtype of Neuroblastoma Cells with High KIT Expression Are Dependent on KIT and Its Knockdown Induces Compensatory Activation of Pro-Survival Signaling


 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
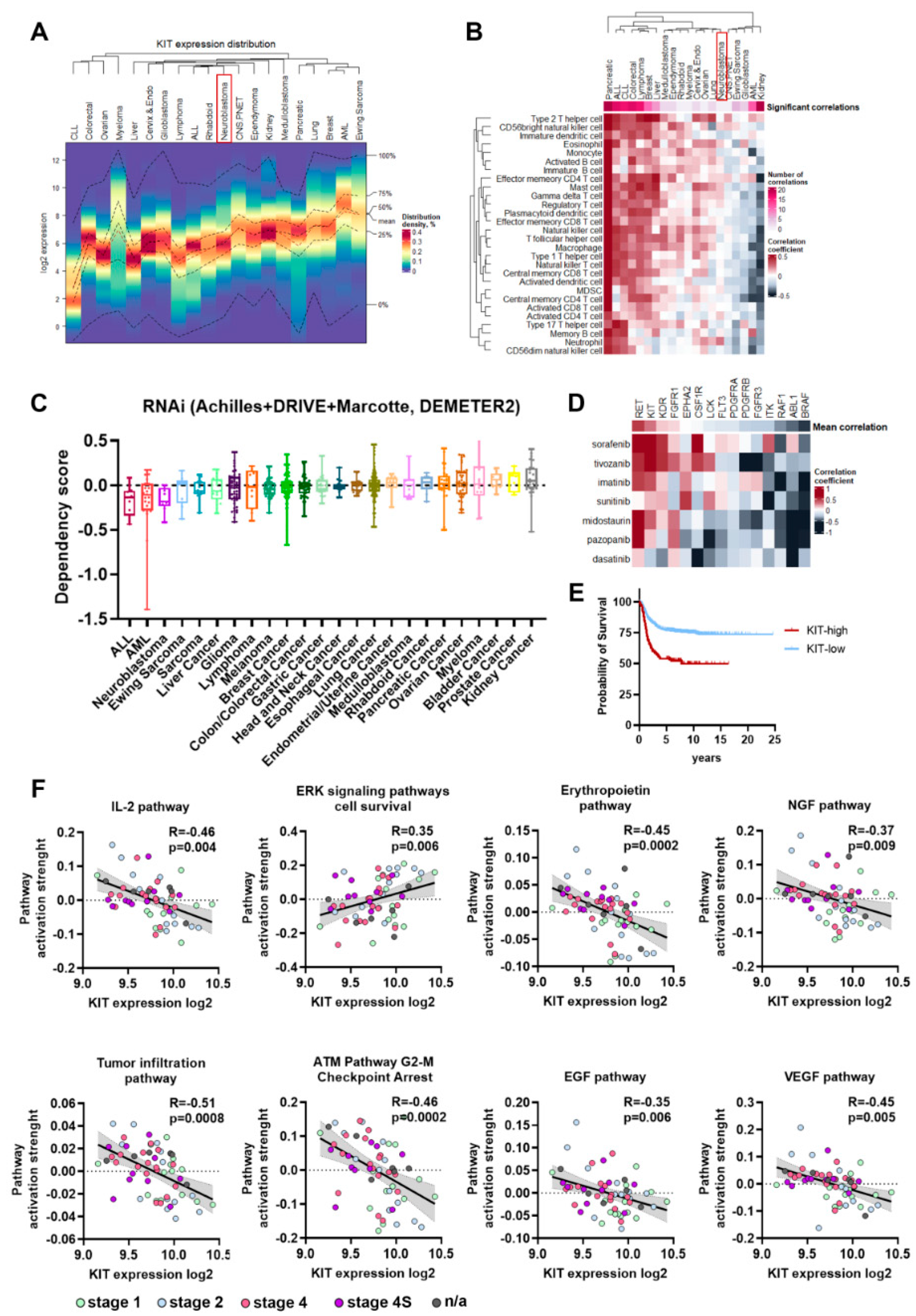
2.1. KIT Expression Hallmarks in NB
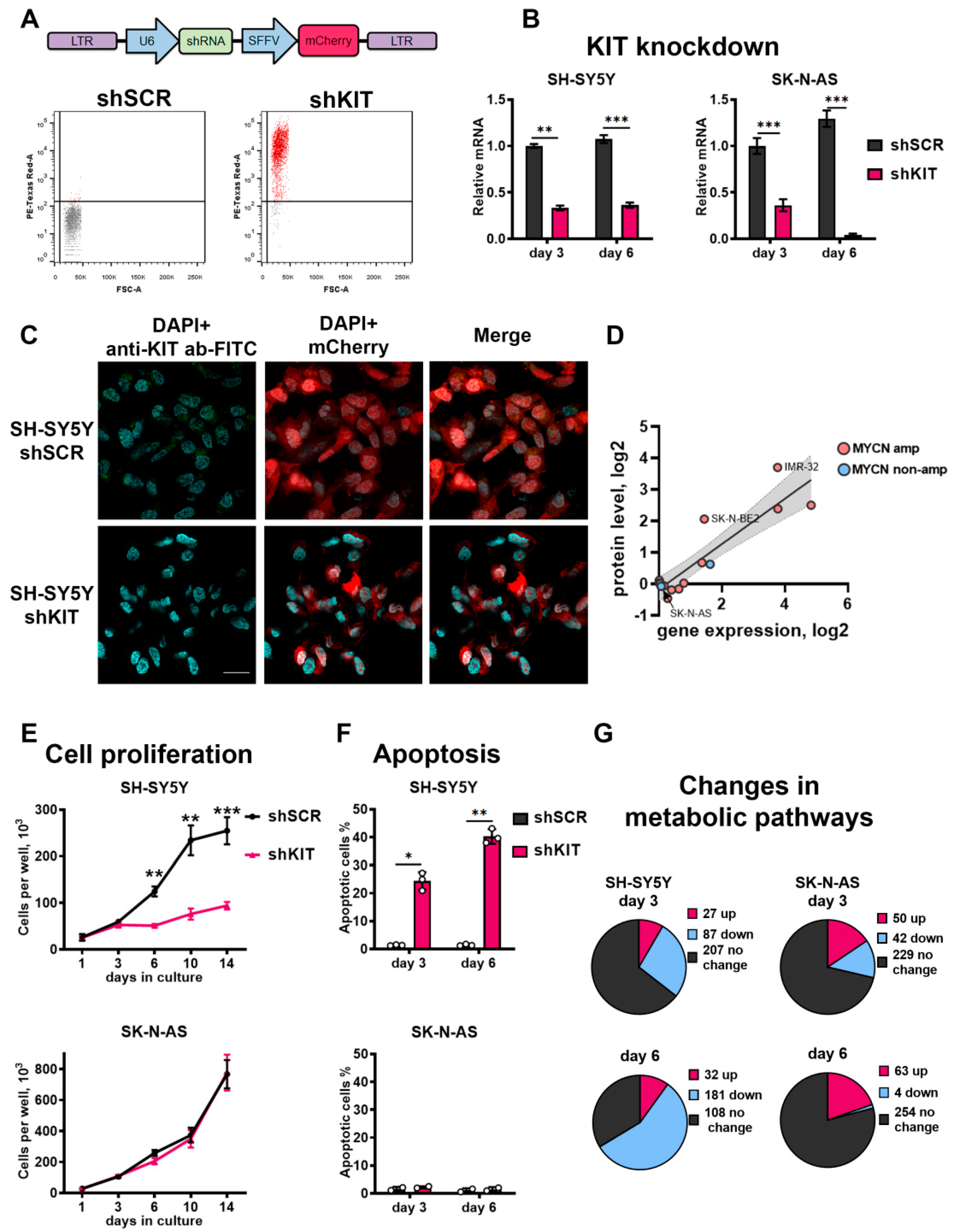
2.2. KIT Knockdown Induces Cell Death via Increased Apoptosis
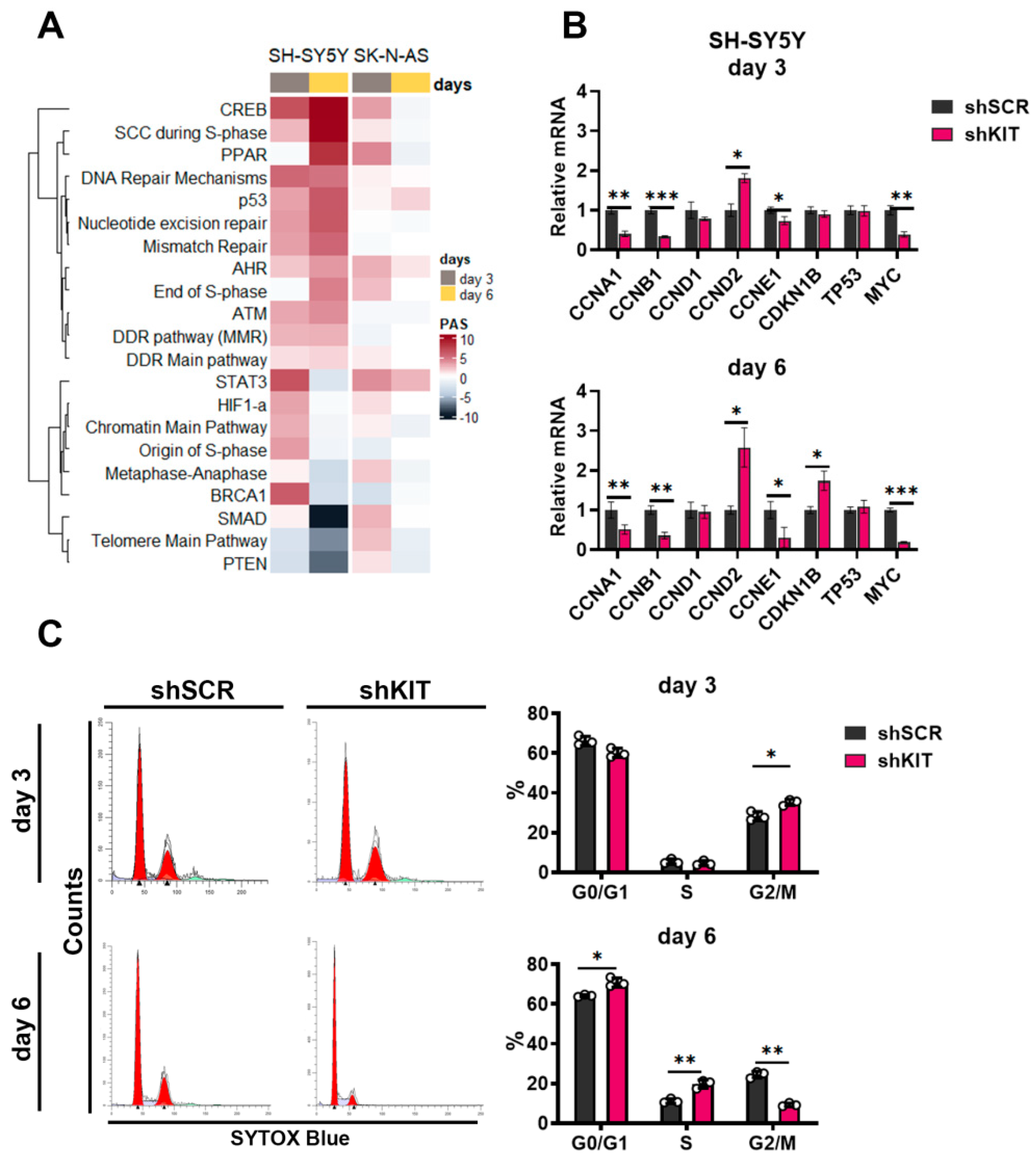
2.3. KIT Knockdown in SH-SY5Y Cells Induces Cell Cycle Arrest
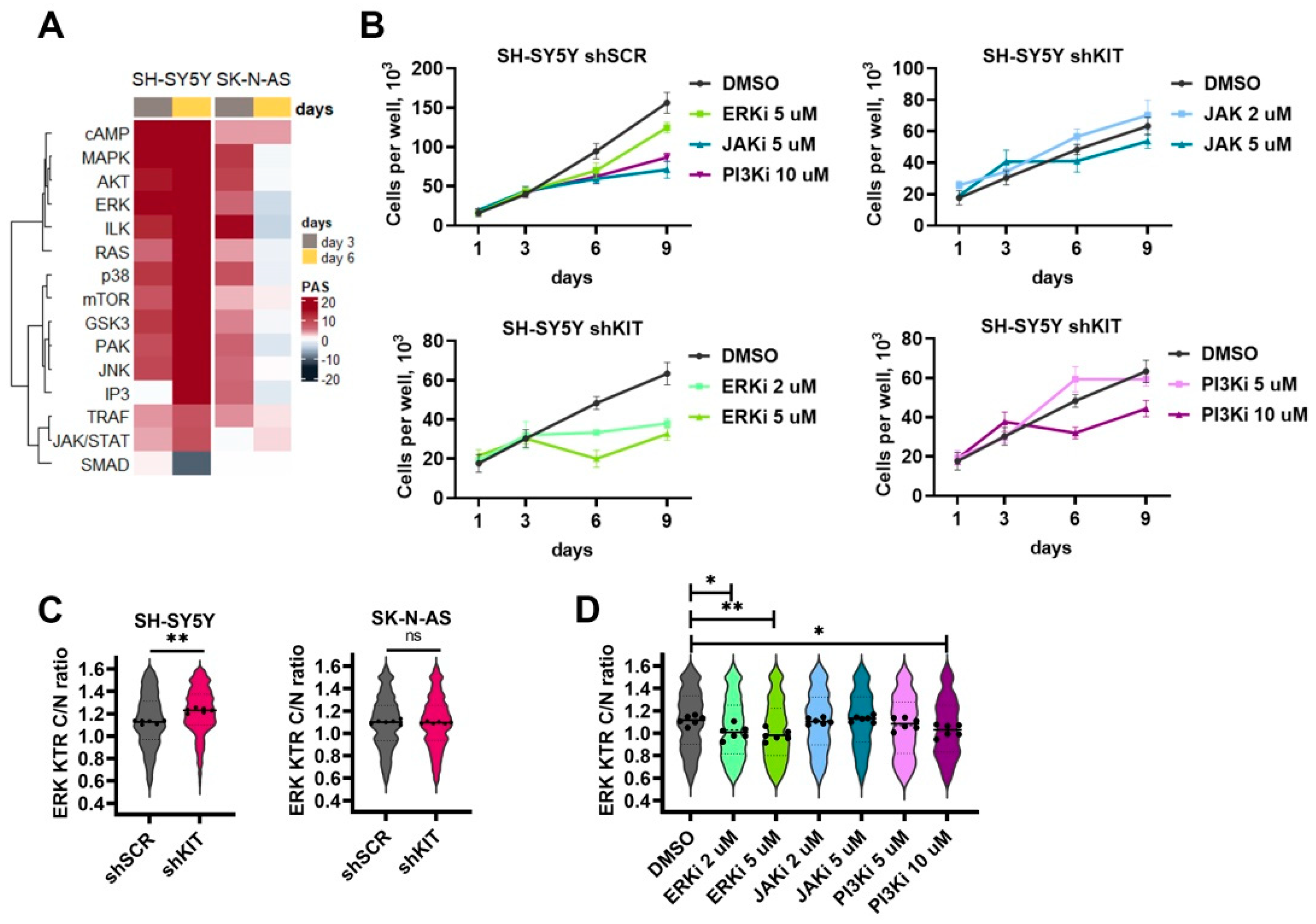
2.4. ERK Pathway Activation Is Essential for NB Cell Survival after KIT Knockdown
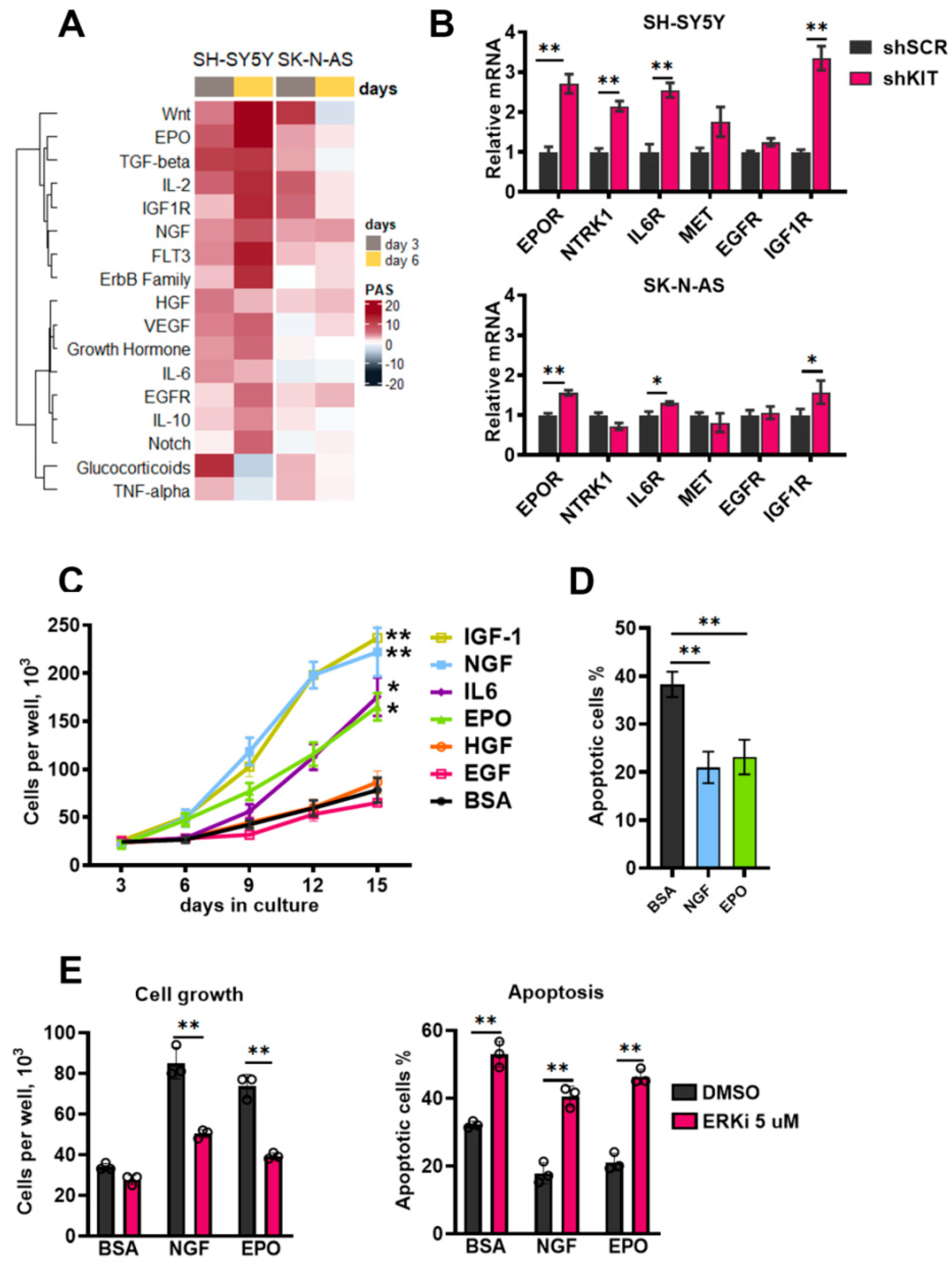
2.5. Compensatory Activation of Growth Factor Signaling Rescues Cells from KIT Knockdown
3. Discussion
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shohet, J.; Foster, J. Neuroblastoma. BMJ 2017, 357, j1863. [Google Scholar] [CrossRef] [PubMed]
- Yalcin, B.; Kremer, L.C.; van Dalen, E.C. High-dose chemotherapy and autologous haematopoietic stem cell rescue for children with high-risk neuroblastoma. Cochrane Database Syst. Rev. 2015, 10, CD006301. [Google Scholar] [CrossRef]
- Cole, K.A.; Maris, J.M. New strategies in refractory and recurrent neuroblastoma: Translational opportunities to impact patient outcome. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 2423–2428. [Google Scholar] [CrossRef] [Green Version]
- Garaventa, A.; Parodi, S.; De Bernardi, B.; Dau, D.; Manzitti, C.; Conte, M.; Casale, F.; Viscardi, E.; Bianchi, M.; D’Angelo, P.; et al. Outcome of children with neuroblastoma after progression or relapse. A retrospective study of the Italian neuroblastoma registry. Eur. J. Cancer 2009, 45, 2835–2842. [Google Scholar] [CrossRef]
- Cohen, P.S.; Chan, J.P.; Lipkunskaya, M.; Biedler, J.L.; Seeger, R.C. Expression of stem cell factor and c-kit in human neuroblastoma. The Children′s Cancer Group. Blood 1994, 84, 3465–3472. [Google Scholar] [CrossRef] [Green Version]
- Lau, S.T.; Hansford, L.M.; Chan, W.K.; Chan, G.C.; Wan, T.S.; Wong, K.K.; Kaplan, D.R.; Tam, P.K.; Ngan, E.S. Prokineticin signaling is required for the maintenance of a de novo population of c-KIT(+) cells to sustain neuroblastoma progression. Oncogene 2015, 34, 1019–1034. [Google Scholar] [CrossRef]
