
c-Kit Induces Migration of Triple-Negative Breast Cancer Cells and Is a Promising Target for Tyrosine Kinase Inhibitor Treatment


 ,
,
Abstract
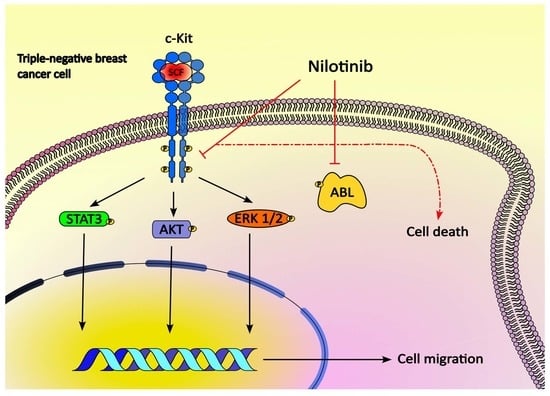
:
1. Introduction
2. Results
2.1. TNBC Cells Express Different Levels of Membrane c-Kit
2.2. SCF Induces Activation of c-Kit in TNBC Cells and Promotes Cell Migration
2.3. SCF Induces Activation of STAT3, Akt and ERK1/2
2.4. The c-Kit/SCF Axis Has No Effect on TNBC Cell Survival
2.5. Responses of TNBC Cells to Tyrosine Kinases Inhibitors
2.6. c-Kit Is a Dominant Targetable RTK in TNBC Tumors
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Reverse Transcriptase-PCR (RT-PCR)
4.3. Analysis of Protein by Western Blot
4.4. Flow Cytometry
4.5. c-Kit DNA Sequencing
4.6. Proliferation Assay
4.7. Migration Assay
4.8. Cytotoxicity Assays
4.9. Publicly Available Data Sets Analysis
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Thakur, K.K.; Bordoloi, D.; Kunnumakkara, A.B. Alarming burden of triple-negative breast cancer in India. Clin. Breast Cancer 2018, 18, e393–e399. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, B.D.; Jovanovic, B.; Chen, X.; Estrada, M.V.; Johnson, K.N.; Shyr, Y.; Moses, H.L.; Sanders, M.E.; Pietenpol, J.A. Refinement of triple-negative breast cancer molecular subtypes: Implications for neoadyuvant chemotherapy selection. PLoS ONE 2016, 11, e0157368. [Google Scholar] [CrossRef] [PubMed]
- Burstein, M.D.; Tsimelzon, A.; Poage, G.M.; Covington, K.R.; Contreras, A.; Fuqua, S.A.W.; Savage, M.I.; Osborne, C.K.; Hilsenbeck, S.G.; Chang, J.C.; et al. Comprehensive genomic analysis identifies novel subtypes and targets of triple-negative breast cancer. Clin. Cancer Res. 2015, 21, 1688–1698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, Z.; Lovly, C.M. Mechanisms of receptor tyrosine kinase activation in cancer. Mol. Cancer 2018, 17, 58. [Google Scholar] [CrossRef]
- Pathania, S.; Pentikäinen, O.T.; Singh, P.K. A holistic view on c-Kit in cancer: Structure, signaling, pathophysiology and its inhibitors. Biochim. Biophys. Acta Rev. Cancer 2021, 1876, 188631. [Google Scholar] [CrossRef]
- Edling, C.E.; Pedersen, M.; Carlsson, L.; Rönnstrand, L.; Palmer, R.H.; Hallberg, B. Haematopoietic progenitor cells utilize conventional PKC to suppress PKB/Akt activity in response to c-Kit stimulation. Br. J. Haematol. 2007, 136, 260–268. [Google Scholar] [CrossRef] [PubMed]
- Wehrle-Haller, B. The role of kit-ligand in melanocyte development and epidermal homeostasis. Pigment Cell Res. 2003, 16, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Todd, J.R.; Scurr, L.L.; Becker, T.M.; Kefford, R.F.; Rizos, H. The MAPK pathway functions as a redundant survival signal that reinforces the PI3K cascade in c-Kit mutant melanoma. Oncogene 2014, 33, 236–245. [Google Scholar] [CrossRef] [Green Version]
- Delyon, J.; Chevret, S.; Jouary, T.; Dalac, S.; Dalle, S.; Guillot, B.; Arnault, J.-P.; Avril, M.-F.; Bedane, C.; Bens, G.; et al. STAT3 mediates nilotinib response in KIT-altered melanoma: A phase II multicenter trial of the French Skin Cancer Network. J. Investig. Dermatol. 2018, 138, 58–67. [Google Scholar] [CrossRef] [Green Version]
