
Effect of Hypoxia on Pulmonary Endothelial Cells from Bleomycin-Induced Pulmonary Fibrosis Model Mice

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Results
2.1. Intratracheal Administration of Bleomycin Induced Inflammatory Cell Infiltration and Pulmonary Fibrosis
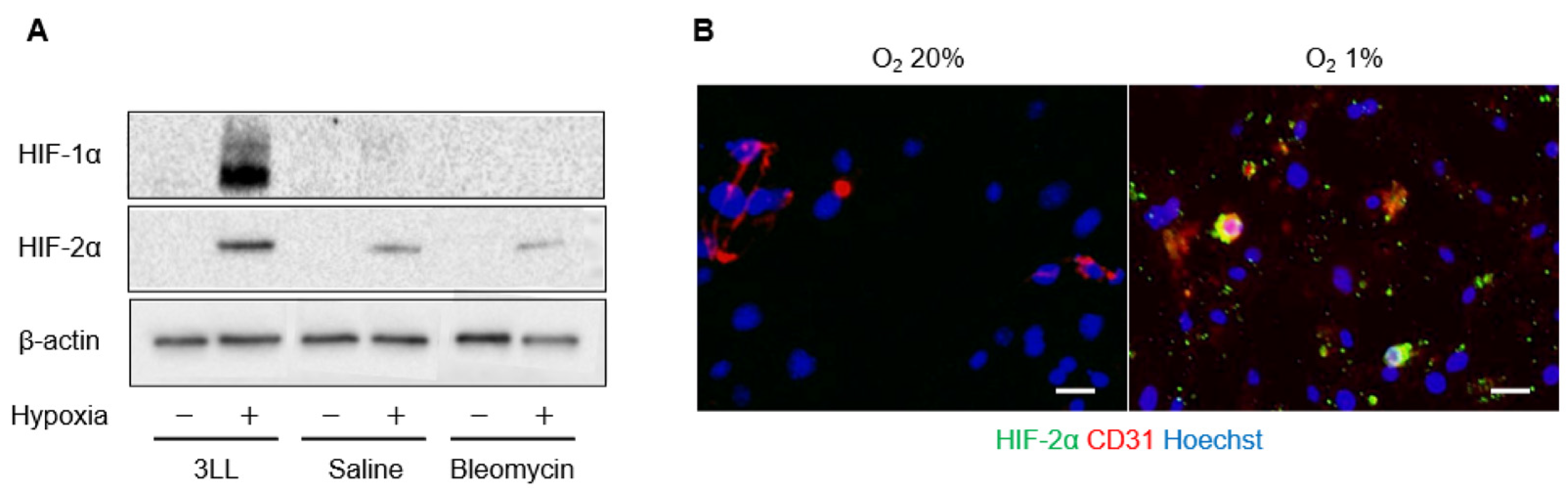
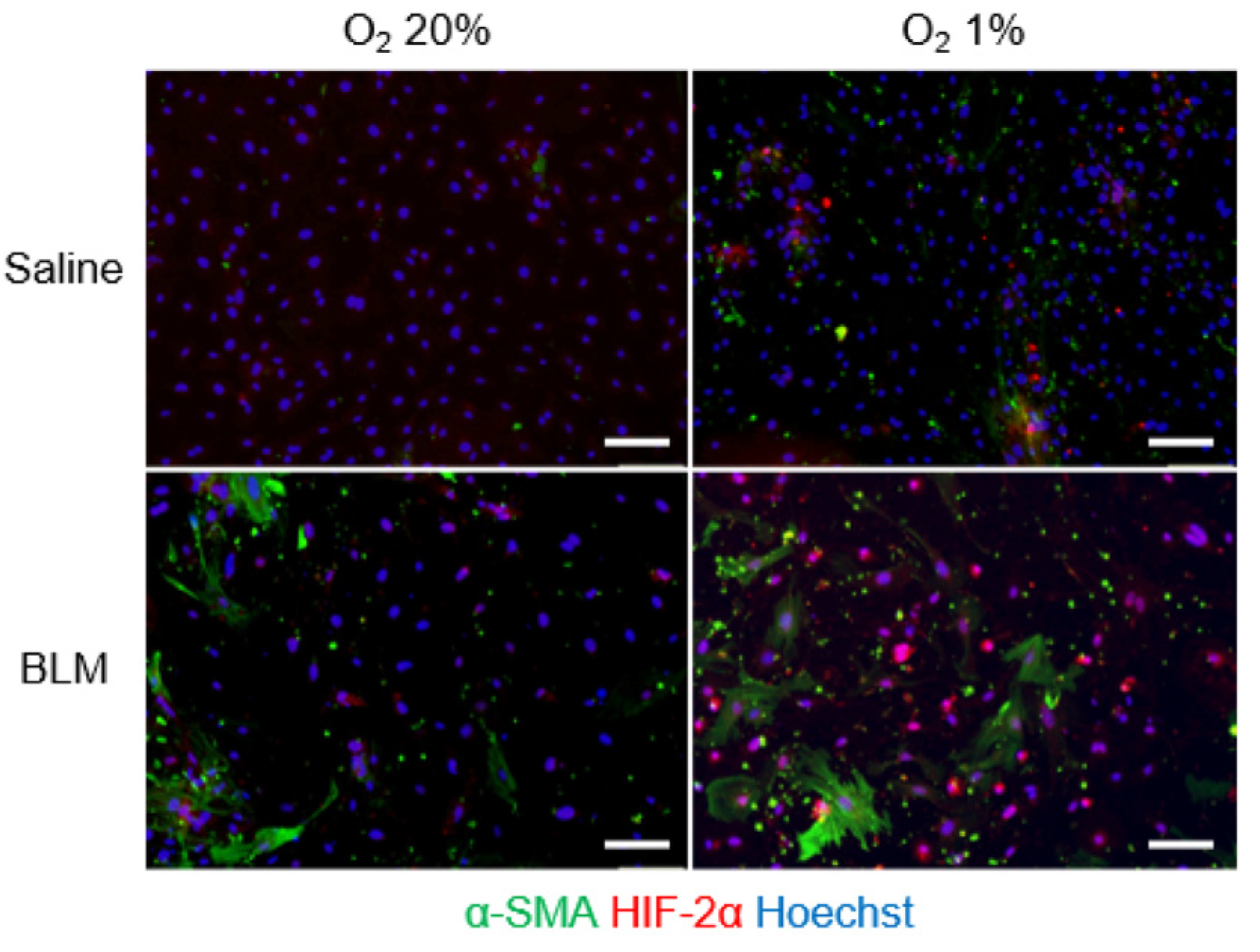
2.2. Expression of HIF-α in Pulmonary Endothelial Cells
2.3. Pulmonary Endothelial Cells Are Injured by Bleomycin Administration and Exposure to Hypoxia
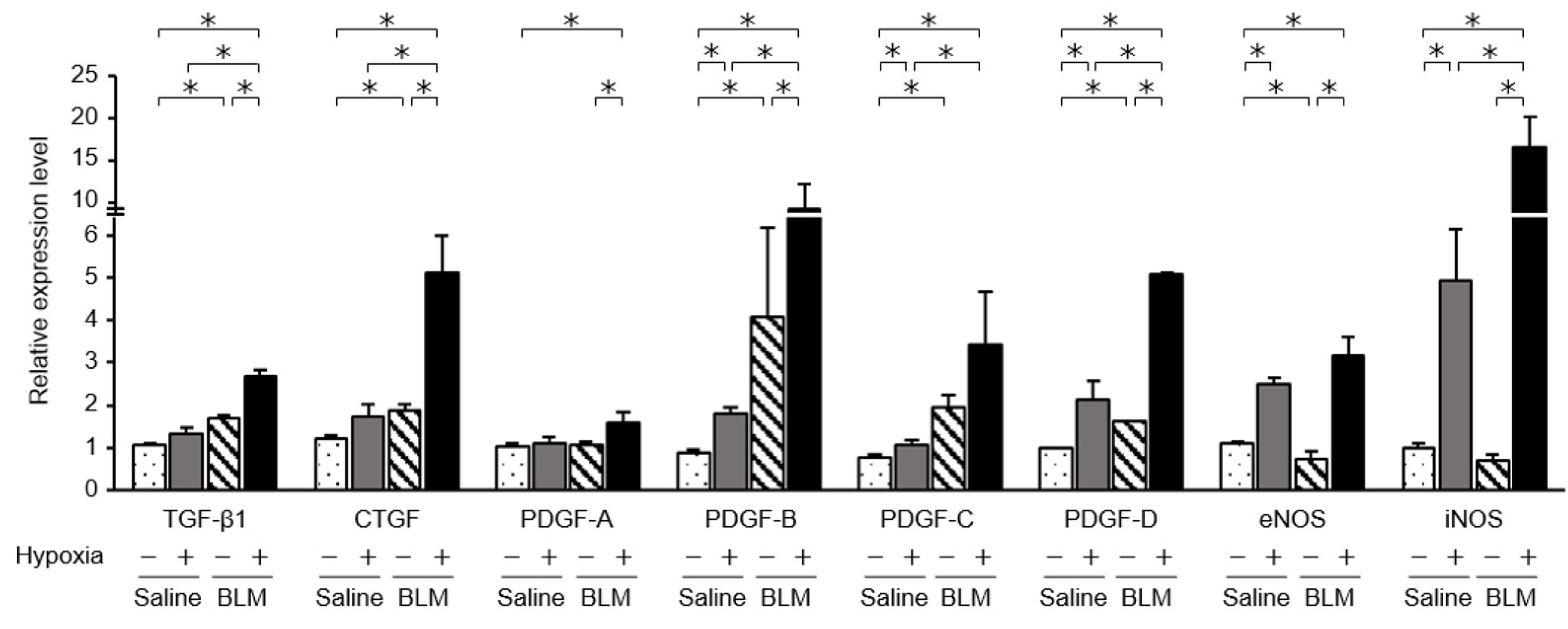
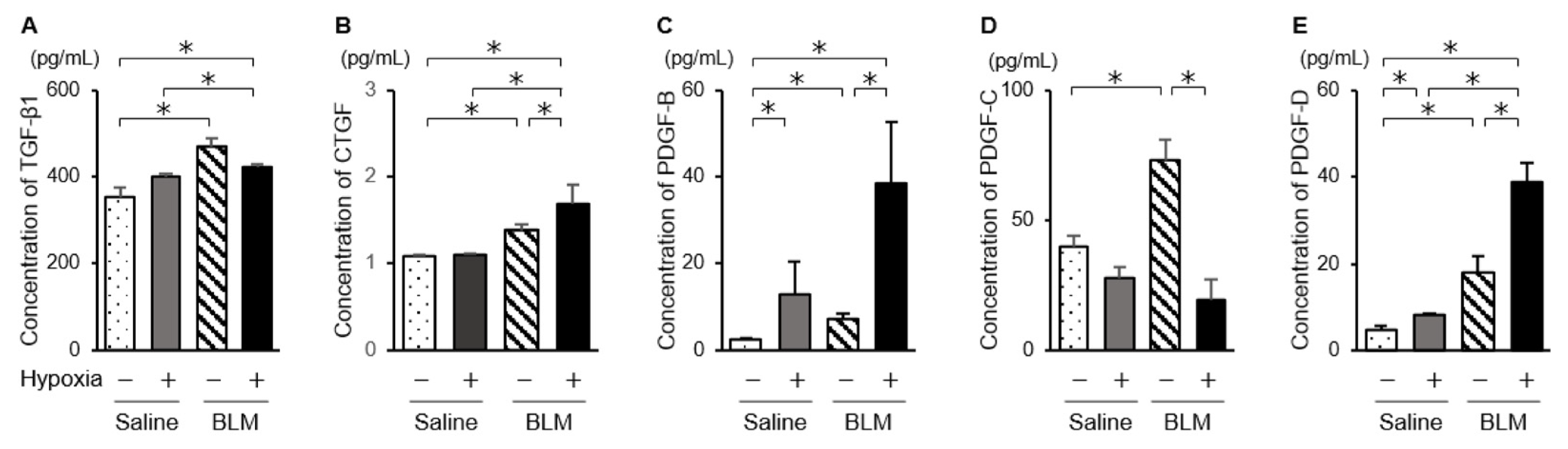
2.4. Expression of Fibrotic Mediators and NOSs in Endothelial Cells Exposed to Hypoxic Conditions
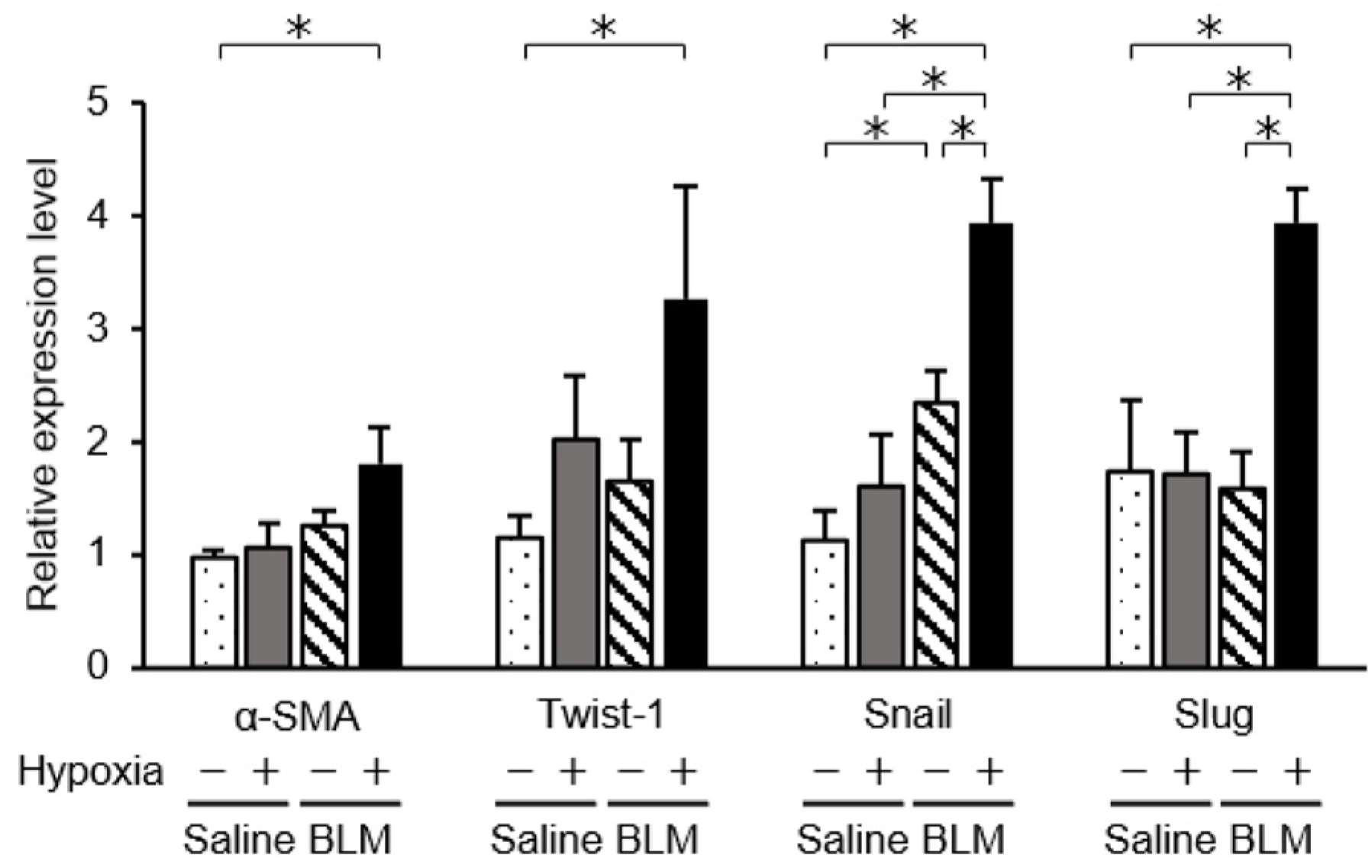
2.5. Phenotypic Changes in Endothelial Cells Cultured under Hypoxic Conditions
3. Discussion
4. Materials and Methods
4.1. Bleomycin-Induced Pulmonary Fibrosis Mouse Model
4.2. Histopathology
4.3. Isolation of Mouse Pulmonary Endothelial Cells
4.4. Cell Culture
4.5. Quantitative Real-Time PCR PCR (qRT-PCR)
4.6. Quantification of Proteins
4.7. Western Blot Analysis
4.8. Immunofluorescence Staining
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- American Thoracic Society. European Respiratory S: American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. This joint statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was adopted by the ATS board of directors, June 2001 and by the ERS Executive Committee, June 2001. Am. J. Respir. Crit. Care Med. 2002, 165, 277–304. [Google Scholar]
- King, T.E., Jr.; Pardo, A.; Selman, M. Idiopathic pulmonary fibrosis. Lancet 2011, 378, 1949–1961. [Google Scholar] [CrossRef]
