
Combined QTL Mapping across Multiple Environments and Co-Expression Network Analysis Identified Key Genes for Embryogenic Callus Induction from Immature Maize Embryos

 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. Phenotypic Performances of EC Induction Traits under Three Environments
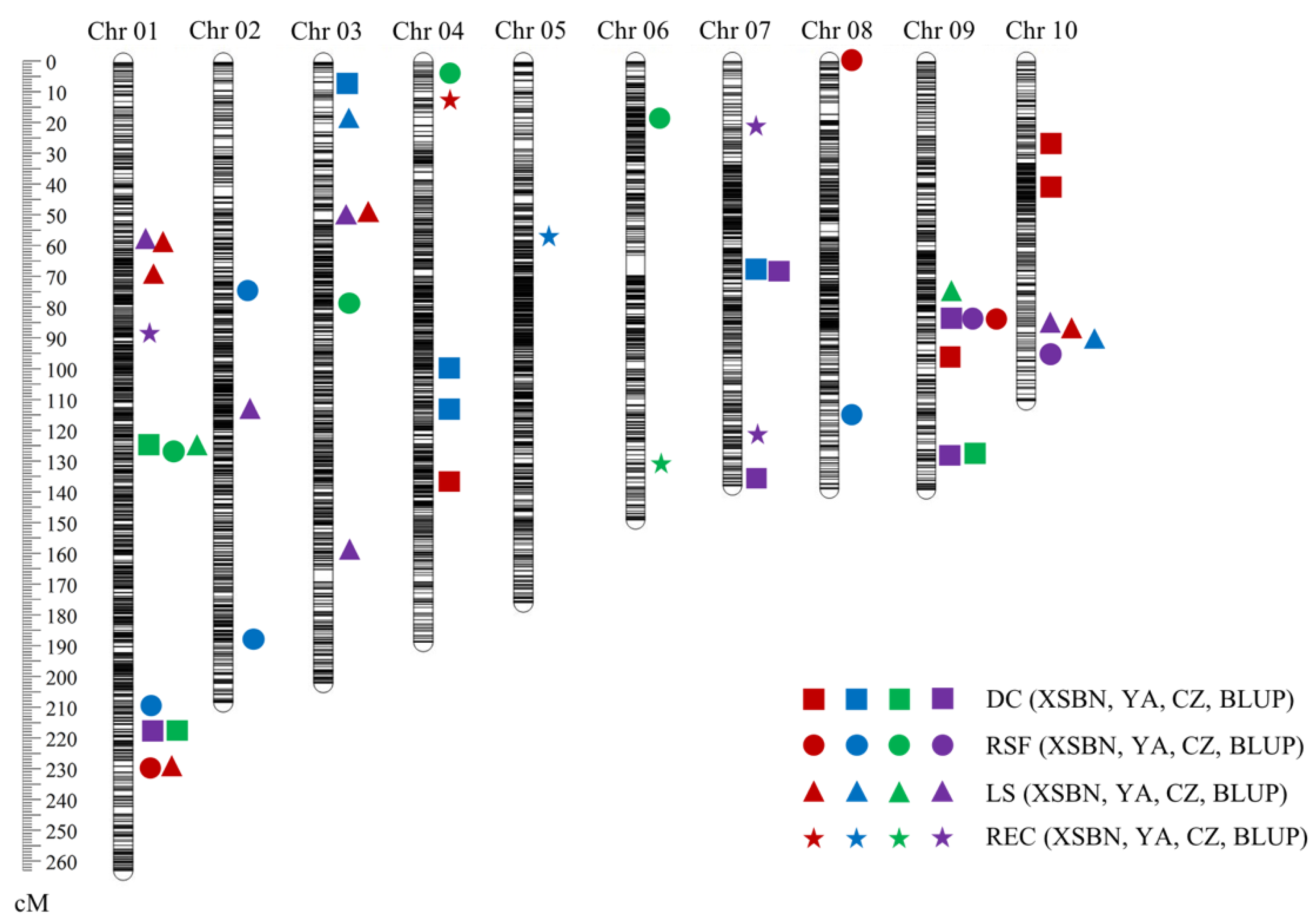
2.2. QTL Responsible for EC Induction
2.2.1. DC
2.2.2. RSF
2.2.3. LS
2.2.4. REC
2.3. QTL Clusters for Embryogenic Callus Induction-Related Traits
2.4. Validation of QTL Intervals
2.5. Candidate Genes and Co-Expression Networks
2.6. Hub Gene-Based Association Mapping
3. Discussion
3.1. Use of IBM Syn10 DH Population for Mapping Embryogenic Callus Induction-Related Traits
3.2. Maize EC Induction Is Probably Controlled by a Few Major Genes
3.3. QTLs for Embryogenic Callus Induction-Related Traits
3.4. Candidate Genes Involved in Embryogenic Callus Induction
4. Materials and Methods
4.1. Plant Materials and Field Trials
4.2. Immature Embryo Culture and Callus Induction
4.3. Phenotype Investigation
4.4. Phenotypic Data Analysis
4.5. QTL Analysis
4.6. DNA Extraction and Variation Validation
4.7. WGCNA
4.8. Association Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ahmar, S.; Gill, R.A.; Jung, K.-H.; Faheem, A.; Qasim, M.U.; Mubeen, M.; Zhou, W. Conventional and molecular techniques from simple breeding to speed breeding in crop plants: Recent advances and future outlook. Int. J. Mol. Sci. 2020, 21, 2590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; He, S.; Long, Y.; Zhang, X.; Zhang, X.; Hu, H.; Li, Z.; Hou, F.; Ge, F.; Gao, S. Genetic variations in ZmSAUR15 contribute to the formation of immature embryo-derived embryonic calluses in maize. Plant J. 2022, 109, 980–991. [Google Scholar] [CrossRef]
- Satish, L.; Rathinapriya, P.; Ceasar, S.A.; Rency, A.S.; Pandian, S.; Rameshkumar, R.; Subramanian, A.; Ramesh, M. Effects of cefotaxime, amino acids and carbon source on somatic embryogenesis and plant regeneration in four Indian genotypes of foxtail millet (Setaria italica L.). In Vitr. Cell. Dev. Biol. Plant 2015, 52, 140–153. [Google Scholar] [CrossRef]
- Saha, S.; Islam, Z.; Islam, S.; Hassan, M.F.; Hossain, M.S.; Islam, S.M.S. Enhancement of Somatic Embryogenesis by Mature and Immature Seeds in Wheat (Triticum aestivum L.). J. Biol. Life Sci. 2017, 8, 20. [Google Scholar] [CrossRef] [Green Version]
- Ana, L.M.L.; Ivone, B.d.O.E.; Carlos, A.S.; Claudete, A.M.; Maria, d.F.P.S.M. Somatic embryogenesis and plant regeneration in popcorn (Zea mays L.) inbred lines. Afr. J. Biotechnol. 2017, 16, 1738–1742. [Google Scholar] [CrossRef] [Green Version]
- Abe, T. Genotypic variability for callus formation and plant regeneration in rice. Theor. Appl. Genet. 1986, 72, 3–10. [Google Scholar] [CrossRef]