- Gromova, I.; Espinoza, J.A.; Grauslund, M.; Santoni-Rugiu, E.; Moller Talman, M.-L.; van Oostrum, J.; Moreira, J.M.A. Functional proteomic profiling of triple-negative breast cancer. Cells 2021, 10, 2768. [Google Scholar] [CrossRef]
- Voigtlaender, M.; Schneider-Merck, T.; Trepel, M. Lapatinib. Recent Results Cancer Res. 2018, 211, 19–44. [Google Scholar] [CrossRef] [PubMed]
- Lennartsson, J.; Rönnstrand, L. Stem cell factor/c-Kit: From basic science to clinical implications. Physiol. Rev. 2012, 92, 1619–1649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Sign. 2013, 6, 11. [Google Scholar] [CrossRef] [Green Version]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef] [Green Version]
- Krasagakis, K.; Fragiadaki, I.; Metaxari, M.; Krüger-Krasagakis, S.; Tzanakakis, G.N.; Stathopoulos, E.N.; Eberle, J.; Tavernarakis, N.; Tosca, A.D. KIT receptor activation by autocrine and paracrine stem cell factor stimulates growth of merkel cell carcinoma in vitro. J. Cell Physiol. 2011, 266, 1099–1109. [Google Scholar] [CrossRef]
- Lebron, M.B.; Brennan, L.; Damoci, C.B.; Prewett, M.C.; O’Mahony, M.; Duignan, I.J.; Credille, K.M.; DeLigio, J.T.; Starodubtsev, M.; Amatulli, M.; et al. A human monoclonal antibody targeting the stem cell factor receptor (c-Kit) blocks tumor cell signaling and inhibits tumor growth. Biol. Ther. 2014, 15, 1208–1218. [Google Scholar] [CrossRef] [Green Version]
- Aguilar, C.; Aguilar, C.; Lopez-Marure, R.; Jiménez-Sánchez, A.; Rocha-Zavaleta, L. Co-stimulation with stem cell factor and erythropoietin enhances migration of c-Kit expressing cervical cancer cells through the sustained activation of ERK1/2. Mol. Med. Rep. 2014, 9, 1895–1902. [Google Scholar] [CrossRef]
- Sundström, M.; Alfredsson, J.; Olsson, N.; Nilsson, G. Stem cell factor-induced migration of mast cells requires p38 mitogen-activated protein kinase activity. Exp. Cell Res. 2001, 267, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K pathway in human disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef] [Green Version]
- Pan, L.; Chen, X.; Fu, S.; Yu, W.; Li, C.; Wang, T.; Lo, H.-W.; Lin, J. LLY17, a novel small molecule STAT3 inhibitor induces apoptosis and suppresses cell migration and tumor growth in triple-negative breast cancer. Breast Cancer Res. Treat. 2020, 181, 31–41. [Google Scholar] [CrossRef]
- Moon, S.Y.; Lee, H.; Kim, S.; Hong, J.H.; Chun, S.H.; Lee, H.Y.; Kang, K.; Kim, H.S.; Won, H.S.; Ko, Y.H. Inhibition of STAT3 enhances sensitivity to tamoxifen in tamoxifen-resistant breast cancer cells. BMC Cancer 2021, 21, 931. [Google Scholar] [CrossRef] [PubMed]
- Hirota, S.; Isozaki, K. Pathology of gastrointestinal stromal tumors. Pathol. Int. 2006, 56, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, R.; Ud Din, H.; Akhtar, F.; Afzal, S.; Muhammad, I.; Hashmi, S.N. Immunohistochemical expression of epidermal growth factor receptor and c-Kit in triple negative breast cancer. J. Coll. Physicians Surg. Pak. 2016, 26, 570–572. [Google Scholar]
- Zhu, Y.; Wang, Y.; Guan, B.; Rao, Q.; Wang, J.; Ma, H.; Zhang, Z.; Zhou, X. c-Kit and PDGFRA gene mutations in triple negative breast cancer. Int. J. Clin. Exp. Pathol. 2014, 7, 4280–4285. [Google Scholar]