- Lebedev, T.D.; Vagapova, E.R.; Popenko, V.I.; Leonova, O.G.; Spirin, P.V.; Prassolov, V.S. Two Receptors, Two Isoforms, Two Cancers: Comprehensive Analysis of KIT and TrkA Expression in Neuroblastoma and Acute Myeloid Leukemia. Front. Oncol. 2019, 9, 1046. [Google Scholar] [CrossRef] [PubMed]
- Uccini, S.; Mannarino, O.; McDowell, H.P.; Pauser, U.; Vitali, R.; Natali, P.G.; Altavista, P.; Andreano, T.; Coco, S.; Boldrini, R.; et al. Clinical and molecular evidence for c-kit receptor as a therapeutic target in neuroblastic tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2005, 11, 380–389. [Google Scholar] [CrossRef]
- Vitali, R.; Cesi, V.; Nicotra, M.R.; McDowell, H.P.; Donfrancesco, A.; Mannarino, O.; Natali, P.G.; Raschella, G.; Dominici, C. c-Kit is preferentially expressed in MYCN-amplified neuroblastoma and its effect on cell proliferation is inhibited in vitro by STI-571. Int. J. Cancer 2003, 106, 147–152. [Google Scholar] [CrossRef]
- Backman, U.; Christofferson, R. The selective class III/V receptor tyrosine kinase inhibitor SU11657 inhibits tumor growth and angiogenesis in experimental neuroblastomas grown in mice. Pediatric Res. 2005, 57, 690–695. [Google Scholar] [CrossRef] [Green Version]
- Moawad, E.Y. Predicting Effectiveness of Imatinib Mesylate in Tumors Expressing Platelet-Derived Growth Factors (PDGF-AA, PDGF-BB), Stem Cell Factor Ligands and Their Respective Receptors (PDGFR-alpha, PDGFR-beta, and c-kit). J. Gastrointest. Cancer 2015, 46, 272–283. [Google Scholar] [CrossRef] [PubMed]
- Neumann, I.; Foell, J.L.; Bremer, M.; Volkmer, I.; Korholz, D.; Burdach, S.; Staege, M.S. Retinoic acid enhances sensitivity of neuroblastoma cells for imatinib mesylate. Pediatric Blood Cancer 2010, 55, 464–470. [Google Scholar] [CrossRef] [PubMed]
- Lebedev, T.D.; Spirin, P.V.; Suntsova, M.V.; Ivanova, A.V.; Buzdin, A.A.; Prokofjeva, M.M.; Rubtsov, P.M.; Prassolov, V.S. Receptor tyrosine kinase KIT may regulate expression of genes involved in spontaneous regression of neuroblastoma. Mol. Biol. 2015, 49, 1052–1055. [Google Scholar] [CrossRef]
- Hirschmann-Jax, C.; Foster, A.E.; Wulf, G.G.; Nuchtern, J.G.; Jax, T.W.; Gobel, U.; Goodell, M.A.; Brenner, M.K. A distinct "side population" of cells with high drug efflux capacity in human tumor cells. Proc. Natl. Acad. Sci. USA 2004, 101, 14228–14233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walton, J.D.; Kattan, D.R.; Thomas, S.K.; Spengler, B.A.; Guo, H.F.; Biedler, J.L.; Cheung, N.K.; Ross, R.A. Characteristics of stem cells from human neuroblastoma cell lines and in tumors. Neoplasia 2004, 6, 838–845. [Google Scholar] [CrossRef] [Green Version]
