
The Chloronium Cation [(C2H3)2Cl+] and Unsaturated C4-Carbocations with C=C and C≡C Bonds in Their Solid Salts and in Solutions: An H1/C13 NMR and Infrared Spectroscopic Study



Abstract
:1. Introduction
2. Results and Discussion
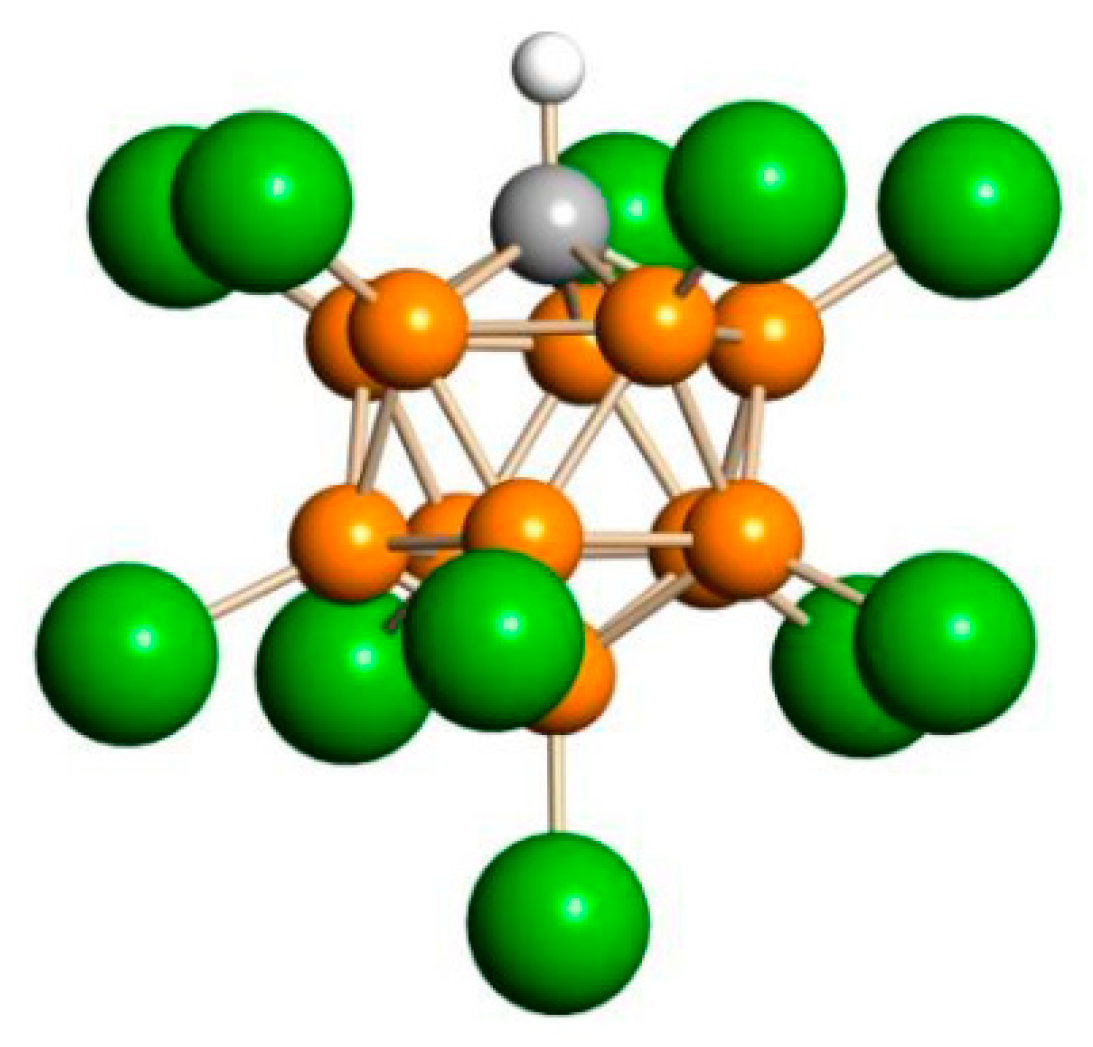
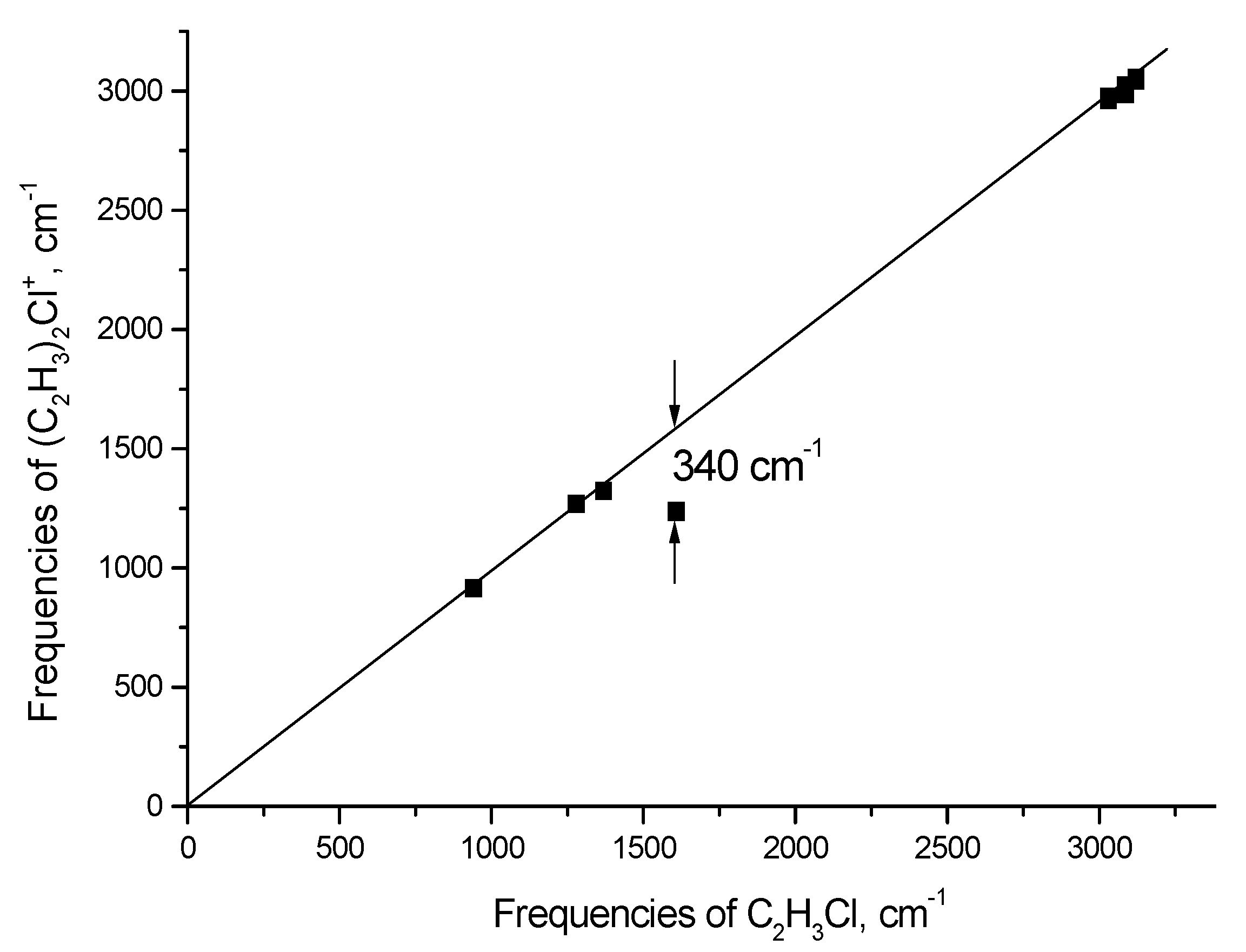
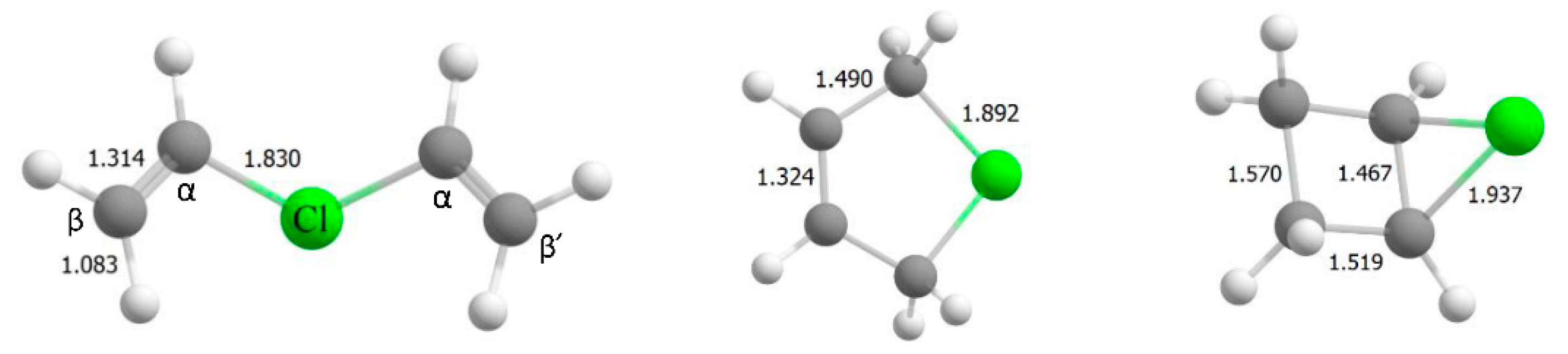