- Bantsimba-Malanda, C.; Marchal-Somme, J.; Goven, D.; Freynet, O.; Michel, L.; Crestani, B.; Soler, P. A role for dendritic cells in bleomycin-induced pulmonary fibrosis in mice? Am. J. Respir. Crit. Care Med. 2010, 182, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Wolters, P.J.; Collard, H.R.; Jones, K.D. Pathogenesis of idiopathic pulmonary fibrosis. Annu. Rev. Pathol. 2014, 9, 157–179. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, I.E.; Eickelberg, O. The impact of TGF-beta on lung fibrosis: From targeting to biomarkers. Proc. Am. Thorac. Soc. 2012, 9, 111–116. [Google Scholar] [CrossRef]
- Inui, N.; Sakai, S.; Kitagawa, M. Molecular Pathogenesis of Pulmonary Fibrosis, with Focus on Pathways Related to TGF-beta and the Ubiquitin-Proteasome Pathway. Int. J. Mol. Sci. 2021, 22, 6107. [Google Scholar] [CrossRef]
- Rockey, D.C.; Bell, P.D.; Hill, J.A. Fibrosis—A common pathway to organ injury and failure. N. Engl. J. Med. 2015, 372, 1138–1149. [Google Scholar] [CrossRef] [PubMed]
- Bagnato, G.; Harari, S. Cellular interactions in the pathogenesis of interstitial lung diseases. Eur. Respir. Rev. 2015, 24, 102–114. [Google Scholar] [CrossRef]
- Yanagihara, T.; Sato, S.; Upagupta, C.; Kolb, M. What have we learned from basic science studies on idiopathic pulmonary fibrosis? Eur. Respir. Rev. 2019, 28, 190029. [Google Scholar] [CrossRef]
- Akamatsu, T.; Arai, Y.; Kosugi, I.; Kawasaki, H.; Meguro, S.; Sakao, M.; Shibata, K.; Suda, T.; Chida, K.; Iwashita, T. Direct isolation of myofibroblasts and fibroblasts from bleomycin-injured lungs reveals their functional similarities and differences. Fibrogenesis Tissue Repair 2013, 6, 15. [Google Scholar] [CrossRef] [PubMed]
- Thannickal, V.J.; Lee, D.Y.; White, E.S.; Cui, Z.; Larios, J.M.; Chacon, R.; Horowitz, J.C.; Day, R.M.; Thomas, P.E. Myofibroblast differentiation by transforming growth factor-beta1 is dependent on cell adhesion and integrin signaling via focal adhesion kinase. J. Biol. Chem. 2003, 278, 12384–12389. [Google Scholar] [CrossRef] [PubMed]
- Piera-Velazquez, S.; Li, Z.; Jimenez, S.A. Role of endothelial-mesenchymal transition (EndoMT) in the pathogenesis of fibrotic disorders. Am. J. Pathol. 2011, 179, 1074–1080. [Google Scholar] [CrossRef]