- Ross, R.A.; Walton, J.D.; Han, D.; Guo, H.F.; Cheung, N.K. A distinct gene expression signature characterizes human neuroblastoma cancer stem cells. Stem Cell Res. 2015, 15, 419–426. [Google Scholar] [CrossRef] [Green Version]
- Vitali, R.; Mancini, C.; Cesi, V.; Tanno, B.; Piscitelli, M.; Mancuso, M.; Sesti, F.; Pasquali, E.; Calabretta, B.; Dominici, C.; et al. Activity of tyrosine kinase inhibitor Dasatinib in neuroblastoma cells in vitro and in orthotopic mouse model. Int. J. Cancer 2009, 125, 2547–2555. [Google Scholar] [CrossRef]
- Ricotti, E.; Fagioli, F.; Garelli, E.; Linari, C.; Crescenzio, N.; Horenstein, A.L.; Pistamiglio, P.; Vai, S.; Berger, M.; di Montezemolo, L.C.; et al. c-kit is expressed in soft tissue sarcoma of neuroectodermic origin and its ligand prevents apoptosis of neoplastic cells. Blood 1998, 91, 2397–2405. [Google Scholar] [CrossRef]
- Choudhary, S.; Pardo, A.; Rosinke, R.; Batra, J.K.; Barth, S.; Verma, R.S. Targeting c-kit receptor in neuroblastomas and colorectal cancers using stem cell factor (SCF)-based recombinant bacterial toxins. Appl. Microbiol. Biotechnol. 2016, 100, 263–277. [Google Scholar] [CrossRef]
- Abbaspour Babaei, M.; Kamalidehghan, B.; Saleem, M.; Huri, H.Z.; Ahmadipour, F. Receptor tyrosine kinase (c-Kit) inhibitors: A potential therapeutic target in cancer cells. Drug Des. Dev. Ther. 2016, 10, 2443–2459. [Google Scholar] [CrossRef] [Green Version]
- Lebedev, T.; Vagapova, E.; Spirin, P.; Rubtsov, P.; Astashkova, O.; Mikheeva, A.; Sorokin, M.; Vladimirova, U.; Suntsova, M.; Konovalov, D.; et al. Growth factor signaling predicts therapy resistance mechanisms and defines neuroblastoma subtypes. Oncogene 2021, 40, 6258–6272. [Google Scholar] [CrossRef]