2.1. The Chloronium Cation
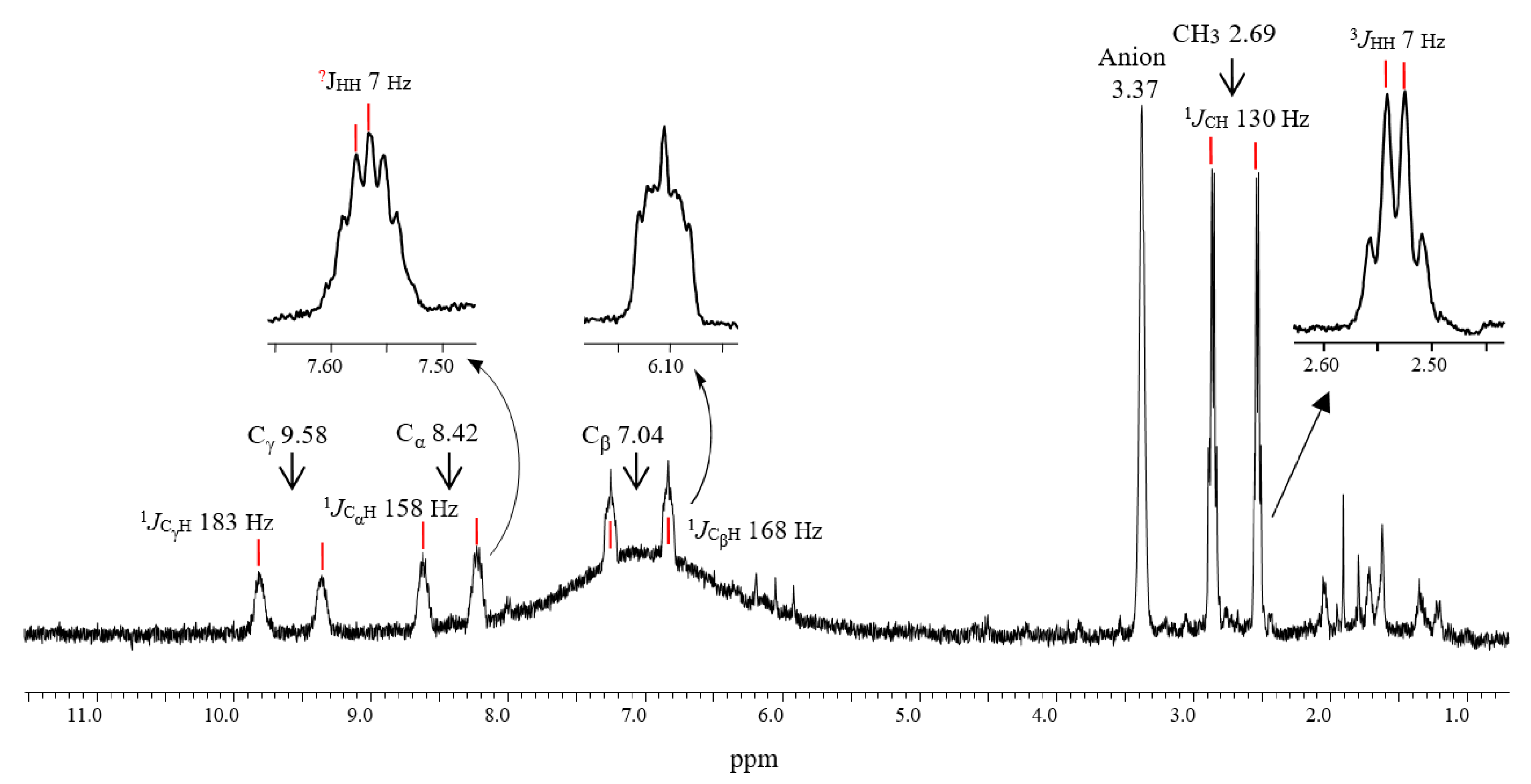
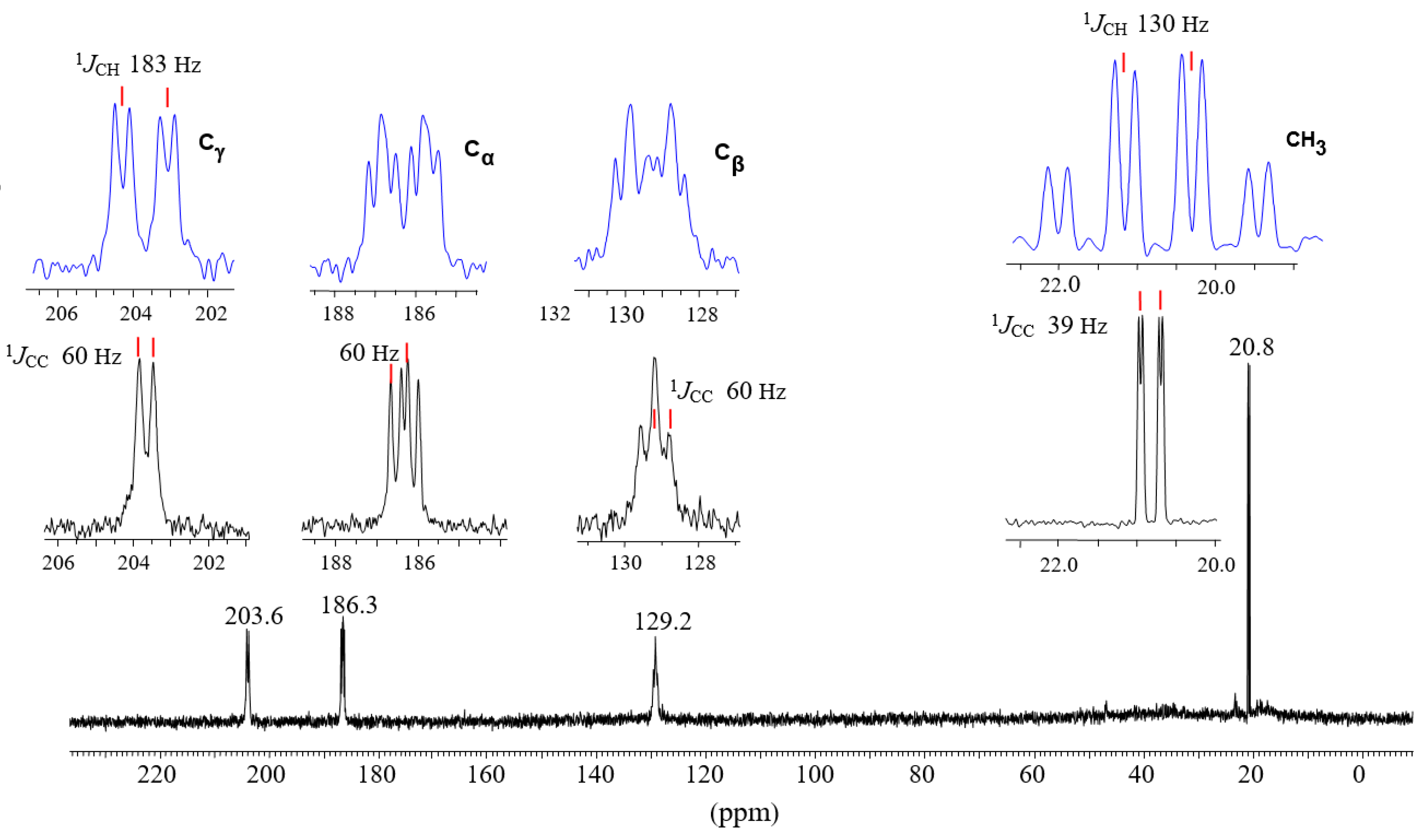
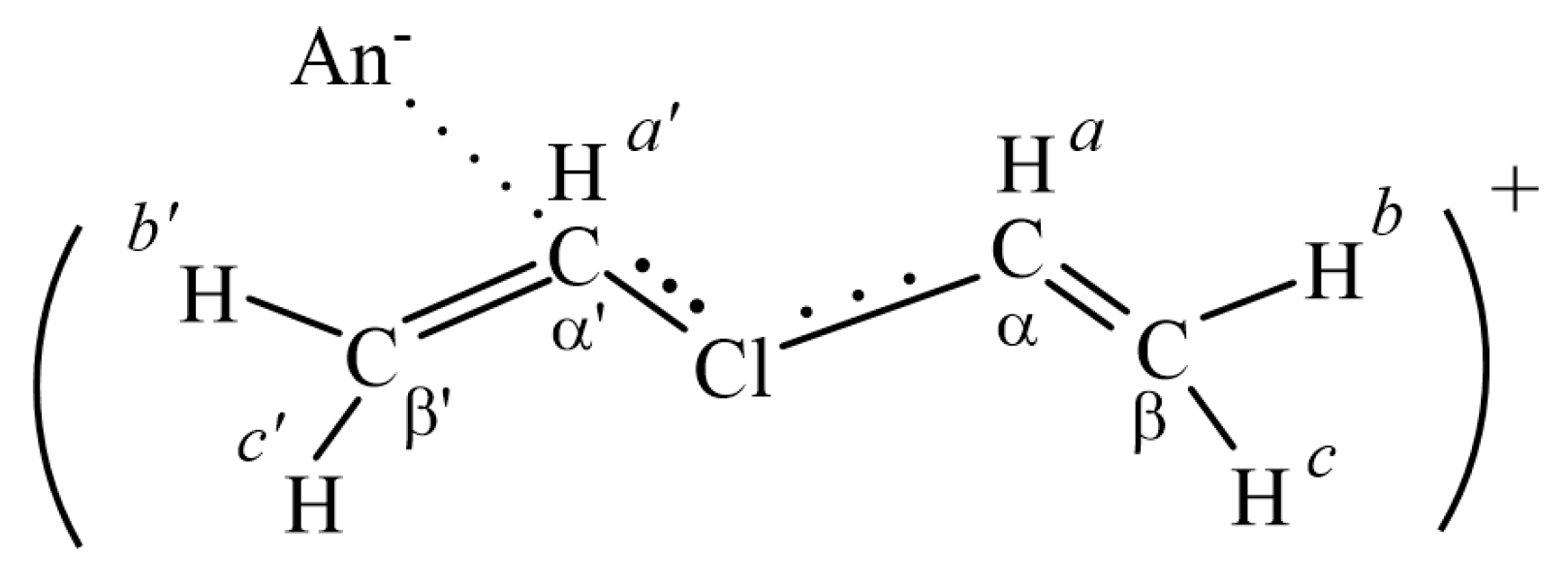
