
The Conservation of Long Intergenic Non-Coding RNAs and Their Response to Verticillium dahliae Infection in Cotton




Abstract
:1. Introduction
2. Results
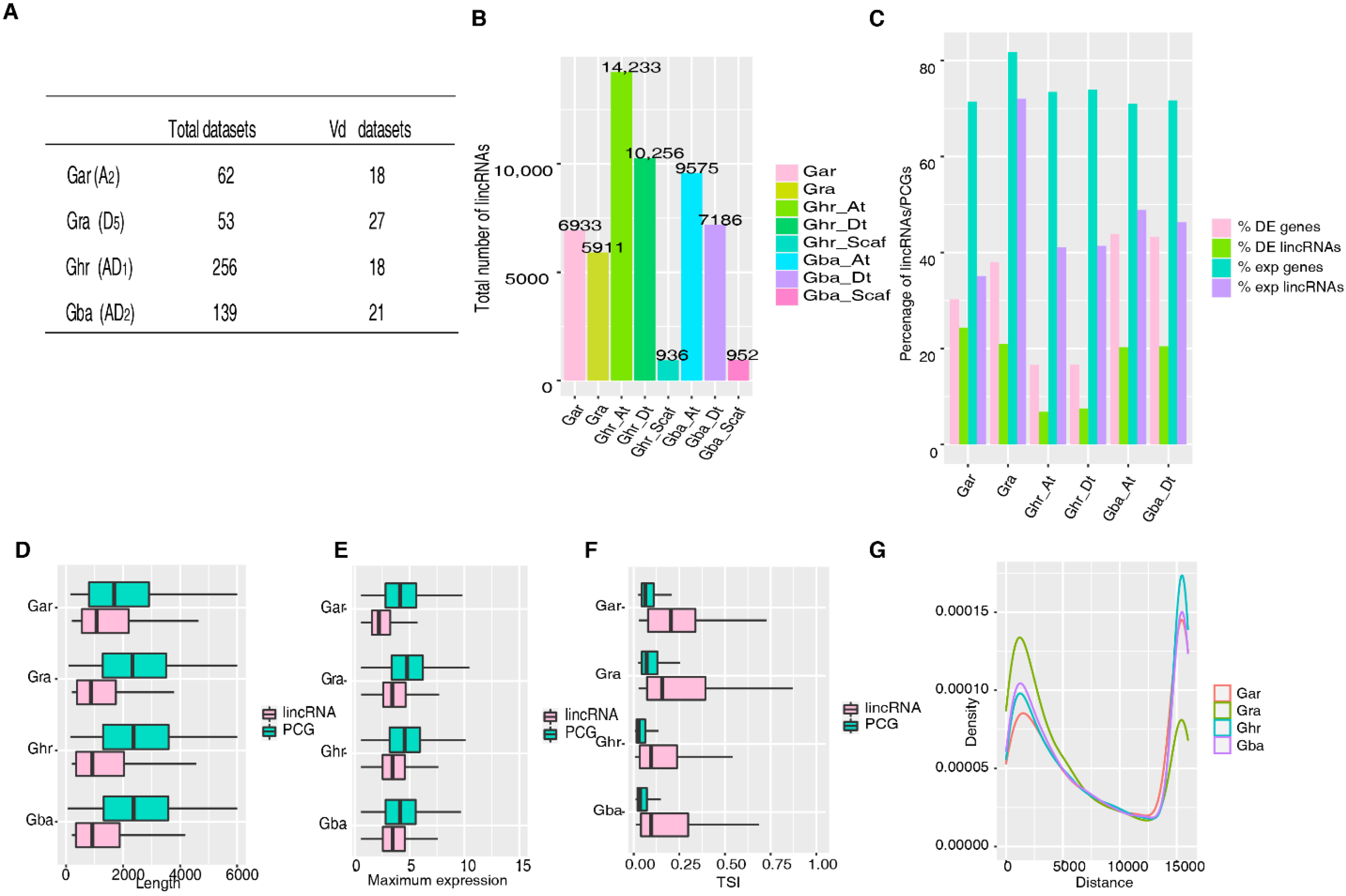
2.1. Identification of lincRNAs in Diploid and Allotetraploid Cotton Species
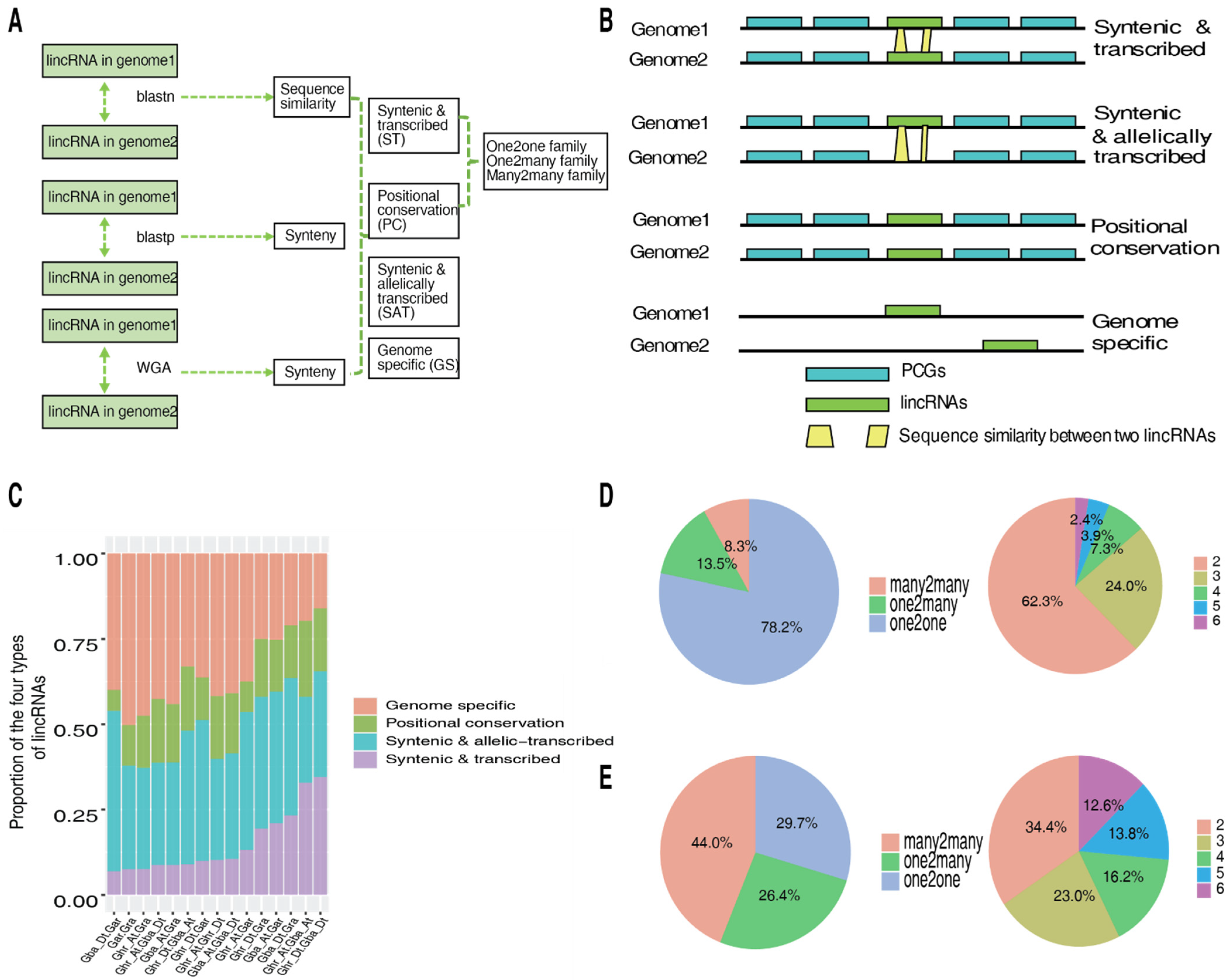
2.2. Conservation of lincRNAs in Diploid and Allotetraploid Cotton Species
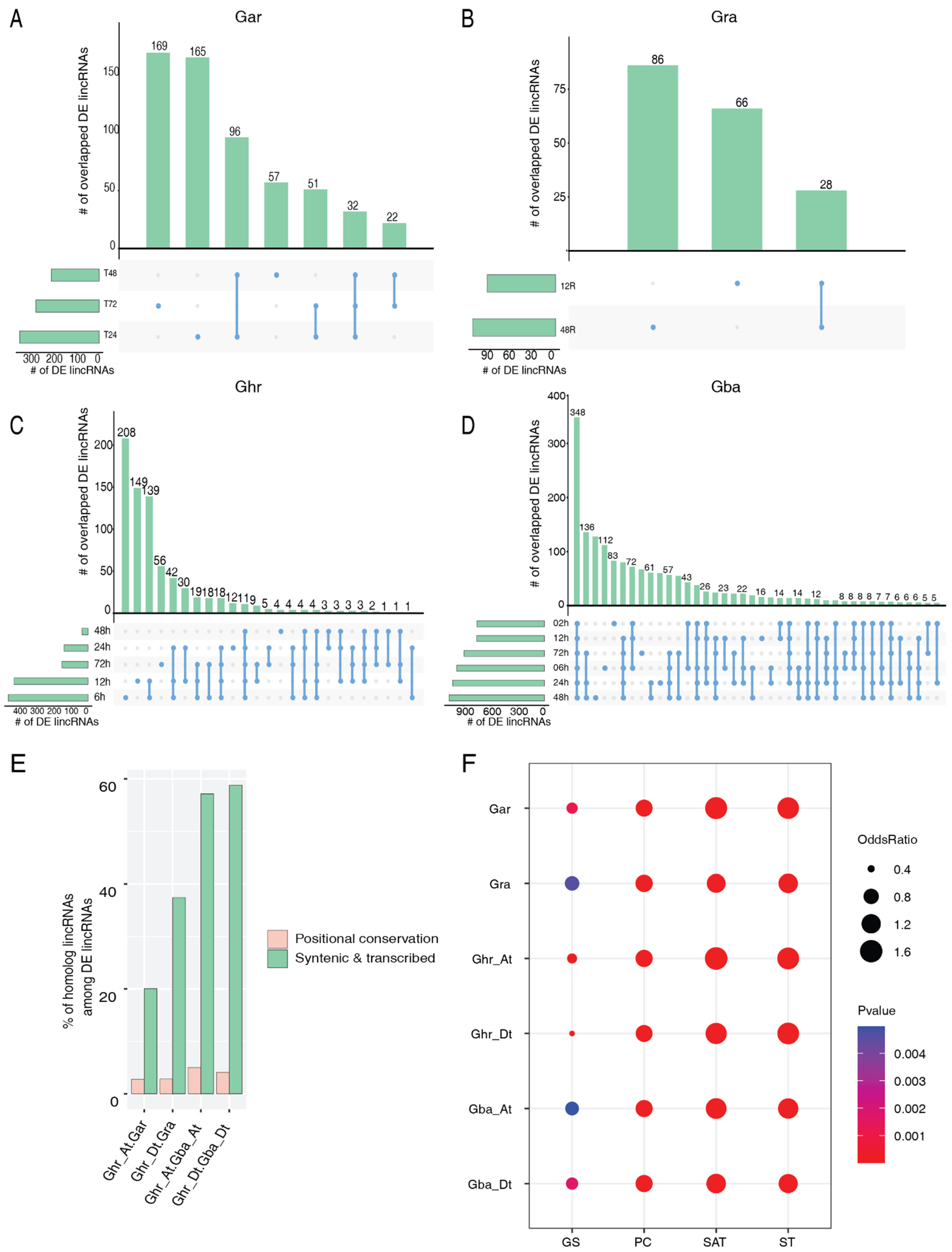
2.3. Relationship between Conservation of lincRNAs and Their Vd Responsiveness
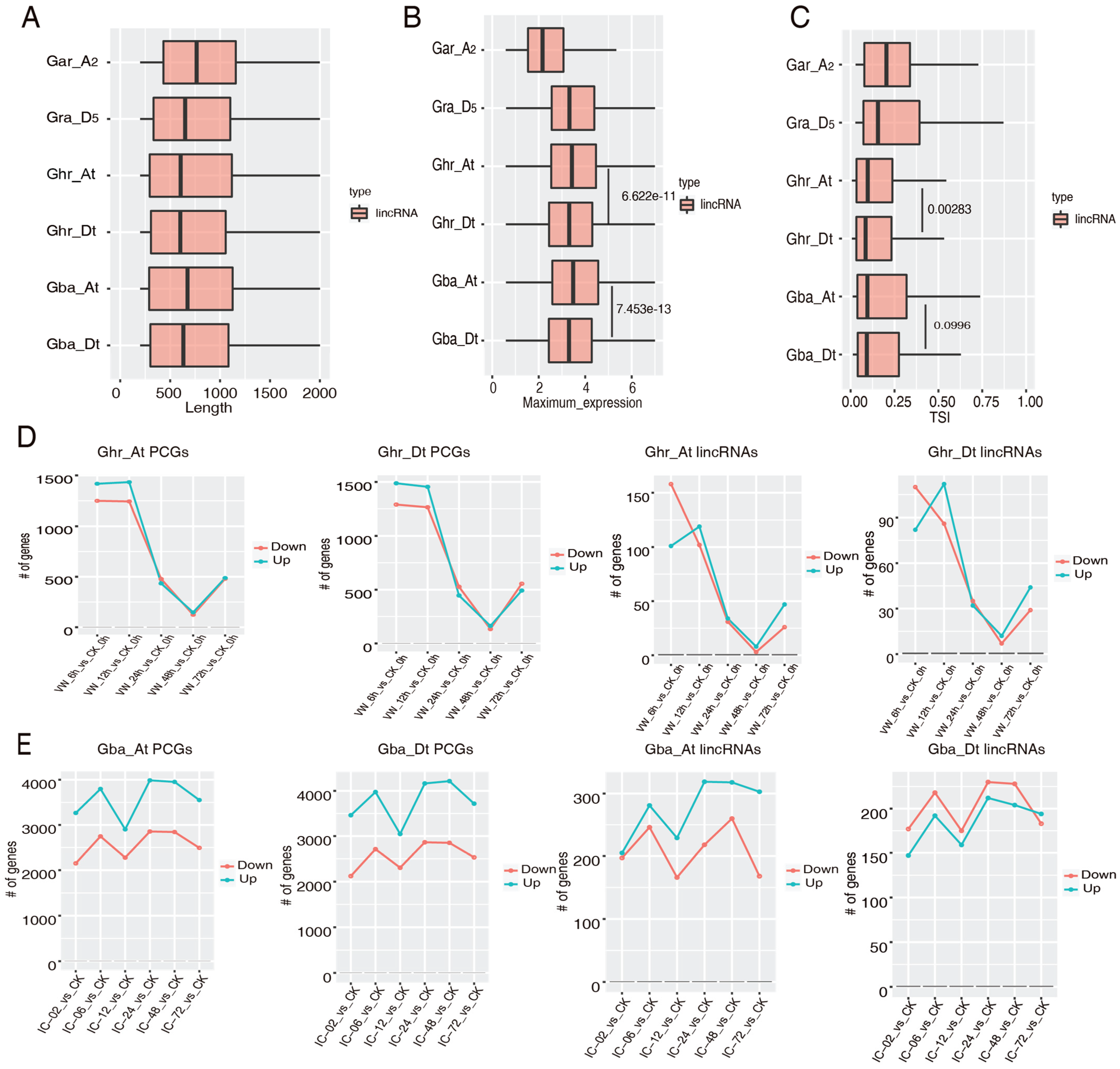
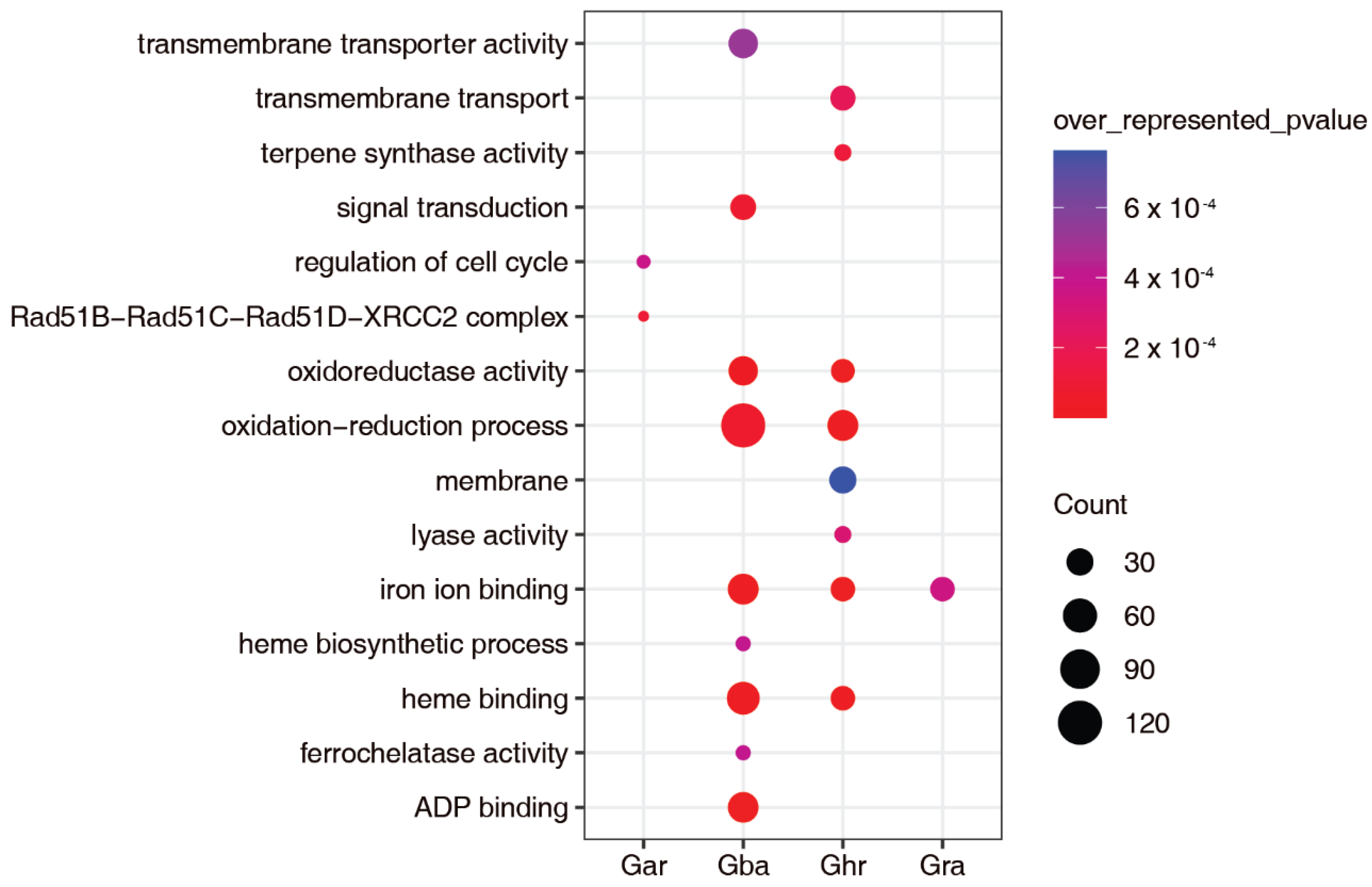
2.4. Subgenome Dominance of the Vd-Responsive lincRNAs
2.5. Cis-Regulatory Role of the Vd-Responsive lincRNAs in Cotton
2.6. Overlapping between Vd-Responsive lincRNAs and QTL
2.7. LincRNAs as Potential Target Mimicry of miR482/2118
3. Discussion
4. Materials and Methods
4.1. Identification of lincRNAs in Diploid and Allotetraploid Cotton Species
4.2. Identification of Homologous lincRNAs
4.3. Quantification of lincRNA Expression and Identification of Vd-Responsive lincRNAs
4.4. Analysis of Cis Targets of Vd-Responsive lincRNAs
4.5. Analysis of Association between Conservation of lincRNAs and Their Vd-Responsiveness
4.6. Identification of Endogenous Target Mimicry of miR482/2118
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- St Laurent, G.; Wahlestedt, C.; Kapranov, P. The Landscape of long noncoding RNA classification. Trends Genet. 2015, 31, 239–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Q.H.; Wang, M.B. Molecular functions of long non-coding RNAs in plants. Genes 2012, 3, 176–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapranov, P.; Willingham, A.T.; Gingeras, T.R. Genome-wide transcription and the implications for genomic organization. Nat. Rev. Genet. 2007, 8, 413–423. [Google Scholar] [CrossRef]
- Chen, L.; Zhu, Q.H.; Kaufmann, K. Long non-coding RNAs in plants: Emerging modulators of gene activity in development and stress responses. Planta 2020, 252, 92. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Zhang, Y.; Chen, X.; Chen, Y. Plant noncoding RNAs: Hidden players in development and stress responses. Annu. Rev. Cell Dev. Biol. 2019, 35, 407–431. [Google Scholar] [CrossRef]
- Wierzbicki, A.T.; Blevins, T.; Swiezewski, S. Long noncoding RNAs in plants. Annu. Rev. Plant Biol. 2021, 72, 245–271. [Google Scholar] [CrossRef]
- Ransohoff, J.D.; Wei, Y.; Khavari, P.A. The functions and unique features of long intergenic non-coding RNA. Nat. Rev. Mol. Cell Biol. 2018, 19, 143–157. [Google Scholar] [CrossRef] [PubMed]
- Washietl, S.; Kellis, M.; Garber, M. Evolutionary dynamics and tissue specificity of human long noncoding RNAs in six mammals. Genome Res. 2014, 24, 616–628. [Google Scholar] [CrossRef] [Green Version]
- Hezroni, H.; Koppstein, D.; Schwartz, M.G.; Avrutin, A.; Bartel, D.P.; Ulitsky, I. Principles of long noncoding RNA evolution derived from direct comparison of transcriptomes in 17 species. Cell Rep. 2015, 11, 1110–1122. [Google Scholar] [CrossRef] [Green Version]