- Huang, S.K.; Scruggs, A.M.; McEachin, R.C.; White, E.S.; Peters-Golden, M. Lung Fibroblasts from Patients with Idiopathic Pulmonary Fibrosis Exhibit Genome-Wide Differences in DNA Methylation Compared to Fibroblasts from Nonfibrotic Lung. PLoS ONE 2014, 9, e107055. [Google Scholar] [CrossRef]
- Aird, W.C. Phenotypic heterogeneity of the endothelium: I. Structure, function, and mechanisms. Circ. Res. 2007, 100, 158–173. [Google Scholar] [CrossRef]
- Takabatake, N.; Arao, T.; Sata, M.; Abe, S.; Inoue, S.; Shibata, Y.; Takeishi, Y.; Kubota, I. Involvement of pulmonary endothelial cell injury in the pathogenesis of pulmonary fibrosis: Clinical assessment by 123I-MIBG lung scintigraphy. Eur. J. Nucl. Med. Mol. Imaging 2005, 32, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Magro, C.M.; Waldman, W.J.; Knight, D.A.; Allen, J.N.; Nadasdy, T.; Frambach, G.E.; Ross, P.; Marsh, C.B. Idiopathic pulmonary fibrosis related to endothelial injury and antiendothelial cell antibodies. Hum. Immunol. 2006, 67, 284–297. [Google Scholar] [CrossRef]
- Kato, S.; Inui, N.; Hakamata, A.; Suzuki, Y.; Enomoto, N.; Fujisawa, T.; Nakamura, Y.; Watanabe, H.; Suda, T. Changes in pulmonary endothelial cell properties during bleomycin-induced pulmonary fibrosis. Respir. Res. 2018, 19, 127. [Google Scholar] [CrossRef]
- Mammoto, T.; Muyleart, M.; Konduri, G.G.; Mammoto, A. Twist1 in Hypoxia-induced Pulmonary Hypertension through Transforming Growth Factor-beta-Smad Signaling. Am. J. Respir. Cell Mol. Biol. 2018, 58, 194–207. [Google Scholar] [CrossRef]
- Senavirathna, L.K.; Huang, C.; Yang, X.; Munteanu, M.C.; Sathiaseelan, R.; Xu, D.; Henke, C.A.; Liu, L. Hypoxia induces pulmonary fibroblast proliferation through NFAT signaling. Sci. Rep. 2018, 8, 2709. [Google Scholar] [CrossRef]