- Charoentong, P.; Finotello, F.; Angelova, M.; Mayer, C.; Efremova, M.; Rieder, D.; Hackl, H.; Trajanoski, Z. Pan-cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade. Cell Rep. 2017, 18, 248–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McFarland, J.M.; Ho, Z.V.; Kugener, G.; Dempster, J.M.; Montgomery, P.G.; Bryan, J.G.; Krill-Burger, J.M.; Green, T.M.; Vazquez, F.; Boehm, J.S.; et al. Improved estimation of cancer dependencies from large-scale RNAi screens using model-based normalization and data integration. Nat. Commun. 2018, 9, 4610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, E.R., 3rd; de Weck, A.; Schlabach, M.R.; Billy, E.; Mavrakis, K.J.; Hoffman, G.R.; Belur, D.; Castelletti, D.; Frias, E.; Gampa, K.; et al. Project DRIVE: A Compendium of Cancer Dependencies and Synthetic Lethal Relationships Uncovered by Large-Scale, Deep RNAi Screening. Cell 2017, 170, 577–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behan, F.M.; Iorio, F.; Picco, G.; Goncalves, E.; Beaver, C.M.; Migliardi, G.; Santos, R.; Rao, Y.; Sassi, F.; Pinnelli, M.; et al. Prioritization of cancer therapeutic targets using CRISPR-Cas9 screens. Nature 2019, 568, 511–516. [Google Scholar] [CrossRef]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrian-Uhalte, E.; et al. The ChEMBL database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef]
- Yu, C.; Mannan, A.M.; Yvone, G.M.; Ross, K.N.; Zhang, Y.L.; Marton, M.A.; Taylor, B.R.; Crenshaw, A.; Gould, J.Z.; Tamayo, P.; et al. High-throughput identification of genotype-specific cancer vulnerabilities in mixtures of barcoded tumor cell lines. Nat. Biotechnol. 2016, 34, 419–423. [Google Scholar] [CrossRef]
- Borisov, N.; Sorokin, M.; Garazha, A.; Buzdin, A. Quantitation of Molecular Pathway Activation Using RNA Sequencing Data. Methods Mol. Biol. 2020, 2063, 189–206. [Google Scholar] [CrossRef]
- Petrov, I.; Suntsova, M.; Ilnitskaya, E.; Roumiantsev, S.; Sorokin, M.; Garazha, A.; Spirin, P.; Lebedev, T.; Gaifullin, N.; Larin, S.; et al. Gene expression and molecular pathway activation signatures of MYCN-amplified neuroblastomas. Oncotarget 2017, 8, 83768–83780. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Zhao, W.; Akbani, R.; Liu, W.; Ju, Z.; Ling, S.; Vellano, C.P.; Roebuck, P.; Yu, Q.; Eterovic, A.K.; et al. Characterization of Human Cancer Cell Lines by Reverse-phase Protein Arrays. Cancer Cell 2017, 31, 225–239. [Google Scholar] [CrossRef] [Green Version]