- Ge, F.; Qu, J.; Liu, P.; Pan, L.; Zou, C.; Yuan, G.; Yang, C.; Pan, G.; Huang, J.; Ma, L. Genome assembly of the maize inbred line A188 provides a new reference genome for functional genomics. Crop J. 2022, 10, 47–55. [Google Scholar] [CrossRef]
- Jia, H.; Yi, D.; Yu, J.; Xue, S.; Ma, Z. Mapping QTLs for tissue culture response of mature wheat embryos. Mol. Cells 2007, 23, 323–330. [Google Scholar]
- Zhao, L.; Manli, Y.U.; Guo, H.; Jiayu, G.U.; Xie, Y.; Zhao, S.; Xiong, H.; Liu, L. Preliminary Gene Mapping for Anther Culture Ability Based on a New Wheat Germplasm with High Anther Culture Ability. J. Nucl. Agric. Sci. 2018, 32, 2305–2312. [Google Scholar]
- Kwon, Y.S.; Kim, K.M.; Eun, M.Y.; Sohn, J.K. Quantitative trait loci mapping associated with plant regeneration ability from seed derived calli in rice (Oryza sativa L.). Mol. Cells 2001, 11, 64–67. [Google Scholar]
- Pan, G.; Zhang, Z.; Wei, X.; Song, Y.; Zhao, M.; Xia, Y.; Rong, T. QTL Analysis of Maize (Zea may L.) Embryo Culturing Capacity. Acta Agron. Sin. 2006, 32, 7–13. [Google Scholar]
- Zhang, H.; Liu, Y.; Guo, X.; Zhang, F.; Li, J.; Tan, Z.; Chen, G.; Sun, D. QTL mapping for callus induction and plant regeneration in maize immature embryos. Acta Agron. Sin. 2006, 32, 385–389. [Google Scholar]
- Salvo, S.; Cook, J.; Carlson, A.R.; Hirsch, C.N.; Kaeppler, S.M.; Kaeppler, H.F. Genetic Fine-Mapping of a Quantitative Trait Locus (QTL) Associated with Embryogenic Tissue Culture Response and Plant Regeneration Ability in Maize (Zea mays L.). Plant Genome 2018, 11, 170111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, L.; An, R.; Jiang, L.; Zhang, C.; Li, Z.; Zou, C.; Yang, C.; Pan, G.; Lübberstedt, T.; Shen, Y. Effects of ZmHIPP on lead tolerance in maize seedlings: Novel ideas for soil bioremediation. J. Hazard. Mater. 2022, 430, 128457. [Google Scholar] [CrossRef]
- Ledwoń, A.; Gaj, M.D. LEAFY COTYLEDON2 gene expression and auxin treatment in relation to embryogenic capacity of Arabidopsis somatic cells. Plant Cell Rep. 2009, 28, 1677. [Google Scholar] [CrossRef] [PubMed]
- Lowe, K.; Wu, E.; Wang, N.; Hoerster, G.; Hastings, C.; Cho, M.-J.; Scelonge, C.; Lenderts, B.; Chamberlin, M.; Cushatt, J.; et al. Morphogenic Regulators Baby boom and Wuschel Improve Monocot Transformation. Plant Cell 2016, 28, 1998–2015. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.-Q.; Lian, H.; Zhou, C.-M.; Xu, L.; Jiao, Y.; Wang, J.-W. A Two-Step Model for de Novo Activation of WUSCHEL during Plant Shoot Regeneration. Plant Cell 2017, 29, 1073–1087. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Liu, M.; Yan, Y.; Qing, C.; Zhang, X.; Zhang, Y.; Long, Y.; Wang, L.; Pan, L.; Zou, C. Genetic dissection of maize embryonic callus regenerative capacity using multi-locus genome-wide association studies. Front. Plant Sci. 2018, 9, 561. [Google Scholar] [CrossRef] [Green Version]