- Jansson, S.; Bendahl, P.-O.; Grabau, D.A.; Falck, A.-K.; Fernö, M.; Aaltonen, K.; Rydén, L. The three receptor tyrosine kinases c-Kit, VEGFR2 and PDGFRα, closely spaced at 4q12, show increased protein expression in triple-negative breast cancer. PLoS ONE 2014, 9, e102176. [Google Scholar] [CrossRef]
- Kanapathy Pillai, S.K.; Tay, A.; Nair, S.; Leong, C.-O. Triple-negative breast cancer is associated with EGFR, CK5/6 and c-KIT expression in Malaysian women. BMC Clin. Pathol. 2012, 12, 18. [Google Scholar] [CrossRef] [Green Version]
- Janostiak, R.; Vyas, M.; Cicek, A.F.; Wajapeyee, N.; Harigopal, M. Loss of c-Kit expression in breast cancer correlates with malignant transformation of breast epithelium and is mediated by KIT gene promoter DNA hypermethylation. Exp. Mol. Pathol. 2018, 105, 41–49. [Google Scholar] [CrossRef]
- Wu, W.; Hou, B.; Tang, C.; Liu, F.; Yang, J.; Pan, T.; Si, K.; Lu, D.; Wang, X.; Wang, J.; et al. (+)-Usnic acid inhibits migration of c-Kit positive cells in human colorectal cancer. Evid. Based Complement. Alternat. Med. 2018, 2018, 5149436. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Yan, J.; Cao, Y.; Yan, Y.; Shen, X.; Yu, B.; Tao, L.; Wang, S. Proliferation, migration and invasion of triple negative breast cancer cells are suppressed by berbamine via the PI3K/AkT/MDM2/p53 and PI3K/Akt/mTOR signaling pathways. Oncol. Lett. 2021, 21, 70. [Google Scholar] [CrossRef]
- Bouaouiche, S.; Ghione, S.; Sghaier, R.; Burgy, O.; Racoeur, C.; Derangere, V.; Bettaieb, A.; Plenchette, S. Nitric oxide-releasing drug glyceryl trinitrate targets JAK2/STAT3 signaling, migration and invasion of triple-negative breast cancer cells. Int. J. Mol. Sci. 2021, 22, 8449. [Google Scholar] [CrossRef] [PubMed]
- Constantinou, C.; Papadopoulos, S.; Karyda, E.; Alexopoulos, A.; Agnati, N.; Batistatou, A.; Harisis, H. Expression and clinical significance of claudin-7, PDL-1, PTEN, c-Kit, c-Met, c-Myc, ALK, CK5/6, CK17, p53, EGFR, Ki67, p63 in triple-negative breast cancer- a single centre prospective observational study. Vivo 2018, 32, 303–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.H.; Qin, L.; Li, X. Role of STAT3 signaling pathway in breast cancer. Cell Commun. Signal 2020, 18, 33. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, T.; Oda, G.; Kawachi, H.; Ishikawa, T.; Okamoto, K.; Uetake, H. Nuclear expression of p-STAT3 is associated with poor prognosis in ER(-) breast cancer. Clin. Pract. 2022, 12, 157–167. [Google Scholar] [CrossRef]
- Blanchard, Z.; Mullins, N.; Ellipeddi, P.; Lage, J.M.; McKinney, S.; El-Etriby, R.; Zhang, X.; Isokpehi, R.; Hernandez, B.; Elshamy, W.M. Geminin overexpression promotes imatinib sensitive breast cancer: A novel treatment approach for aggressive breast cancers, including a subset of triple negative. PLoS ONE 2014, 9, e95663. [Google Scholar] [CrossRef] [Green Version]
- Bahman, F.; Pittala, V.; Haider, M.; Greish, K. Enhanced anticancer activity of nanoformulation of dasatinib against triple-negative breast cancer. J. Pers. Med. 2021, 11, 559. [Google Scholar] [CrossRef]
- Chinchar, E.; Makey, K.L.; Gibson, J.; Chen, F.; Cole, S.A.; Megason, G.C.; Vijayakumar, S.; Miele, L.; Gu, J.-W. Sunitinib significantly suppresses the proliferation, migration, apoptosis resistance, tumor angiogenesis and growth of triple-negative breast cancers but increases breast cancer stem cells. Vasc. Cell 2014, 6, 12. [Google Scholar] [CrossRef] [Green Version]