- Ulitsky, I. Evolution to the rescue: Using comparative genomics to understand long non-coding RNAs. Nat. Rev. Genet. 2016, 17, 601–614. [Google Scholar] [CrossRef]
- Wang, H.; Niu, Q.W.; Wu, H.W.; Liu, J.; Ye, J.; Yu, N.; Chua, N.H. Analysis of non-coding transcriptome in rice and maize uncovers roles of conserved lncRNAs associated with agriculture traits. Plant J. Cell Mol. Biol. 2015, 84, 404–416. [Google Scholar] [CrossRef]
- Zhang, Y.C.; Liao, J.Y.; Li, Z.Y.; Yu, Y.; Zhang, J.P.; Li, Q.F.; Qu, L.H.; Shu, W.S.; Chen, Y.Q. Genome-wide screening and functional analysis identify a large number of long noncoding RNAs involved in the sexual reproduction of rice. Genome Biol. 2014, 15, 512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palazzo, A.F.; Koonin, E.V. Functional long non-coding RNAs evolve from junk transcripts. Cell 2020, 183, 1151–1161. [Google Scholar] [CrossRef]
- Zhao, T.; Tao, X.; Feng, S.; Wang, L.; Hong, H.; Ma, W.; Shang, G.; Guo, S.; He, Y.; Zhou, B.; et al. LncRNAs in polyploid cotton interspecific hybrids are derived from transposon neofunctionalization. Genome Biol. 2018, 19, 195. [Google Scholar] [CrossRef] [Green Version]
- Lv, Y.; Hu, F.; Zhou, Y.; Wu, F.; Gaut, B.S. Maize transposable elements contribute to long non-coding RNAs that are regulatory hubs for abiotic stress response. BMC Genom. 2019, 20, 864. [Google Scholar] [CrossRef]
- Wendel, J.F.; Grove, C.E. Taxonomy and evolution of the cotton genus, Gossypium. In Cotton, 2nd ed.; Agronomy Monograph 57; Fang, D.D., Percey, R.G., Eds.; American Society of Agronomy, Inc.: Madison, WI, USA; Crop Science Society of America, Inc.: Madison, WI, USA; Soil Science Society of America, Inc.: Madison, WI, USA, 2015; Volume 57, pp. 25–44. [Google Scholar]
- Huang, G.; Wu, Z.; Percy, R.G.; Bai, M.; Li, Y.; Frelichowski, J.E.; Hu, J.; Wang, K.; Yu, J.Z.; Zhu, Y. Genome sequence of Gossypium herbaceum and genome updates of Gossypium arboreum and Gossypium hirsutum provide insights into cotton A-genome evolution. Nat. Genet. 2020, 52, 516–524. [Google Scholar] [CrossRef] [Green Version]
- Cho, J. Transposon-derived non-coding RNAs and their function in plants. Front. Plant Sci. 2018, 9, 600. [Google Scholar] [CrossRef]
- Wang, X.; Ai, G.; Zhang, C.; Cui, L.; Wang, J.; Li, H.; Zhang, J.; Ye, Z. Expression and diversification analysis reveals transposable elements play important roles in the origin of Lycopersicon-specific lncRNAs in tomato. New Phytol. 2016, 209, 1442–1455. [Google Scholar] [CrossRef] [PubMed]
- Deng, F.; Zhang, X.; Wang, W.; Yuan, R.; Shen, F. Identification of Gossypium hirsutum long non-coding RNAs (lncRNAs) under salt stress. BMC Plant Biol. 2018, 18, 23. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Chen, X.; Mu, M.; Wang, J.; Wang, X.; Wang, D.; Yin, Z.; Fan, W.; Wang, S.; Guo, L.; et al. Genome-wide analysis of long noncoding RNAs and their responses to drought stress in cotton (Gossypium hirsutum L.). PLoS ONE 2016, 11, e0156723. [Google Scholar] [CrossRef] [Green Version]