- Ge, F.; Luo, X.; Huang, X.; Zhang, Y.; He, X.; Liu, M.; Lin, H.; Peng, H.; Li, L.; Zhang, Z. Genome-wide analysis of transcription factors involved in maize embryonic callus formation. Physiol. Plant. 2016, 158, 452–462. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Jansen, C.; Zhang, Y.; Liu, H.; Gonzalez-Portilla, P.J.; Lauter, N.; Kumar, B.; Trucillo-Silva, I.; Martin, J.P.S.; Lee, M.; Simcox, K.; et al. Genetic and agronomic assessment of cob traits in corn under low and normal nitrogen management conditions. Theor. Appl. Genet. 2015, 128, 1231–1242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morino, K.; Matsuda, F.; Miyazawa, H.; Sukegawa, A.; Miyagawa, H.; Wakasa, K. Metabolic profiling of tryptophan-overproducing rice calli that express a feedback-insensitive α subunit of anthranilate synthase. Plant Cell Physiol. 2005, 46, 514–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, F.; Hu, H.; Huang, X.; Zhang, Y.; Wang, Y.; Li, Z.; Zou, C.; Peng, H.; Li, L.; Gao, S. Metabolomic and proteomic analysis of maize embryonic callus induced from immature embryo. Sci. Rep. 2017, 7, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, X.; Fang, T.; Liu, Y.; Huang, L.; Zang, M.; Wang, G.; Liu, Y.; Fu, J. Transcriptome profiling predicts new genes to promote maize callus formation and transformation. Front. Plant Sci. 2019, 10, 1633. [Google Scholar] [CrossRef]
- Roos, D.; de Boer, M. Mutations in cis that affect mRNA synthesis, processing and translation. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2021, 1867, 166166. [Google Scholar] [CrossRef]
- Haddad, L.A. Protein-Coding Genes. In Human Genome Structure, Function and Clinical Considerations; Haddad, L.A., Ed.; Springer International Publishing: Cham, Switzerland, 2021; pp. 93–138. [Google Scholar]
- Li, H.; Zhang, L.; Wang, J. Analysis and answers to frequently asked questions in quantitative trait locus mapping. Acta Agron. Sin. 2010, 36, 918–931. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.-K. Inclusive composite interval mapping of quantitative trait genes. Acta Agron. Sin. 2009, 35, 239–245. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Li, H.; Li, Z.; Wang, J. Interactions Between Markers Can Be Caused by the Dominance Effect of Quantitative Trait Loci. Genetics 2008, 180, 1177–1190. [Google Scholar] [CrossRef] [Green Version]
- Krakowsky, M.D.; Lee, M.; Garay, L.; Woodman-Clikeman, W.; Long, M.J.; Sharopova, N.; Frame, B.; Wang, K. Quantitative trait loci for callus initiation and totipotency in maize (Zea mays L.). Theor. Appl. Genet. 2006, 113, 821–830. [Google Scholar] [CrossRef] [Green Version]