- Raghu, G.; Remy-Jardin, M.; Myers, J.L.; Richeldi, L.; Ryerson, C.J.; Lederer, D.J.; Behr, J.; Cottin, V.; Danoff, S.K.; Morell, F.; et al. Diagnosis of Idiopathic Pulmonary Fibrosis An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2018, 198, E44–E68. [Google Scholar] [CrossRef]
- Flaherty, K.R.; Andrei, A.C.; Murray, S.; Fraley, C.; Colby, T.V.; Travis, W.D.; Lama, V.; Kazerooni, E.A.; Gross, B.H.; Toews, G.B.; et al. Idiopathic pulmonary fibrosis: Prognostic value of changes in physiology and six-minute-walk test. Am. J. Respir. Crit. Care Med. 2006, 174, 803–809. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Life with oxygen. Science 2007, 318, 62–64. [Google Scholar] [CrossRef]
- Semenza, G.L. Oxygen sensing, homeostasis, and disease. N. Engl. J. Med. 2011, 365, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Eltzschig, H.K.; Carmeliet, P. Hypoxia and inflammation. N. Engl. J. Med. 2011, 364, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Krick, S.; Eul, B.G.; Hanze, J.; Savai, R.; Grimminger, F.; Seeger, W.; Rose, F. Role of hypoxia-inducible factor-1alpha in hypoxia-induced apoptosis of primary alveolar epithelial type II cells. Am. J. Respir. Cell Mol. Biol. 2005, 32, 395–403. [Google Scholar] [CrossRef]
- Moore, B.B.; Lawson, W.E.; Oury, T.D.; Sisson, T.H.; Raghavendran, K.; Hogaboam, C.M. Animal models of fibrotic lung disease. Am. J. Respir. Cell Mol. Biol. 2013, 49, 167–179. [Google Scholar] [CrossRef]
- Piera-Velazquez, S.; Jimenez, S.A. Endothelial to Mesenchymal Transition: Role in Physiology and in the Pathogenesis of Human Diseases. Physiol. Rev. 2019, 99, 1281–1324. [Google Scholar] [CrossRef]
- Lasky, J.A.; Ortiz, L.A.; Tonthat, B.; Hoyle, G.W.; Corti, M.; Athas, G.; Lungarella, G.; Brody, A.; Friedman, M. Connective tissue growth factor mRNA expression is upregulated in bleomycin-induced lung fibrosis. Am. J. Physiol. 1998, 275, L365–L371. [Google Scholar] [CrossRef]