- Luu, T.; Frankel, P.; Chung, C.; Chow, W.; Mortimer, J.; Hurria, A.; Somlo, G. Phase I/II trial vinorelbine and sorafenib in metastatic breast cancer. Clin. Breast Cancer 2014, 14, 94–100. [Google Scholar] [CrossRef]
- Chevalier, C.; Cannet, A.; Descamps, S.; Sirvent, A.; Simon, V.; Roche, S.; Benistant, C. ABL tyrosine kinase inhibition variable effects on the invasive properties of different triple negative breast cancer cell lines. PLoS ONE 2015, 10, e0118854. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Xie, Y.; Bao, A.; Li, J.; Ye, T.; Yang, C.; Yu, S. Nilotinib, a discoidin domain receptor 1 (DDR1) inhibitor, induces apoptosis and inhibits migration in breast cancer. Neoplasma 2021, 68, 975–982. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.S.; Sultana, A.; Reza, M.S.; Amanullah, M.; Kabir, S.R.; Mollah, M.N.H. Integrated bioinformatics and statistical approaches to explore molecular biomarkers for breast cancer diagnosis, prognosis and therapies. PLoS ONE 2022, 17, e0268967. [Google Scholar] [CrossRef] [PubMed]
- Ulivi, O.; Zoli, W.; Medri, L.; Amadori, D.; Saragoni, L.; Barbanti, F.; Calistri, D.; Silvestrini, R. c-Kit and SCF expression in normal and tumor breast tissue. Breast Cancer Res. Treat. 2004, 83, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Ono, A.; Hirooka, K.; Nakano, Y.; Nitta, E.; Nishiyama, A.; Tsujikawa, A. Gene expression changes in the retina after systemic administration of aldosterone. Jpn. J. Ophtalmol. 2018, 62, 499–507. [Google Scholar] [CrossRef]
- Xue, F.; Jia, Y.; Zhao, J. Overexpression of FYN suppresses the epithelial-to-mesenchymal transition through down-regulating PI3K/AKT pathway in lung adenocarcinoma. Surg. Oncol. 2020, 33, 108–117. [Google Scholar] [CrossRef]
- Tahiri, A.; Tekpli, X.; Satheesh, S.V.; DeWijn, R.; Lüders, T.; Bukholm, I.R.; Hurtado, A.; Geisler, J.; Kristensen, V.N. Loss of progesterone receptor is associated with distinct tyrosine kinase profiles in breast cancer. Breast Cancer Res. Treat. 2020, 183, 585–598. [Google Scholar] [CrossRef]
- Chen, J.; Li, Y.; Xie, X. MicroRNA-425 inhibits proliferation of chronic lymphocytic leukaemia cells through regulation of the Bruton’s tyrosine kinase/phospholipase Cγ2 signalling pathway. Exp. Ther. Med. 2020, 20, 1169–1175. [Google Scholar] [CrossRef]
- Sen, P.; Ghosal, S.; Hazra, R.; Arega, S.; Mohanty, R.; Kulkarni, K.K.; Budhwar, R.; Ganguly, N. Transcriptomic analyses of gene expression by CRISPR knockout of miR-214 in cervical cancer cells. Genomics 2020, 112, 1490–1499. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Ringnér, M.; Fredlund, E.; Häkkinen, J.; Borg, A.; Staaf, J. GOBO: Gene expression-based outcome for breast cancer online. PLoS ONE 2011, 6, e17911. [Google Scholar] [CrossRef] [PubMed]
- Fredlund, E.; Staaf, J.; Rantala, J.K.; Kallioniemi, O.; Borg, A.; Ringnér, M. The gene expression landscape of breast cancer is shaped by tumor protein p53 status and epithelial-mesenchymal transition. Breast Cancer Res. 2012, 14, R113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, J.S.; Mullins, M.; Cheang, M.C.; Leung, S.; Voduc, D.; Vickery, T.; Davies, S.; Fauron, C.; He, X.; Hu, Z.; et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol. 2009, 27, 1160–1167. [Google Scholar] [CrossRef]