- Taguchi-Shiobara, F.; Yamamoto, T.; Yano, M.; Oka, S. Mapping QTLs that control the performance of rice tissue culture and evaluation of derived near-isogenic lines. Theor. Appl. Genet. 2006, 112, 968–976. [Google Scholar] [CrossRef]
- Li, S.; Yan, S.; Wang, A.h.; Zou, G.; Huang, X.; Han, B.; Qian, Q.; Tao, Y. Identification of QTLs associated with tissue culture response through sequencing-based genotyping of RILs derived from 93-11 × Nipponbare in rice (Oryza sativa). Plant Cell Rep. 2013, 32, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Fehér, A. Callus, Dedifferentiation, Totipotency, Somatic Embryogenesis: What These Terms Mean in the Era of Molecular Plant Biology? Front. Plant Sci. 2019, 10, 536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Etedali, F.; Khossroshahli, M.; Motallebi-Azar, A.; Valizadeh, M.; Kazemiani, S. Gene action and heterosis for callus induction and growth from mature embryo culture of rapeseed (Brassica napus L.). Int. Res. J. Agric. Sci. Soil Sci. 2011, 1, 402–407. [Google Scholar]
- Chang, L.; He, K.; Cui, T.; Xue, J.; Liu, J. QTL mapping and QTL × environment interaction analysis of kernel ratio in maize (Zea mays). J. Agric. Biotechnol. 2017, 25, 517–525. [Google Scholar]
- van Eeuwijk, F.A.; Bink, M.C.A.M.; Chenu, K.; Chapman, S.C. Detection and use of QTL for complex traits in multiple environments. Curr. Opin. Plant Biol. 2010, 13, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Rocheford, T.R.; Widholm, J.M. RFLP analysis to identify putative chromosomal regions involved in the anther culture response and callus formation of maize. Theor. Appl. Genet. 1992, 85, 360–365. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, C.L.; Romero-Severson, J.; Hodges, T.K. Improved tissue culture response of an elite maize inbred through backcross breeding, and identification of chromosomal regions important for regeneration by RFLP analysis. Theor. Appl. Genet. 1992, 84, 755–762. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, X.; Lin, Y.; Xie, G.; Fu, F.; Liu, H.; Wang, J.; Gao, S.; Lan, H.; Rong, T. Characterization of a ZmSERK gene and its relationship to somatic embryogenesis in a maize culture. Plant Cell Tissue Organ Cult. 2011, 105, 29–37. [Google Scholar] [CrossRef]
- Glawischnig, E.; Tomas, A.; Eisenreich, W.; Spiteller, P.; Bacher, A.; Gierl, A. Auxin biosynthesis in maize kernels. Plant Physiol. 2000, 123, 1109–1120. [Google Scholar] [CrossRef] [Green Version]
- Guilfoyle, T.; Hagen, G. Auxin signal transduction. Essays Biochem. 2015, 58, 1–12. [Google Scholar] [CrossRef]
- Su, Y.H.; Zhao, X.Y.; Liu, Y.B.; Zhang, C.L.; O’Neill, S.D.; Zhang, X.S. Auxin-induced WUS expression is essential for embryonic stem cell renewal during somatic embryogenesis in Arabidopsis. Plant J. 2009, 59, 448–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nic-Can, G.I.; Loyola-Vargas, V.M. The Role of the Auxins During Somatic Embryogenesis. In Somatic Embryogenesis: Fundamental Aspects and Applications; Loyola-Vargas, V.M., Ochoa-Alejo, N., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 171–182. [Google Scholar]