- Dang, C.V. c-Myc target genes involved in cell growth, apoptosis, and metabolism. Mol. Cell. Biol. 1999, 19, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Delisle, L.; Pierre-Eugene, C.; Louis-Brennetot, C.; Surdez, D.; Raynal, V.; Baulande, S.; Boeva, V.; Grossetete-Lalami, S.; Combaret, V.; Peuchmaur, M.; et al. Activated ALK signals through the ERK-ETV5-RET pathway to drive neuroblastoma oncogenesis. Oncogene 2018, 37, 1417–1429. [Google Scholar] [CrossRef] [Green Version]
- Vagapova, E.; Kozlov, M.; Lebedev, T.; Ivanenko, K.; Leonova, O.; Popenko, V.; Spirin, P.; Kochetkov, S.; Prassolov, V. Selective Inhibition of HDAC Class I Sensitizes Leukemia and Neuroblastoma Cells to Anticancer Drugs. Biomedicines 2021, 9, 1846. [Google Scholar] [CrossRef]
- Yan, S.; Li, Z.; Thiele, C.J. Inhibition of STAT3 with orally active JAK inhibitor, AZD1480, decreases tumor growth in Neuroblastoma and Pediatric Sarcomas In vitro and In vivo. Oncotarget 2013, 4, 433–445. [Google Scholar] [CrossRef] [Green Version]
- Lebedev, T.D.; Vagapova, E.R.; Astashkova, O.O.; Spirin, P.V.; Prassolov, V.S. Inhibition of Non-Receptor Tyrosine Kinase JAK2 Reduces Neuroblastoma Cell Growth and Enhances the Action of Doxorubicin. Mol. Biol. 2020, 54, 293–299. [Google Scholar] [CrossRef]
- Chilamakuri, R.; Rouse, D.C.; Yu, Y.; Kabir, A.S.; Muth, A.; Yang, J.; Lipton, J.M.; Agarwal, S. BX-795 inhibits neuroblastoma growth and enhances sensitivity towards chemotherapy. Transl. Oncol. 2022, 15, 101272. [Google Scholar] [CrossRef]
- Lebedev, T.D.; Vagapova, E.R.; Prassolov, V.S. The Different Impact of ERK Inhibition on Neuroblastoma, Astrocytoma, and Rhabdomyosarcoma Cell Differentiation. Acta Nat. 2021, 13, 69–77. [Google Scholar] [CrossRef]
- Regot, S.; Hughey, J.J.; Bajar, B.T.; Carrasco, S.; Covert, M.W. High-sensitivity measurements of multiple kinase activities in live single cells. Cell 2014, 157, 1724–1734. [Google Scholar] [CrossRef] [Green Version]
- Enguita-German, M.; Gurrea, M.; Schiapparelli, P.; Zhu, T.S.; Crowley, J.G.; Hamm, L.L.; Costello, M.A.; He, X.; Talsma, C.E.; Flack, C.G.; et al. KIT expression and methylation in medulloblastoma and PNET cell lines and tumors. J. Neuro-Oncol. 2011, 103, 247–253. [Google Scholar] [CrossRef]
- Gonzalez, I.; Andreu, E.J.; Panizo, A.; Inoges, S.; Fontalba, A.; Fernandez-Luna, J.L.; Gaboli, M.; Sierrasesumaga, L.; Martin-Algarra, S.; Pardo, J.; et al. Imatinib inhibits proliferation of Ewing tumor cells mediated by the stem cell factor/KIT receptor pathway, and sensitizes cells to vincristine and doxorubicin-induced apoptosis. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2004, 10, 751–761. [Google Scholar] [CrossRef] [Green Version]