- Salih, H.; Gong, W.; He, S.; Xia, W.; Odongo, M.R.; Du, X. Long non-coding RNAs and their potential functions in Ligon-lintless-1 mutant cotton during fiber development. BMC Genom. 2019, 20, 661. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Yuan, D.; Tu, L.; Gao, W.; He, Y.; Hu, H.; Wang, P.; Liu, N.; Lindsey, K.; Zhang, X. Long noncoding RNAs and their proposed functions in fibre development of cotton (Gossypium spp.). New Phytol. 2015, 207, 1181–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; Wang, X.; Zhang, Y.; Yang, J.; Li, Z.; Wu, L.; Wu, J.; Wu, N.; Liu, L.; Liu, Z.; et al. Dynamic characteristics and functional analysis provide new insights into long non-coding RNA responsive to Verticillium dahliae infection in Gossypium hirsutum. BMC Plant Biol. 2021, 21, 68. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Han, J.; Lu, K.; Li, M.; Gao, M.; Cao, Z.; Zhao, T.; Chen, X.; Tao, X.; Chen, Q.; et al. Functional examination of lncRNAs in allotetraploid Gossypium hirsutum. BMC Genom. 2021, 22, 443. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, M.; Li, N.; Wang, H.; Qiu, P.; Pei, L.; Xu, Z.; Wang, T.; Gao, E.; Liu, J.; et al. Long noncoding RNAs involve in resistance to Verticillium dahliae, a fungal disease in cotton. Plant Biotechnol. J. 2018, 16, 1172–1185. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Li, J.; Saeed, S.; Batchelor, W.D.; Alariqi, M.; Meng, Q.; Zhu, F.; Zou, J.; Xu, Z.; Si, H.; et al. Identification and functional analysis of lncRNA by CRISPR/Cas9 during the cotton response to sap-sucking insect infestation. Front. Plant Sci. 2022, 13, 784511. [Google Scholar] [CrossRef]
- Hu, H.; Wang, M.; Ding, Y.; Zhu, S.; Zhao, G.; Tu, L.; Zhang, X. Transcriptomic repertoires depict the initiation of lint and fuzz fibres in cotton (Gossypium hirsutum L.). Plant Biotechnol. J. 2018, 16, 1002–1012. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Li, L.; Yang, Z.; Zhao, G.; Zhang, X.; Wang, L.; Zheng, L.; Zhuo, F.; Yin, H.; Ge, X.; et al. Target of Rapamycin (TOR) regulates the expression of lncRNAs in response to abiotic stresses in cotton. Front. Genet. 2018, 9, 690. [Google Scholar] [CrossRef] [Green Version]
- Tao, X.; Li, M.; Zhao, T.; Feng, S.; Zhang, H.; Wang, L.; Han, J.; Gao, M.; Lu, K.; Chen, Q.; et al. Neofunctionalization of a polyploidization-activated cotton long intergenic non-coding RNA DAN1 during drought stress regulation. Plant Physiol. 2021, 186, 2152–2168. [Google Scholar] [CrossRef]
- Zhang, X.; Dong, J.; Deng, F.; Wang, W.; Cheng, Y.; Song, L.; Hu, M.; Shen, J.; Xu, Q.; Shen, F. The long non-coding RNA lncRNA973 is involved in cotton response to salt stress. BMC Plant Biol. 2019, 19, 459. [Google Scholar] [CrossRef]
- Zhang, X.; Shen, J.; Xu, Q.; Dong, J.; Song, L.; Wang, W.; Shen, F. Long noncoding RNA lncRNA354 functions as a competing endogenous RNA of miR160b to regulate ARF genes in response to salt stress in upland cotton. Plant Cell Environ. 2021, 44, 3302–3321. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, J.; Cheng, J.; Sun, Q.; Zhang, Y.; Liu, J.; Li, H.; Zhang, Z.; Wang, P.; Cai, C.; et al. lncRNA7 and lncRNA2 modulate cell wall defense genes to regulate cotton resistance to Verticillium wilt. Plant Physiol. 2022, 189, 264–284. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Tu, L.; Yuan, D.; Zhu, D.; Shen, C.; Li, J.; Liu, F.; Pei, L.; Wang, P.; Zhao, G.; et al. Reference genome sequences of two cultivated allotetraploid cottons, Gossypium hirsutum and Gossypium barbadense. Nat. Genet. 2019, 51, 224–229. [Google Scholar] [CrossRef] [Green Version]