- Wójcik, A.M.; Wójcikowska, B.; Gaj, M.D. Current Perspectives on the Auxin-Mediated Genetic Network that Controls the Induction of Somatic Embryogenesis in Plants. Int. J. Mol. Sci. 2020, 21, 1333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, S.; Dwivedi, U.N. Plant regeneration from callus of Cuscuta reflexa–an angiospermic parasite and modulation of catalase and peroxidase activity by salicylic acid and naphthalene acetic acid. Plant Physiol. Biochem. 2001, 39, 529–538. [Google Scholar] [CrossRef]
- Hao, L.; Zhou, L.; Xu, X.; Cao, J.; Xi, T. The role of salicylic acid and carrot embryogenic callus extracts in somatic embryogenesis of naked oat (Avena nuda). Plant Cell Tissue Organ Cult. 2006, 85, 109–113. [Google Scholar] [CrossRef]
- Gadzovska, S.; Maury, S.; Delaunay, A.; Spasenoski, M.; Hagège, D.; Courtois, D.; Joseph, C. The influence of salicylic acid elicitation of shoots, callus, and cell suspension cultures on production of naphtodianthrones and phenylpropanoids in Hypericum perforatum L. Plant Cell Tissue Organ Cult. 2013, 113, 25–39. [Google Scholar] [CrossRef]
- Armstrong, C.; Green, C. Establishment and maintenance of friable, embryogenic maize callus and the involvement of L-proline. Planta 1985, 164, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Knapp, S.; Stroup, W.; Ross, W. Exact confidence intervals for heritability on a progeny mean basis 1. Crop Sci. 1985, 25, 192–194. [Google Scholar] [CrossRef]
- Liu, H.; Niu, Y.; Gonzalez-Portilla, P.J.; Zhou, H.; Wang, L.; Zuo, T.; Qin, C.; Tai, S.; Jansen, C.; Shen, Y.; et al. An ultra-high-density map as a community resource for discerning the genetic basis of quantitative traits in maize. BMC Genom. 2015, 16, 1078. [Google Scholar] [CrossRef] [Green Version]
- Meng, L.; Li, H.; Zhang, L.; Wang, J. QTL IciMapping: Integrated software for genetic linkage map construction and quantitative trait locus mapping in biparental populations. Crop J. 2015, 3, 269–283. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.; Li, H.; Zhang, L.; Wang, J. Properties of the test statistic under null hypothesis and the calculation of LOD threshold in quantitative trait loci (QTL) mapping. Acta Agron. Sin. 2013, 39, 1–11. [Google Scholar] [CrossRef]
- Saghai-Maroof, M.A.; Soliman, K.M.; Jorgensen, R.A.; Allard, R. Ribosomal DNA spacer-length polymorphisms in barley: Mendelian inheritance, chromosomal location, and population dynamics. Proc. Natl. Acad. Sci. USA 1984, 81, 8014–8018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genom. Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait a | Environment b | Parents (n = 6, t-Test) | IBM Syn10 DH Population | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Mo17 ± SD | B73 ± SD | Range | Average | SD | CV% | Skewness | Kurtosis | ||