- Ergun, S.; Altay, D.U.; Gunes, S.; Buyukalpelli, R.; Karahan, S.C.; Tomak, L.; Abur, U. Tr-KIT/c-KIT ratio in renal cell carcinoma. Mol. Biol. Rep. 2019, 46, 5287–5294. [Google Scholar] [CrossRef]
- Shimada, A.; Hirato, J.; Kuroiwa, M.; Kikuchi, A.; Hanada, R.; Wakai, K.; Hayashi, Y. Expression of KIT and PDGFR is associated with a good prognosis in neuroblastoma. Pediatric Blood Cancer 2008, 50, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Ootsuka, S.; Asami, S.; Sasaki, T.; Yoshida, Y.; Nemoto, N.; Shichino, H.; Chin, M.; Mugishima, H.; Suzuki, T. Analyses of novel prognostic factors in neuroblastoma patients. Biol. Pharm. Bull. 2007, 30, 2294–2299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krams, M.; Parwaresch, R.; Sipos, B.; Heidorn, K.; Harms, D.; Rudolph, P. Expression of the c-kit receptor characterizes a subset of neuroblastomas with favorable prognosis. Oncogene 2004, 23, 588–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, S.; Zheng, J.; Zhu, H.; Yang, L.; Xiao, X. Identification of neuroblastoma stem cells by characterization of side population cells in the human neuroblastoma SK-N-SH cell line. J. Pediatric Surg. 2010, 45, 2305–2311. [Google Scholar] [CrossRef]
- Yu, U.Y.; Cha, J.E.; Ju, S.Y.; Cho, K.A.; Yoo, E.S.; Ryu, K.H.; Woo, S.Y. Effect on cell cycle progression by N-Myc knockdown in SK-N-BE(2) neuroblastoma cell line and cytotoxicity with STI-571 compound. Cancer Res. Treat. 2008, 40, 27–32. [Google Scholar] [CrossRef] [Green Version]
- Munugalavadla, V.; Kapur, R. Role of c-Kit and erythropoietin receptor in erythropoiesis. Crit. Rev. Oncol. Hematol. 2005, 54, 63–75. [Google Scholar] [CrossRef]
- Aguilar, C.; Lopez-Marure, R.; Jimenez-Sanchez, A.; Rocha-Zavaleta, L. Co-stimulation with stem cell factor and erythropoietin enhances migration of c-Kit expressing cervical cancer cells through the sustained activation of ERK1/2. Mol. Med. Rep. 2014, 9, 1895–1902. [Google Scholar] [CrossRef]
- Wiedenmann, T.; Ehrhardt, S.; Cerny, D.; Hildebrand, D.; Klein, S.; Heeg, K.; Kubatzky, K.F. Erythropoietin acts as an anti-inflammatory signal on murine mast cells. Mol. Immunol. 2015, 65, 68–76. [Google Scholar] [CrossRef]