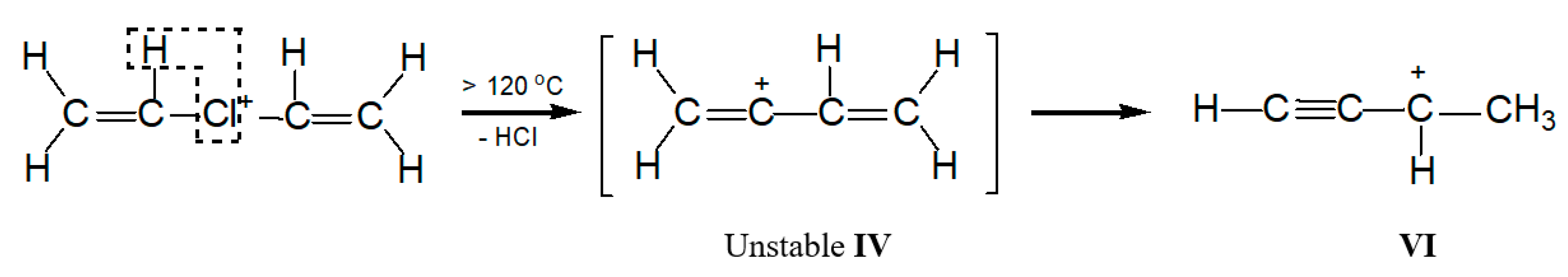
2.2. The C4-Carbocations
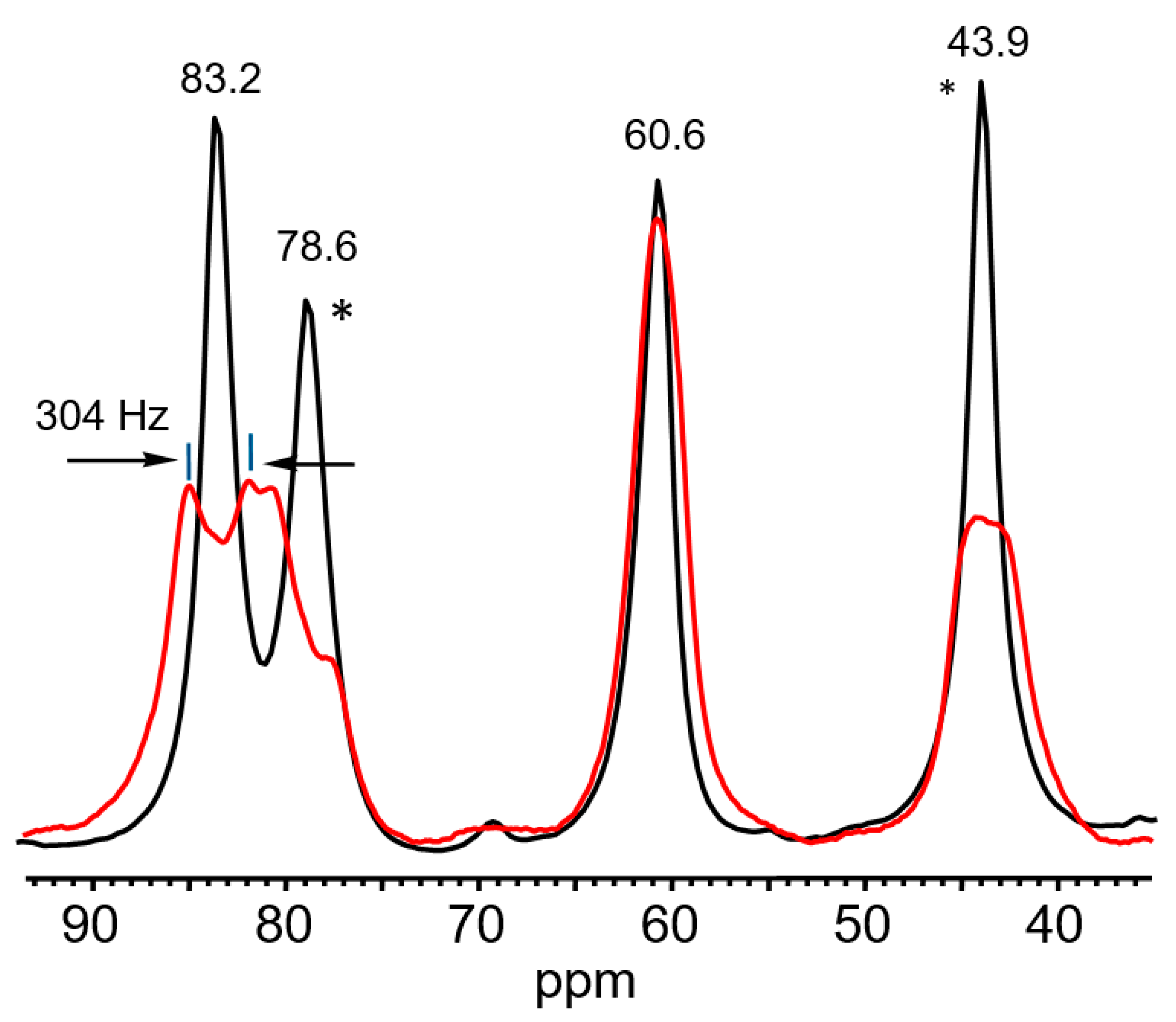
 of both cations was similar. Based on this similarity, it is possible to interpret the 13C signals of cation VI (Table 5).