- Zhuo, Y.; Zhang, J.; Laboy, M.; Lasky, J.A. Modulation of PDGF-C and PDGF-D expression during bleomycin-induced lung fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 286, L182–L188. [Google Scholar] [CrossRef]
- Jiang, X.; Tian, W.; Tu, A.B.; Pasupneti, S.; Shuffle, E.; Dahms, P.; Zhang, P.; Cai, H.; Dinh, T.T.; Liu, B.; et al. Endothelial Hypoxia-Inducible Factor-2alpha Is Required for the Maintenance of Airway Microvasculature. Circulation 2019, 139, 502–517. [Google Scholar] [CrossRef]
- Shimoda, L.A.; Semenza, G.L. HIF and the lung: Role of hypoxia-inducible factors in pulmonary development and disease. Am. J. Respir. Crit. Care Med. 2011, 183, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Wiesener, M.S.; Jurgensen, J.S.; Rosenberger, C.; Scholze, C.K.; Horstrup, J.H.; Warnecke, C.; Mandriota, S.; Bechmann, I.; Frei, U.A.; Pugh, C.W.; et al. Widespread hypoxia-inducible expression of HIF-2alpha in distinct cell populations of different organs. FASEB J. 2003, 17, 271–273. [Google Scholar] [CrossRef] [PubMed]
- Bartoszewski, R.; Moszynska, A.; Serocki, M.; Cabaj, A.; Polten, A.; Ochocka, R.; Dell’Italia, L.; Bartoszewska, S.; Kroliczewski, J.; Dabrowski, M.; et al. Primary endothelial cell-specific regulation of hypoxia-inducible factor (HIF)-1 and HIF-2 and their target gene expression profiles during hypoxia. FASEB J. 2019, 33, 7929–7941. [Google Scholar] [CrossRef]
- Bodempudi, V.; Hergert, P.; Smith, K.; Xia, H.; Herrera, J.; Peterson, M.; Khalil, W.; Kahm, J.; Bitterman, P.B.; Henke, C.A. miR-210 promotes IPF fibroblast proliferation in response to hypoxia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L283–L294. [Google Scholar] [CrossRef] [PubMed]
- Franke, K.; Gassmann, M.; Wielockx, B. Erythrocytosis: The HIF pathway in control. Blood 2013, 122, 1122–1128. [Google Scholar] [CrossRef]
- Stroka, D.M.; Burkhardt, T.; Desbaillets, I.; Wenger, R.H.; Neil, D.A.; Bauer, C.; Gassmann, M.; Candinas, D. HIF-1 is expressed in normoxic tissue and displays an organ-specific regulation under systemic hypoxia. FASEB J. 2001, 15, 2445–2453. [Google Scholar] [CrossRef]