- Kosmider, O.; Buet, D.; Gallais, I.; Denis, N.; Moreau-Gachelin, F. Erythropoietin down-regulates stem cell factor receptor (Kit) expression in the leukemic proerythroblast: Role of Lyn kinase. PLoS ONE 2009, 4, e5721. [Google Scholar] [CrossRef] [Green Version]
- Mirkina, I.; Hadzijusufovic, E.; Krepler, C.; Mikula, M.; Mechtcheriakova, D.; Strommer, S.; Stella, A.; Jensen-Jarolim, E.; Holler, C.; Wacheck, V.; et al. Phenotyping of human melanoma cells reveals a unique composition of receptor targets and a subpopulation co-expressing ErbB4, EPO-R and NGF-R. PLoS ONE 2014, 9, e84417. [Google Scholar] [CrossRef] [Green Version]
- Duncan, J.S.; Whittle, M.C.; Nakamura, K.; Abell, A.N.; Midland, A.A.; Zawistowski, J.S.; Johnson, N.L.; Granger, D.A.; Jordan, N.V.; Darr, D.B.; et al. Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer. Cell 2012, 149, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Marampon, F.; Ciccarelli, C.; Zani, B.M. Down-regulation of c-Myc following MEK/ERK inhibition halts the expression of malignant phenotype in rhabdomyosarcoma and in non muscle-derived human tumors. Mol. Cancer 2006, 5, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chappell, J.; Sun, Y.; Singh, A.; Dalton, S. MYC/MAX control ERK signaling and pluripotency by regulation of dual-specificity phosphatases 2 and 7. Genes Dev. 2013, 27, 725–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, K.; Bartsch, U.; Stocking, C.; Fehse, B. A multicolor panel of novel lentiviral “gene ontology” (LeGO) vectors for functional gene analysis. Mol. Ther. J. Am. Soc. Gene Ther. 2008, 16, 698–706. [Google Scholar] [CrossRef] [PubMed]
- Spirin, P.V.; Lebedev, T.D.; Orlova, N.N.; Gornostaeva, A.S.; Prokofjeva, M.M.; Nikitenko, N.A.; Dmitriev, S.E.; Buzdin, A.A.; Borisov, N.M.; Aliper, A.M.; et al. Silencing AML1-ETO gene expression leads to simultaneous activation of both pro-apoptotic and proliferation signaling. Leukemia 2014, 28, 2222–2228. [Google Scholar] [CrossRef] [PubMed]
- Spirin, P.V.; Nikitenko, N.A.; Lebedev, T.D.; Rubtsov, P.M.; Stocking, C.; Prasolov, V.S. Modulation of activated oncogene c-kit expression with RNA-interference. Mol. Biol. 2011, 45, 1036–1045. [Google Scholar] [CrossRef]