| RSF (%) | XSBN | 81.34 ± 4.00 | 95.77 ± 0.64 ** | 0.00–99.07 | 67.64 | 24.99 | 0.37 | −0.78 | −0.42 |
| YA | 81.82 ± 6.24 | 96.67 ± 1.96 ** | 0.00–100.00 | 70.84 | 25.53 | 0.36 | −0.86 | −0.25 | |
| CZ | 79.06 ± 2.35 | 94.44 ± 5.01 ** | 0.00–100.00 | 70.27 | 24.87 | 0.35 | −1.02 | 0.26 | |
| LS (mm) | XSBN | 4.0 ± 0.0 | 10.3 ± 0.6 ** | 0.0–15.0 | 5.5 | 2.65 | 0.48 | 0.70 | 0.20 |
| YA | 4.3 ± 0.3 | 11.0 ± 1.0 ** | 0.0–12.0 | 6.0 | 2.54 | 0.42 | 0.26 | −0.32 | |
| CZ | 5.0 ± 0.0 | 10.2 ± 1.8 ** | 0.0–11.7 | 5.3 | 2.29 | 0.43 | 0.38 | 0.13 | |
| DC (mm) | XSBN | 2.7 ± 0.3 | 3.8 ± 0.3 ** | 1.5–4.3 | 2.9 | 0.66 | 0.23 | 0.34 | −0.85 |
| YA | 3.0 ± 0.0 | 3.8 ± 0.3 ** | 2.0–5.0 | 3.0 | 0.56 | 0.19 | 0.33 | −0.50 | |
| CZ | 3.3 ± 0.3 | 3.8 ± 0.3 | 2.0–4.0 | 2.8 | 0.46 | 0.16 | 0.15 | 0.39 | |
| REC (%) | XSBN | 6.94 ± 1.39 | 0.00 ± 0.00 ** | 0.00–52.17 | 4.39 | 8.79 | 2.00 | 3.08 | 10.54 |
| YA | 7.05 ± 1.91 | 0.00 ± 0.00 ** | 0.00–58.61 | 2.74 | 7.95 | 2.90 | 4.48 | 22.56 | |
| CZ | 3.39 ± 1.23 | 0.00 ± 0.00 ** | 0.00–56.48 | 2.27 | 6.03 | 2.67 | 4.84 | 32.70 | |
| Environment | Trait | REC (n = 210) | SC (n = 210) | LS (n = 210) |
|---|---|---|---|---|
| XSBN | RSF | −0.310 ** | 0.531 ** | 0.692 ** |
| LS | −0.263 ** | 0.725 ** | ||
| DC | −0.304 ** | |||
| YA | RSF | −0.342 ** | 0.482 ** | 0.693 ** |
| LS | −0.243 ** | 0.612 ** | ||
| DC | −0.318 ** | |||
| CZ | RSF | −0.105 | 0.571 ** | 0.737 ** |
| LS | −0.134 * | 0.639 ** | ||
| DC | −0.177 * |
| Trait | Source of Variation | df | Mean Square | Significance | H2 (%) |
|---|---|---|---|---|---|
| RSF | Genotype(G) | 209 | 2092.792 | <0.01 ** | 84.02 |
| Environment(E) | 2 | 1136.991 | <0.01 ** | ||
| G × E | 418 | 1147.495 | <0.01 ** | ||
| Error | 1260 | 140.281 | |||
| LS | Genotype(G) | 209 | 22.363 | <0.01 ** | 85.64 |
| Environment(E) | 2 | 40.006 | <0.01 ** | ||
| G × E | 418 | 10.892 | <0.01 ** | ||
| Error | 1260 | 1.067 | |||
| DC | Genotype(G) | 209 | 1.076 | <0.01 ** | 83.17 |
| Environment(E) | 2 | 3.584 | <0.01 ** | ||
| G × E | 418 | 0.633 | <0.01 ** | ||
| Error | 1260 | 0.061 | |||
| REC | Genotype(G) | 209 | 194.825 | <0.01 ** | 88.27 |
| Environment(E) | 2 | 369.202 | <0.01 ** | ||
| G × E | 418 | 73.082 | <0.01 ** | ||
| Error | 1260 | 13.689 |
| Trait | Name | Env. | Chr. | Genetic Position (cM) | Physical Position (Mb) | LOD a | PVE b | ADD c |
|---|---|---|---|---|---|---|---|---|
| DC | qDC1-1 | CZ | 1 | 125.84 | 191.425–191.600 | 3.02 | 7.10 | −0.1174 |
| qDC1-2 | CZ | 1 | 217.21 | 286.200–286.300 | 2.61 | 5.73 | 0.1089 | |