- Franco-Zorrilla, J.M.; Valli, A.; Todesco, M.; Mateos, I.; Puga, M.I.; Rubio-Somoza, I.; Leyva, A.; Weigel, D.; Garcia, J.A.; Paz-Ares, J. Target mimicry provides a new mechanism for regulation of microRNA activity. Nat. Genet. 2007, 39, 1033–1037. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, S.; Park, G.; Atamian, H.S.; Han, C.S.; Stajich, J.E.; Kaloshian, I.; Borkovich, K.A. MicroRNAs suppress NB domain genes in tomato that confer resistance to Fusarium oxysporum. PLoS Pathog. 2014, 10, e1004464. [Google Scholar] [CrossRef]
- Shivaprasad, P.V.; Chen, H.M.; Patel, K.; Bond, D.M.; Santos, B.A.; Baulcombe, D.C. A microRNA superfamily regulates nucleotide binding site-leucine-rich repeats and other mRNAs. Plant Cell 2012, 24, 859–874. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Q.H.; Fan, L.; Liu, Y.; Xu, H.; Llewellyn, D.; Wilson, I. miR482 regulation of NBS-LRR defense genes during fungal pathogen infection in cotton. PLoS ONE 2013, 8, e84390. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Jung, C.; Xu, J.; Wang, H.; Deng, S.; Bernad, L.; Arenas-Huertero, C.; Chua, N.H. Genome-wide analysis uncovers regulation of long intergenic noncoding RNAs in Arabidopsis. Plant Cell 2012, 24, 4333–4345. [Google Scholar] [CrossRef] [Green Version]
- Mohammadin, S.; Edger, P.P.; Pires, J.C.; Schranz, M.E. Positionally-conserved but sequence-diverged: Identification of long non-coding RNAs in the Brassicaceae and Cleomaceae. BMC Plant Biol. 2015, 15, 217. [Google Scholar]
- Shen, E.; Zhu, X.; Hua, S.; Chen, H.; Ye, C.; Zhou, L.; Liu, Q.; Zhu, Q.H.; Fan, L.; Chen, X. Genome-wide identification of oil biosynthesis-related long non-coding RNAs in allopolyploid Brassica napus. BMC Genom. 2018, 19, 745. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.; Li, Z.; Ramanujan, K.; Clay, I.; Zhang, Y.; Lemire-Brachat, S.; Glass, D.J. A long non-coding RNA, LncMyoD, regulates skeletal muscle differentiation by blocking IMP2-mediated mRNA translation. Dev. Cell 2015, 34, 181–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ke, L.; Zhou, Z.; Xu, X.W.; Wang, X.; Liu, Y.; Xu, Y.; Huang, Y.; Wang, S.; Deng, X.; Chen, L.L.; et al. Evolutionary dynamics of lincRNA transcription in nine citrus species. Plant J. Cell Mol. Biol. 2019, 98, 912–927. [Google Scholar] [CrossRef] [PubMed]
- Lucero, L.; Ferrero, L.; Fonouni-Farde, C.; Ariel, F. Functional classification of plant long noncoding RNAs: A transcript is known by the company it keeps. New Phytol. 2021, 229, 1251–1260. [Google Scholar] [CrossRef]
- Zhu, Q.H.; Stephen, S.; Taylor, J.; Helliwell, C.A.; Wang, M.B. Long noncoding RNAs responsive to Fusarium oxysporum infection in Arabidopsis thaliana. New Phytol. 2014, 201, 574–584. [Google Scholar] [CrossRef]