- Bolstad, B.M.; Irizarry, R.A.; Astrand, M.; Speed, T.P. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 2003, 19, 185–193. [Google Scholar] [CrossRef] [Green Version]
- Sorokin, M.; Borisov, N.; Kuzmin, D.; Gudkov, A.; Zolotovskaia, M.; Garazha, A.; Buzdin, A. Algorithmic Annotation of Functional Roles for Components of 3044 Human Molecular Pathways. Front. Genet. 2021, 12, 617059. [Google Scholar] [CrossRef]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [Green Version]
- Kamentsky, L.; Jones, T.R.; Fraser, A.; Bray, M.A.; Logan, D.J.; Madden, K.L.; Ljosa, V.; Rueden, C.; Eliceiri, K.W.; Carpenter, A.E. Improved structure, function and compatibility for CellProfiler: Modular high-throughput image analysis software. Bioinformatics 2011, 27, 1179–1180. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lebedev, T.; Buzdin, A.; Khabusheva, E.; Spirin, P.; Suntsova, M.; Sorokin, M.; Popenko, V.; Rubtsov, P.; Prassolov, V. Subtype of Neuroblastoma Cells with High KIT Expression Are Dependent on KIT and Its Knockdown Induces Compensatory Activation of Pro-Survival Signaling. Int. J. Mol. Sci. 2022, 23, 7724. https://doi.org/10.3390/ijms23147724
Lebedev T, Buzdin A, Khabusheva E, Spirin P, Suntsova M, Sorokin M, Popenko V, Rubtsov P, Prassolov V. Subtype of Neuroblastoma Cells with High KIT Expression Are Dependent on KIT and Its Knockdown Induces Compensatory Activation of Pro-Survival Signaling. International Journal of Molecular Sciences. 2022; 23(14):7724. https://doi.org/10.3390/ijms23147724
Chicago/Turabian StyleLebedev, Timofey, Anton Buzdin, Elmira Khabusheva, Pavel Spirin, Maria Suntsova, Maxim Sorokin, Vladimir Popenko, Petr Rubtsov, and Vladimir Prassolov. 2022. "Subtype of Neuroblastoma Cells with High KIT Expression Are Dependent on KIT and Its Knockdown Induces Compensatory Activation of Pro-Survival Signaling" International Journal of Molecular Sciences 23, no. 14: 7724. https://doi.org/10.3390/ijms23147724
APA StyleLebedev, T., Buzdin, A., Khabusheva, E., Spirin, P., Suntsova, M., Sorokin, M., Popenko, V., Rubtsov, P., & Prassolov, V. (2022). Subtype of Neuroblastoma Cells with High KIT Expression Are Dependent on KIT and Its Knockdown Induces Compensatory Activation of Pro-Survival Signaling. International Journal of Molecular Sciences, 23(14), 7724. https://doi.org/10.3390/ijms23147724