| qDC1-2 | BLUP | 1 | 217.49 | 286.400–286.550 | 3.55 | 4.64 | 0.0398 | |
| qDC3-1 | YA | 3 | 9.85 | 2.700–2.800 | 3.19 | 6.59 | −0.1322 | |
| qDC4-1 | YA | 4 | 101.97 | 170.300–170.675 | 3.40 | 7.13 | 0.1407 | |
| qDC4-2 | YA | 4 | 116.7 | 180.300–180.400 | 4.76 | 9.98 | −0.1705 | |
| qDC4-3 | XSBN | 4 | 138.78 | 210.875–211.000 | 2.85 | 5.30 | 0.1734 | |
| qDC7-1 | YA | 7 | 71.29 | 132.275–132.525 | 3.21 | 6.62 | 0.1437 | |
| qDC7-1 | BLUP | 7 | 72.01 | 132.800–132.900 | 4.84 | 6.40 | 0.0501 | |
| qDC7-2 | BLUP | 7 | 140.95 | 174.175–174.300 | 3.42 | 4.61 | −0.0391 | |
| qDC9-1 | BLUP | 9 | 86.05 | 133.775–133.900 | 5.29 | 7.13 | 0.0504 | |
| qDC9-2 | XSBN | 9 | 99.62 | 141.700–141.950 | 4.14 | 7.89 | 0.1858 | |
| qDC9-3 | CZ | 9 | 131.38 | 152.200–152.300 | 3.92 | 9.49 | 0.1512 | |
| qDC9-3 | BLUP | 9 | 132.23 | 152.600–152.700 | 4.06 | 5.40 | 0.047 | |
| qDC10-1 | XSBN | 10 | 27.82 | 7.400–7.775 | 4.00 | 7.79 | 0.1936 | |
| qDC10-2 | XSBN | 10 | 41.13 | 67.250–68.900 | 3.81 | 7.17 | −0.184 | |
| RSF | qRSF1-1 | CZ | 1 | 127.16 | 192.525–193.650 | 4.29 | 7.37 | −6.7656 |
| qRSF1-2 | YA | 1 | 211.45 | 280.975–281.100 | 3.46 | 7.59 | −6.9403 | |
| qRSF1-3 | XSBN | 1 | 230.48 | 290.700–290.800 | 3.21 | 6.49 | −6.7815 | |
| qRSF2-1 | YA | 2 | 75.79 | 32.650–33.175 | 2.68 | 5.33 | −5.8722 | |
| qRSF2-2 | YA | 2 | 190.64 | 232.500–232.600 | 4.75 | 9.95 | −8.0007 | |
| qRSF3-1 | CZ | 3 | 81.31 | 155.675–155.675 | 5.71 | 9.97 | 8.2628 | |
| qRSF4-1 | CZ | 4 | 5.09 | 2.500–2.600 | 3.77 | 6.36 | −6.4367 | |
| qRSF6-1 | CZ | 6 | 17.13 | 13.600–13.725 | 3.31 | 5.65 | 6.0506 | |
| qRSF8-1 | XSBN | 8 | 0.06 | 0.100–0.350 | 2.82 | 5.55 | 6.1132 | |
| qRSF8-2 | YA | 8 | 115.14 | 169.300–169.400 | 3.30 | 6.61 | −6.8164 | |
| qRSF9-1 | BLUP | 9 | 86.7 | 133.900–134.000 | 4.49 | 6.89 | 2.655 | |
| qRSF9-1 | XSBN | 9 | 87.33 | 134.000–134.100 | 3.73 | 7.46 | 7.3017 | |
| qRSF10-1 | BLUP | 10 | 95.16 | 145.200–145.300 | 4.30 | 7.13 | −2.8252 | |
| LS | qLS1-1 | BLUP | 1 | 58.26 | 23.675–23.925 | 6.83 | 9.74 | 0.3231 |
| qLS1-1 | XSBN | 1 | 60.77 | 24.975–25.225 | 7.70 | 11.54 | 0.946 | |
| qLS1-2 | XSBN | 1 | 71.27 | 35.375–35.625 | 3.15 | 4.44 | −0.5845 | |
| qLS1-3 | CZ | 1 | 127.16 | 192.525–193.650 | 5.59 | 10.68 | −0.8065 | |
| qLS1-4 | XSBN | 1 | 231.2 | 290.800–290.900 | 3.51 | 5.03 | −0.6832 | |
| qLS2-1 | BLUP | 2 | 115.59 | 185.350–185.550 | 3.54 | 4.90 | 0.2473 | |
| qLS3-1 | YA | 3 | 17.74 | 4.100–4.200 | 2.98 | 6.29 | 0.6625 | |
| qLS3-2 | BLUP | 3 | 52.41 | 14.300–14.400 | 6.33 | 9.20 | −0.8419 | |
| qLS3-2 | XSBN | 3 | 52.68 | 14.650–14.850 | 5.14 | 7.34 | −0.2805 | |
| qLS3-3 | BLUP | 3 | 161.37 | 216.325–216.550 | 3.15 | 4.42 | 0.2312 | |
| qLS9-1 | CZ | 9 | 77.71 | 117.825–118.200 | 3.27 | 6.05 | −0.6074 | |