- Shen, E.; Chen, T.; Zhu, X.; Fan, L.; Sun, J.; Llewellyn, D.J.; Wilson, I.; Zhu, Q.H. Expansion of MIR482/2118 by a class-II transposable element in cotton. Plant J. Cell Mol. Biol. 2020, 103, 2084–2099. [Google Scholar] [CrossRef]
- Zheng, X.; Chen, Y.; Zhou, Y.; Shi, K.; Hu, X.; Li, D.; Ye, H.; Zhou, Y.; Wang, K. Full-length annotation with multistrategy RNA-seq uncovers transcriptional regulation of lncRNAs in cotton. Plant Physiol. 2021, 185, 179–195. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Paterson, A.H.; Wendel, J.F.; Gundlach, H.; Guo, H.; Jenkins, J.; Jin, D.; Llewellyn, D.; Showmaker, K.C.; Shu, S.; Udall, J.; et al. Repeated polyploidization of Gossypium genomes and the evolution of spinnable cotton fibres. Nature 2012, 492, 423–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, X.; Huang, G.; He, S.; Yang, Z.; Sun, G.; Ma, X.; Li, N.; Zhang, X.; Sun, J.; Liu, M.; et al. Resequencing of 243 diploid cotton accessions based on an updated A genome identifies the genetic basis of key agronomic traits. Nat. Genet. 2018, 50, 796–802. [Google Scholar] [CrossRef]
- Yu, J.; Jung, S.; Cheng, C.H.; Lee, T.; Zheng, P.; Buble, K.; Crabb, J.; Humann, J.; Hough, H.; Jones, D.; et al. CottonGen: The Community database for cotton genomics, genetics, and breeding research. Plants 2021, 10, 2805. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, Y.J.; Yang, D.C.; Kong, L.; Hou, M.; Meng, Y.Q.; Wei, L.; Gao, G. CPC2: A fast and accurate coding potential calcu lator based on sequence intrinsic features. Nucleic Acids Res. 2017, 45, W12–W16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hezroni, H.; Ben-Tov Perry, R.; Meir, Z.; Housman, G.; Lubelsky, Y.; Ulitsky, I. A subset of conserved mammalian long non-coding RNAs are fossils of ancestral protein-coding genes. Genome Biol. 2017, 18, 162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Tang, H.; Debarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.H.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pegueroles, C.; Iraola-Guzman, S.; Chorostecki, U.; Ksiezopolska, E.; Saus, E.; Gabaldon, T. Transcriptomic analyses reveal groups of co-expressed, syntenic lncRNAs in four species of the genus Caenorhabditis. RNA Biol. 2019, 16, 320–329. [Google Scholar] [CrossRef] [Green Version]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Kryuchkova-Mostacci, N.; Robinson-Rechavi, M. A benchmark of gene expression tissue-specificity metrics. Brief. Bioinform. 2017, 18, 205–214. [Google Scholar] [CrossRef]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [Green Version]
- Ye, C.Y.; Xu, H.; Shen, E.; Liu, Y.; Wang, Y.; Shen, Y.; Qiu, J.; Zhu, Q.H.; Fan, L. Genome-wide identification of non-coding RNAs interacted with microRNAs in soybean. Front. Plant Sci. 2014, 5, 743. [Google Scholar] [CrossRef]