- Ueno, M.; Maeno, T.; Nomura, M.; Aoyagi-Ikeda, K.; Matsui, H.; Hara, K.; Tanaka, T.; Iso, T.; Suga, T.; Kurabayashi, M. Hypoxia-inducible factor-1alpha mediates TGF-beta-induced PAI-1 production in alveolar macrophages in pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 300, L740–L752. [Google Scholar] [CrossRef]
- Higgins, D.F.; Kimura, K.; Bernhardt, W.M.; Shrimanker, N.; Akai, Y.; Hohenstein, B.; Saito, Y.; Johnson, R.S.; Kretzler, M.; Cohen, C.D.; et al. Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J. Clin. Investig. 2007, 117, 3810–3820. [Google Scholar] [CrossRef]
- Bryant, A.J.; Carrick, R.P.; McConaha, M.E.; Jones, B.R.; Shay, S.D.; Moore, C.S.; Blackwell, T.R.; Gladson, S.; Penner, N.L.; Burman, A.; et al. Endothelial HIF signaling regulates pulmonary fibrosis-associated pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 310, L249–L262. [Google Scholar] [CrossRef]
- Higgins, D.F.; Biju, M.P.; Akai, Y.; Wutz, A.; Johnson, R.S.; Haase, V.H. Hypoxic induction of Ctgf is directly mediated by Hif-1. Am. J. Physiol. Renal Physiol. 2004, 287, F1223–F1232. [Google Scholar] [CrossRef]
- Tzouvelekis, A.; Harokopos, V.; Paparountas, T.; Oikonomou, N.; Chatziioannou, A.; Vilaras, G.; Tsiambas, E.; Karameris, A.; Bouros, D.; Aidinis, V. Comparative expression profiling in pulmonary fibrosis suggests a role of hypoxia-inducible factor-1alpha in disease pathogenesis. Am. J. Respir. Crit. Care Med. 2007, 176, 1108–1119. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.J.; Wang, L.Y.; Chodosh, L.A.; Keith, B.; Simon, M.C. Differential roles of hypoxia-inducible factor 1alpha (HIF-1alpha) and HIF-2alpha in hypoxic gene regulation. Mol. Cell. Biol. 2003, 23, 9361–9374. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, J.; Choi, H.; Hsieh, M.H.; Neugent, M.L.; Ahn, J.M.; Hayenga, H.N.; Singh, P.K.; Shackelford, D.B.; Lee, I.K.; Shulaev, V.; et al. Targeting Hypoxia-Inducible Factor-1alpha/Pyruvate Dehydrogenase Kinase 1 Axis by Dichloroacetate Suppresses Bleomycin-induced Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2018, 58, 216–231. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Niu, W.; Dong, H.Y.; Liu, M.L.; Luo, Y.; Li, Z.C. Hypoxia induces endothelial mesenchymal transition in pulmonary vascular remodeling. Int. J. Mol. Med. 2018, 42, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Iyer, A.K.; Ramesh, V.; Castro, C.A.; Kaushik, V.; Kulkarni, Y.M.; Wright, C.A.; Venkatadri, R.; Rojanasakul, Y.; Azad, N. Nitric oxide mediates bleomycin-induced angiogenesis and pulmonary fibrosis via regulation of VEGF. J. Cell. Biochem. 2015, 116, 2484–2493. [Google Scholar] [CrossRef] [PubMed]
- Michiels, C. Endothelial cell functions. J. Cell. Physiol. 2003, 196, 430–443. [Google Scholar] [CrossRef]
- Chung, M.P.; Monick, M.M.; Hamzeh, N.Y.; Butler, N.S.; Powers, L.S.; Hunninghake, G.W. Role of repeated lung injury and genetic background in bleomycin-induced fibrosis. Am. J. Respir. Cell Mol. Biol. 2003, 29, 375–380. [Google Scholar] [CrossRef]
- Branco-Price, C.; Zhang, N.; Schnelle, M.; Evans, C.; Katschinski, D.M.; Liao, D.; Ellies, L.; Johnson, R.S. Endothelial cell HIF-1alpha and HIF-2alpha differentially regulate metastatic success. Cancer Cell 2012, 21, 52–65. [Google Scholar] [CrossRef]
- Fukumura, D.; Gohongi, T.; Kadambi, A.; Izumi, Y.; Ang, J.; Yun, C.O.; Buerk, D.G.; Huang, P.L.; Jain, R.K. Predominant role of endothelial nitric oxide synthase in vascular endothelial growth factor-induced angiogenesis and vascular permeability. Proc. Natl. Acad. Sci. USA 2001, 98, 2604–2609. [Google Scholar] [CrossRef]