| qLS10-1 | BLUP | 10 | 87.58 | 143.675–143.800 | 2.90 | 4.01 | −0.2087 | |
| qLS10-1 | XSBN | 10 | 91.16 | 144.600–144.700 | 3.96 | 5.90 | −0.7004 | |
| qLS10-1 | YA | 10 | 96.74 | 145.400–145.500 | 3.24 | 6.98 | −0.741 | |
| REC | qREC1-1 | BLUP | 1 | 89.61 | 66.800–66.900 | 3.83 | 6.25 | 1.0291 |
| qREC4-1 | XSBN | 4 | 14.29 | 4.400–4.500 | 3.18 | 11.45 | 2.3818 | |
| qREC5-1 | YA | 5 | 58.4 | 21.200–21.300 | 3.13 | 7.73 | −2.2782 | |
| qREC6-1 | CZ | 6 | 132.21 | 164.300–164.400 | 2.55 | 6.00 | −1.5787 | |
| qREC7-1 | BLUP | 7 | 23.37 | 5.600–5.700 | 2.51 | 4.01 | −0.8092 | |
| qREC7-2 | BLUP | 7 | 120.07 | 167.200–167.300 | 3.96 | 6.36 | −0.9828 |
| QTL Cluster Number | Chromosome | Traits a | QTL Names | Position (Mb) | Positive Alleles | Range of Explained Phenotypic Variation (%) |
|---|---|---|---|---|---|---|
| a | 1 | DC + LS + RSF | qDC1-1; qRSF1-1; qLS1-3 | 191.425–193.650 | Mo17 + Mo17 + Mo17 | 7.10–10.68 |
| b | 1 | RSF + LS | qRSF1-3; qLS1-4 | 290.700–290.900 | Mo17 + Mo17 | 5.03–6.49 |
| c | 9 | DC + RSF | qDC9-1; qRSF9-1 | 133.775–134.100 | B73 + B73 + B73 | 6.89–7.46 |
| d | 10 | RSF + LS | qRSF10-1; qLS10-1 | 143.675–145.500 | Mo17 + Mo17 + Mo17 + Mo17 | 4.01–7.13 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Long, Y.; Liang, T.; Ma, L.; Liu, P.; Yang, Y.; Zhang, X.; Zou, C.; Zhang, M.; Ge, F.; Yuan, G.; et al. Combined QTL Mapping across Multiple Environments and Co-Expression Network Analysis Identified Key Genes for Embryogenic Callus Induction from Immature Maize Embryos. Int. J. Mol. Sci. 2022, 23, 8786. https://doi.org/10.3390/ijms23158786
Long Y, Liang T, Ma L, Liu P, Yang Y, Zhang X, Zou C, Zhang M, Ge F, Yuan G, et al. Combined QTL Mapping across Multiple Environments and Co-Expression Network Analysis Identified Key Genes for Embryogenic Callus Induction from Immature Maize Embryos. International Journal of Molecular Sciences. 2022; 23(15):8786. https://doi.org/10.3390/ijms23158786
Chicago/Turabian StyleLong, Yun, Tianhu Liang, Langlang Ma, Peng Liu, Yun Yang, Xiaoling Zhang, Chaoying Zou, Minyan Zhang, Fei Ge, Guangsheng Yuan, and et al. 2022. "Combined QTL Mapping across Multiple Environments and Co-Expression Network Analysis Identified Key Genes for Embryogenic Callus Induction from Immature Maize Embryos" International Journal of Molecular Sciences 23, no. 15: 8786. https://doi.org/10.3390/ijms23158786
APA StyleLong, Y., Liang, T., Ma, L., Liu, P., Yang, Y., Zhang, X., Zou, C., Zhang, M., Ge, F., Yuan, G., Lübberstedt, T., Pan, G., & Shen, Y. (2022). Combined QTL Mapping across Multiple Environments and Co-Expression Network Analysis Identified Key Genes for Embryogenic Callus Induction from Immature Maize Embryos. International Journal of Molecular Sciences, 23(15), 8786. https://doi.org/10.3390/ijms23158786