- Wu, H.J.; Wang, Z.M.; Wang, M.; Wang, X.J. Widespread long noncoding RNAs as endogenous target mimics for microRNAs in plants. Plant Physiol. 2013, 161, 1875–1884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Y.; Cai, X.; Wang, Q.; Wang, P.; Zhang, Y.; Cai, C.; Xu, Y.; Wang, K.; Zhou, Z.; Wang, C.; et al. Genome sequencing of the Australian wild diploid species Goss-ypium australe highlights disease resistance and delayed gland morphogenesis. Plant Biotechnol. J. 2020, 18, 814–828. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Wang, D.; Zheng, X.; Qin, A.; Zhou, J.; Guo, B.; Chen, Y.; Wen, X.; Ye, W.; Zhou, Y.; et al. Multi-strategic RNA-seq analysis reveals a high-resolution transcriptional landscape in cotton. Nat. Commun. 2019, 10, 4714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, Q.; Magwanga, R.O.; Cai, X.; Lu, P.; Kirungu, J.N.; Zhou, Z.; Wang, X.; Wang, X.; Xu, Y.; Hou, Y.; et al. RNA-Sequencing, Physiological and RNAi Analyses Provide Insights into the Response Mechanism of the ABC-Mediated Resistance to Verticillium dahliae Infection in Cotton. Genes 2019, 10, 110. [Google Scholar] [CrossRef] [Green Version]
- Hu, G.; Grover, C.E.; Arick, M.A.; Liu, M.; Peterson, D.G.; Wendel, J.F. Homoeologous gene expression and co-expression network analyses and evolutionary inference in allopolyploids. Brief. Bioinform. 2020, 22, 1819–1835. [Google Scholar] [CrossRef]
- Zhang, T.; Hu, Y.; Jiang, W.; Fang, L.; Guan, X.; Chen, J.; Zhang, J.; Saski, C.A.; Scheffler, B.E.; Stelly, D.M.; et al. Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat. Biotechnol. 2015, 33, 531–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, G.; Li, W.; Wang, G.; Li, L.; Si, Q.; Cai, C.; Guo, W. Genetic Basis of Fiber Improvement and Decreased Stress Tolerance in Culti-vated Versus Semi-Domesticated Upland Cotton. Front. Plant Sci. 2019, 10, 1572. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.Y.; Huang, J.Q.; Li, N.Y.; Ma, X.F.; Wang, J.L.; Liu, C.; Liu, Y.F.; Liang, Y.; Bao, Y.M.; Dai, X.F. Genome-wide analysis of the gene families of resistance gene analogues in cotton and their response to Verticillium wilt. BMC Plant Biol. 2015, 15, 148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdelraheem, A.; Elassbli, H.; Zhu, Y.; Vasu, K.; Hinze, L.; Stelly, D.; Wedegaertner, T.; Zhang, J. A genome-wide association study uncovers consistent quantitative trait loci for resistance to Verticillium wilt and Fusarium wilt race 4 in the US Upland cotton. Theor. Appl. Genet. 2020, 133, 563–577. [Google Scholar] [CrossRef]
- Fang, H.; Zhou, H.; Sanogo, S.; Flynn, R.; Percy, R.G.; Hughs, S.E.; Ulloa, M.; Jones, D.C.; Zhang, J. Quantitative trait locus mapping for Verticillium wilt resistance in a backcross inbred line population of cotton (Gossypium hirsutum × Gossypium barbadense) based on RGA-AFLP analysis. Euphytica 2013, 194, 79–91. [Google Scholar] [CrossRef]
- Fang, H.; Zhou, H.; Sanogo, S.; Lipka, A.E.; Fang, D.D.; Percy, R.G.; Hughs, S.E.; Jones, D.C.; Gore, M.A.; Zhang, J.F. Quantitative trait locus analysis of Verticillium wilt resistance in an introgressed recombinant inbred population of Upland cotton. Mol. Breed. 2014, 33, 709–720. [Google Scholar] [CrossRef]
- Jiang, F.; Zhao, J.; Zhou, L.; Guo, W.Z.; Zhang, T.Z. Molecular mapping of Verticillium wilt resistance QTL clustered on chro-mosomes D7 and D9 in Upland cotton. Sci. China Ser. C-Life Sci. 2009, 52, 872–884. [Google Scholar] [CrossRef] [PubMed]