- Nauta, T.D.; van den Broek, M.; Gibbs, S.; van der Pouw-Kraan, T.C.; Oudejans, C.B.; van Hinsbergh, V.W.; Koolwijk, P. Identification of HIF-2alpha-regulated genes that play a role in human microvascular endothelial sprouting during prolonged hypoxia in vitro. Angiogenesis 2017, 20, 39–54. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Saline | Bleomycin | |||
|---|---|---|---|---|
| 20% O2 | 1% O2 | 20% O2 | 1% O2 | |
| vWF | 1.33 ± 0.16 | 8.79 ± 1.55 * | 3.22 ± 0.57 * | 17.5 ± 0.98 * |
| MMP-12 | 1.37 ± 0.20 | 78.2 ± 5.96 * | 1.98 ± 0.13 | 906 ± 148 * |
| PAI-1 | 1.08 ± 0.26 | 14.4 ± 1.00 * | 3.02 ± 0.21 * | 16.1 ± 2.75 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akahori, D.; Inui, N.; Inoue, Y.; Yasui, H.; Hozumi, H.; Suzuki, Y.; Karayama, M.; Furuhashi, K.; Enomoto, N.; Fujisawa, T.; et al. Effect of Hypoxia on Pulmonary Endothelial Cells from Bleomycin-Induced Pulmonary Fibrosis Model Mice. Int. J. Mol. Sci. 2022, 23, 8996. https://doi.org/10.3390/ijms23168996
Akahori D, Inui N, Inoue Y, Yasui H, Hozumi H, Suzuki Y, Karayama M, Furuhashi K, Enomoto N, Fujisawa T, et al. Effect of Hypoxia on Pulmonary Endothelial Cells from Bleomycin-Induced Pulmonary Fibrosis Model Mice. International Journal of Molecular Sciences. 2022; 23(16):8996. https://doi.org/10.3390/ijms23168996
Chicago/Turabian StyleAkahori, Daisuke, Naoki Inui, Yusuke Inoue, Hideki Yasui, Hironao Hozumi, Yuzo Suzuki, Masato Karayama, Kazuki Furuhashi, Noriyuki Enomoto, Tomoyuki Fujisawa, and et al. 2022. "Effect of Hypoxia on Pulmonary Endothelial Cells from Bleomycin-Induced Pulmonary Fibrosis Model Mice" International Journal of Molecular Sciences 23, no. 16: 8996. https://doi.org/10.3390/ijms23168996
APA StyleAkahori, D., Inui, N., Inoue, Y., Yasui, H., Hozumi, H., Suzuki, Y., Karayama, M., Furuhashi, K., Enomoto, N., Fujisawa, T., & Suda, T. (2022). Effect of Hypoxia on Pulmonary Endothelial Cells from Bleomycin-Induced Pulmonary Fibrosis Model Mice. International Journal of Molecular Sciences, 23(16), 8996. https://doi.org/10.3390/ijms23168996