- Alexandrou, S.; George, S.M.; Ormandy, C.J.; Lim, E.; Oakes, S.R.; Caldon, C.E. The proliferative and apoptotic land-scape of basal-like breast cancer. Int. J. Mol. Sci. 2019, 20, 667. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tyrosine Kinase Inhibitor | HCC-1806 IC50 (nM) ± SD | HCC-1937 IC50 (nM) ± SD | HCC-70 IC50 (nM) ± SD | MDA-MB-468 IC50 (nM) ± SD |
|---|---|---|---|---|
| Imatinib | 51,800 ± 5853 | 24,750 ± 1819 | 24,800 ± 1042 | 24,310 ± 905.1 |
| Dasatinib | 340 ± 30.8 | 3120 ± 153.7 | 10,230 ± 1198 | 9210 ± 226.9 |
| Sorafenib | 250 ± 13.6 | 585 ± 47.3 | 13,660 ± 1119 | 130 ± 11.3 |
| Sunitinib | 3640 ± 550 | 2910 ± 190.6 | 3420 ± 277.6 | 285 ± 13.3 |
| Nilotinib | 46.0 ± 4.3 | 11.0 ± 1.6 | 31.0 ± 2.9 | 23.0 ± 4.1 |
| Lapatinib | 26.0 ± 5.8 | 739.0 ± 35.3 | 80.0 ± 8.9 | 1380 ± 125.4 |
| Doxorubicin | 180.5 ± 19.6 | 191.2 ± 17.6 | 700.0 ± 180 | 127.7 ± 8.0 |
| Tyrosine Kinase | Forward 5′-3′ | Reverse 5′-3′ | Annealing Temperature (°C) | Reference |
|---|---|---|---|---|
| c-Kit | GTTGAGGCAACTTGCTTATGG | GCTTCTGCATGATCTTCCTG | 58 | [44] |
| LCK | CGATCTGGTCCGCCATTACA | ATGGTTTCTGGGGCTTCTGG | 60 | [45] |
| YES | GCGGTAGCAGCGACTCAT | TGCACCTCCAAAAGGCGTTA | 59 | * |
| FYN | AAAACTGACGGAGGAGAGGG | TTCACACCTCCAAAGACGGT | 61 | [46] |
| FRK | CGAGCAGGTGACAAACTTCA | TGCCTGTAGGCTTCTGTCCT | 61 | [47] |
| LYN | TGAAGACTCAACCAGTTCCAGA | GGTGGATGCCATCATAGGGG | 60 | * |
| BTK | GAGAAGCTGGTGCAGTTGTA | GGCCGAAATCAGATACTTTAAC | 51.9 | [48] |
| ABL | AGGTAGCTGAGGAGCTTGGGAGAG | TTTGCTTTCGAGGCAGTGCTGGGG | 66 | [49] |
| GAPDH | CAGCCTCAAGATCATCAGC | ATGATGTTCTGGAGAGCCC | 50 | [46] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
López-Mejía, J.A.; Tallabs-Utrilla, L.F.; Salazar-Sojo, P.; Mantilla-Ollarves, J.C.; Sánchez-Carballido, M.A.; Rocha-Zavaleta, L. c-Kit Induces Migration of Triple-Negative Breast Cancer Cells and Is a Promising Target for Tyrosine Kinase Inhibitor Treatment. Int. J. Mol. Sci. 2022, 23, 8702. https://doi.org/10.3390/ijms23158702
López-Mejía JA, Tallabs-Utrilla LF, Salazar-Sojo P, Mantilla-Ollarves JC, Sánchez-Carballido MA, Rocha-Zavaleta L. c-Kit Induces Migration of Triple-Negative Breast Cancer Cells and Is a Promising Target for Tyrosine Kinase Inhibitor Treatment. International Journal of Molecular Sciences. 2022; 23(15):8702. https://doi.org/10.3390/ijms23158702
Chicago/Turabian StyleLópez-Mejía, José A., Luis F. Tallabs-Utrilla, Pablo Salazar-Sojo, Jessica C. Mantilla-Ollarves, Manuel A. Sánchez-Carballido, and Leticia Rocha-Zavaleta. 2022. "c-Kit Induces Migration of Triple-Negative Breast Cancer Cells and Is a Promising Target for Tyrosine Kinase Inhibitor Treatment" International Journal of Molecular Sciences 23, no. 15: 8702. https://doi.org/10.3390/ijms23158702
APA StyleLópez-Mejía, J. A., Tallabs-Utrilla, L. F., Salazar-Sojo, P., Mantilla-Ollarves, J. C., Sánchez-Carballido, M. A., & Rocha-Zavaleta, L. (2022). c-Kit Induces Migration of Triple-Negative Breast Cancer Cells and Is a Promising Target for Tyrosine Kinase Inhibitor Treatment. International Journal of Molecular Sciences, 23(15), 8702. https://doi.org/10.3390/ijms23158702