- Li, T.G.; Ma, X.F.; Li, N.Y.; Zhou, L.; Liu, Z.; Han, H.Y.; Gui, Y.J.; Bao, Y.; Chen, J.; Dai, X. Genome-wide association study discovered candidate genes of Verticillium wilt resistance in upland cotton (Gossypium hirsutum L.). Plant Biotechnol. J. 2017, 15, 1520–1532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ning, Z.Y.; Zhao, R.; Chen, H.; Ai, N.; Zhang, X.; Zhao, J.; Mei, H.; Wang, P.; Guo, W.; Zhang, T. Molecular tagging of a major quantitative trait locus for broad-spectrum resistance to Verticillium wilt in Upland cotton cultivar Prema. Crop. Sci. 2013, 53, 2304–2312. [Google Scholar]
- Palanga, K.K.; Jamshed, M.; Rashid, M.H.O.; Gong, J.; Li, J.; Iqbal, M.S.; Liu, A.; Shang, H.; Shi, Y.; Chen, T.; et al. Quantitative trait locus mapping for verti-cillium wilt resistance in an upland cotton recombinant inbred line using SNP-based high density genetic map. Front. Plant Sci. 2017, 8, 382. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Zhang, B.; Liu, A.; Li, W.; Li, J.; Lu, Q.; Zhang, Z.; Li, S.; Gong, W.; Shang, H.; et al. Quantitative trait loci analysis of Verticillium wilt resistance in interspecific backcross populations of Gossypium hirsutum × Gossypium barbadense. BMC Genom. 2016, 17, 877. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.R.; Liu, R.Z.; Wang, L.M.; Zhang, C.Y.; Liu, G.D.; Liu, Q.H.; Ma, X.B.; Zhang, J. Molecular marker of Verticillium resistance in upland cotton (Gossypium hirsutum L.) cultivar and their effects on assisted phenotypic selection. Cotton Sci. 2007, 6, 424–430. [Google Scholar]
- Wang, H.M.; Lin, Z.X.; Zhang, X.L.; Chen, W.; Guo, X.P.; Nie, Y.C.; Li, Y.H. Mapping and quantitative trait loci analysis of Verticillium wilt resistance genes in cotton. J. Integr. Plant Biol. 2008, 50, 174–182. [Google Scholar] [CrossRef]
- Yang, C.; Guo, W.; Li, G.; Gao, F.; Lin, S.; Zhang, T. QTLs mapping for Verticillium wilt resistance at seedling and maturity stages in Gossypium barbadense L. Plant Sci. 2008, 174, 290–298. [Google Scholar] [CrossRef]
- Zhang, J.; Yu, J.; Pei, W.; Li, X.; Said, J.; Song, M.; Sanogo, S. Genetic analysis of Verticillium wilt resistance in a back-cross inbred line population and a meta-analysis of quantitative trait loci for disease resistance in cotton. BMC Genom. 2015, 16, 577. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Abdelraheem, A.; Thyssen, G.N.; Fang, D.D.; Jenkins, J.N.; McCarty, J.C.; Wedegaertner, T. Evaluation and genome-wide association study of Verticillium wilt resistance in a MAGIC population derived from intermating of eleven Upland cotton (Gossypium hirsutum) parents. Euphytica 2020, 216, 1–3. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, B.; Sun, Z.; Liu, Z.; Cui, Y.; Ke, H.; Wang, Z.; Wu, L.; Zhang, G.; Wang, G.; et al. A large-scale genomic association analysis identifies a fragment in Dt11 chromosome conferring cotton Verticillium wilt resistance. Plant Biotechnol. J. 2021, 19, 2126–2138. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wang, H.; Chen, W.; Li, Y. Genetic Structure, Linkage Disequilibrium and Association Mapping of Verticillium Wilt Resistance in Elite Cotton (Gossypium hirsutum L.) Germplasm Population. PLoS ONE 2014, 9, e86308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Chen, W.; Cui, Y.; Sang, X.; Lu, J.; Jing, H.; Wang, W.; Zhao, P.; Wang, H. Detection of candidate genes and development of KASP markers for Verticillium wilt resistance by combining genome-wide association study, QTL-seq and transcriptome sequencing in cotton. Theor. Appl. Genet. 2021, 134, 1063–1081. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Gar § | Gra | Ghr_At | Ghr_Dt | Gba_At | Gba_Dt | No. of lincRNAs (%) |
|---|---|---|---|---|---|---|---|
| 1a | √ | √ | √ | √ | √ | √ | 48 (0.7) |
| 1b | √ | √ | Lost in one to all four subgenomes | 214 (3.1) | |||
| 2a | √ | x | √ | √ or x | √ | √ or x | 577 (8.3) |
| 2b | √ | x | √ | √ or x | x | √ or x | 375 (5.4) |
| 2c | √ | x | x | √ or x | √ | √ or x | 233 (3.3) |
| 2d | √ | x | x | √ or x | x | √ or x | 164 (2.4) |
| 3a | x | √ | √ or x | √ | √ or x | √ | 505 (7.3) |
| 3b | x | √ | √ or x | √ | √ or x | x | 397 (5.7) |
| 3c | x | √ | √ or x | x | √ or x | √ | 262 (3.8) |
| 3d | x | √ | √ or x | x | √ or x | x | 233 (3.3) |
| 4 | x | x | Presence in two to all four subgenomes | 3954 (56.8) | |||
| Species | Vd Response | No. of DE lincRNAs | Unique to the Species § | One2One | One2Many | Many2Many |
|---|---|---|---|---|---|---|
| Gar | Up | 2 | 1 (50.0) | 0 | 0 | 1 (50.0) |
| Down | 6 | 2 (33.3) | 3 (50.0) | 0 | 1 (16.7) | |
| Gra | Up | 28 | 14 (50.0) | 5 (17.9) | 4 (14.3) | 5 (17.9) |
| Down | 19 | 12 (63.2) | 5 (26.3) | 0 | 2 (10.5) | |
| Ghr | Up | 33 | 20 (60.6) | 10 (30.3) | 2 (6.1) | 1 (3.0) |
| Down | 48 | 23 (47.9) | 13 (27.1) | 4 (8.3) | 8 (16.7) | |
| Gba | Up | 246 | 118 (48.0) | 78 (31.7) | 18 (7.3) | 32 (13.0) |
| Down | 134 | 63 (47.0) | 41 (30.6) | 18 (13.4) | 12 (9.0) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, L.; Shen, E.; Zhao, Y.; Wang, H.; Wilson, I.; Zhu, Q.-H. The Conservation of Long Intergenic Non-Coding RNAs and Their Response to Verticillium dahliae Infection in Cotton. Int. J. Mol. Sci. 2022, 23, 8594. https://doi.org/10.3390/ijms23158594
Chen L, Shen E, Zhao Y, Wang H, Wilson I, Zhu Q-H. The Conservation of Long Intergenic Non-Coding RNAs and Their Response to Verticillium dahliae Infection in Cotton. International Journal of Molecular Sciences. 2022; 23(15):8594. https://doi.org/10.3390/ijms23158594
Chicago/Turabian StyleChen, Li, Enhui Shen, Yunlei Zhao, Hongmei Wang, Iain Wilson, and Qian-Hao Zhu. 2022. "The Conservation of Long Intergenic Non-Coding RNAs and Their Response to Verticillium dahliae Infection in Cotton" International Journal of Molecular Sciences 23, no. 15: 8594. https://doi.org/10.3390/ijms23158594
APA StyleChen, L., Shen, E., Zhao, Y., Wang, H., Wilson, I., & Zhu, Q. -H. (2022). The Conservation of Long Intergenic Non-Coding RNAs and Their Response to Verticillium dahliae Infection in Cotton. International Journal of Molecular Sciences, 23(15), 8594. https://doi.org/10.3390/ijms23158594