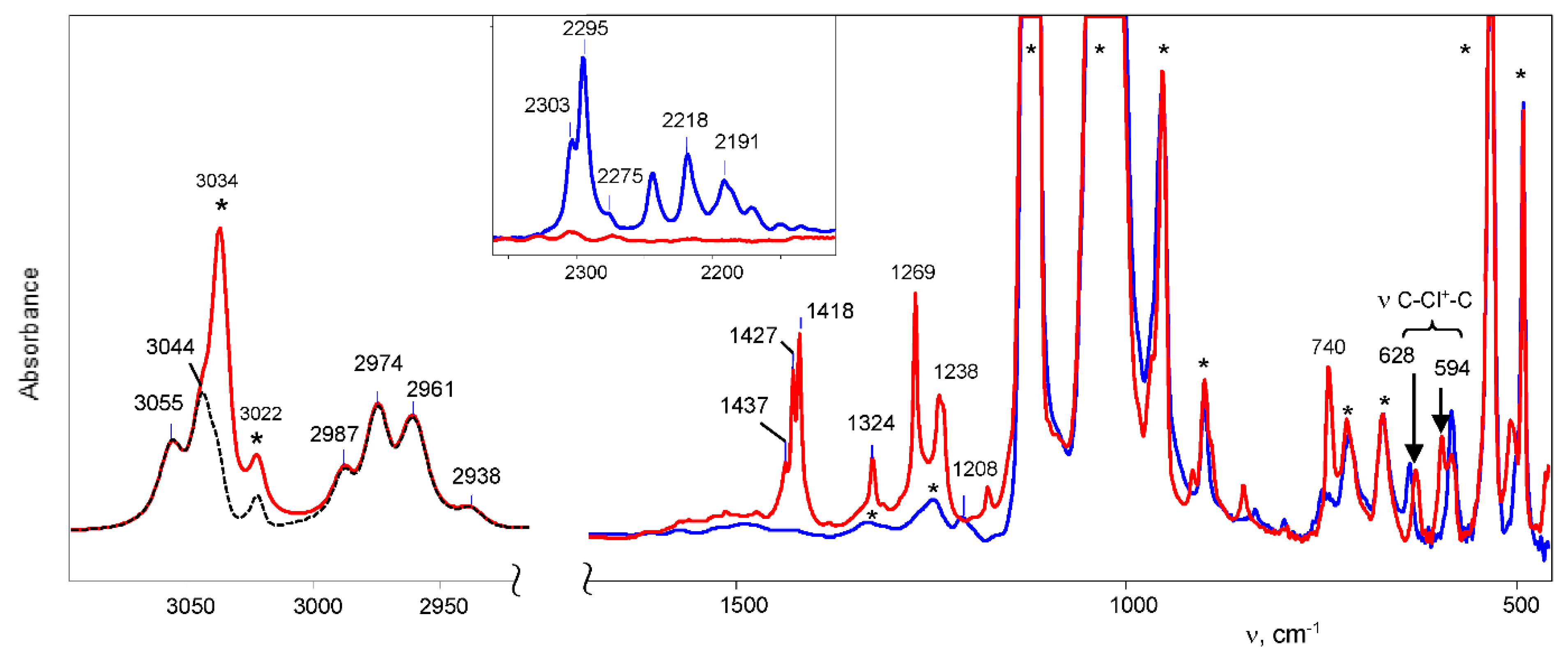
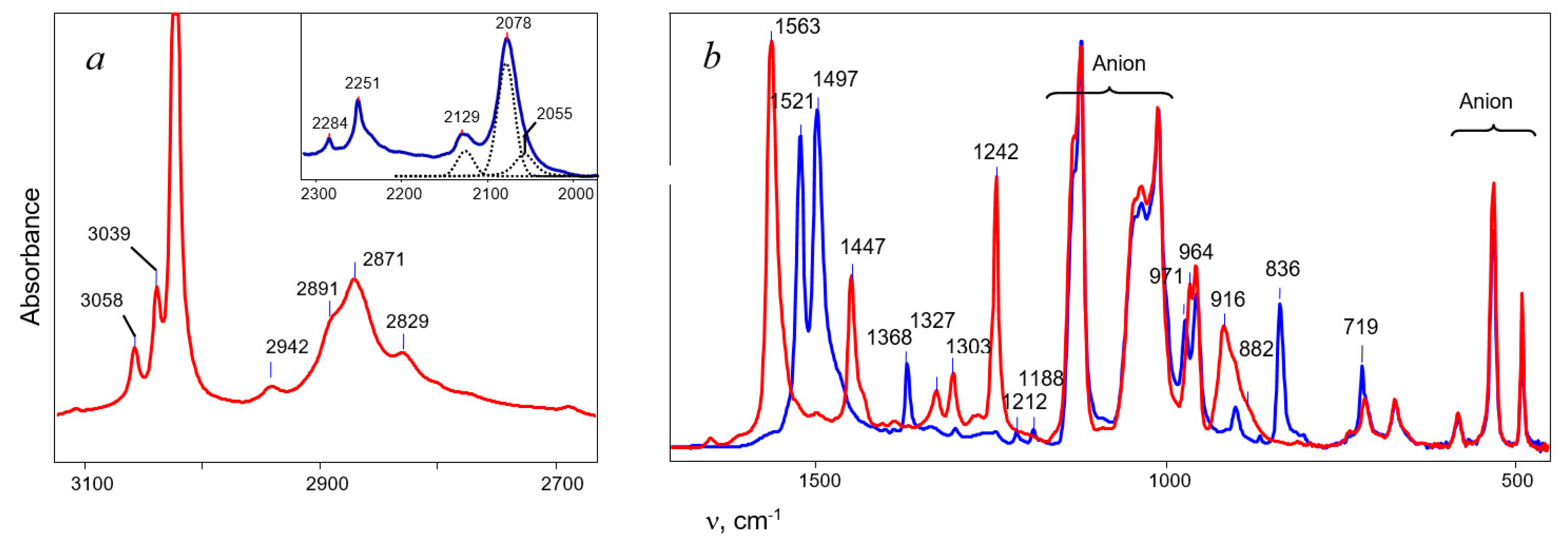
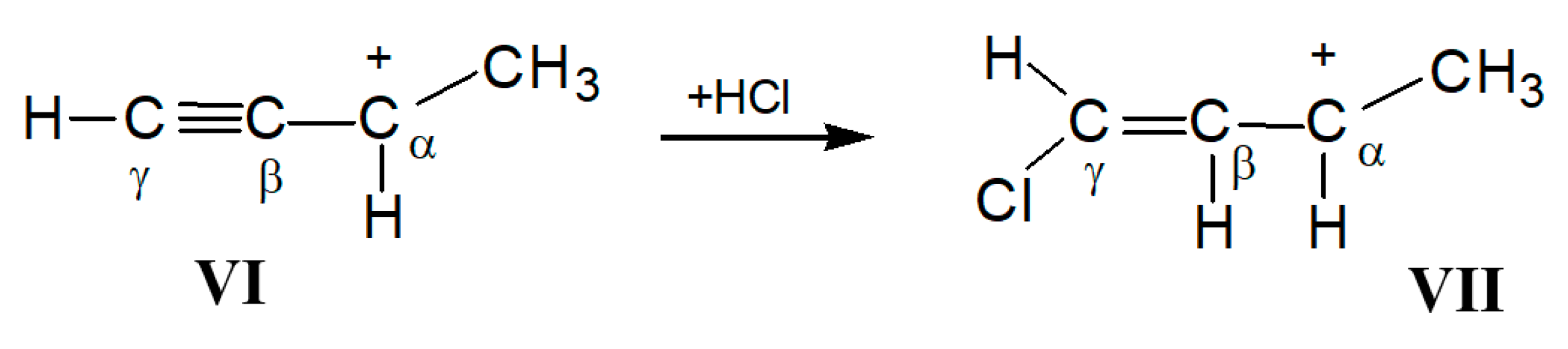
of both cations was similar. Based on this similarity, it is possible to interpret the 13C signals of cation VI (Table 5). . The frequency of the C-Cl stretch vibration is seen at 636 cm−1 (Figure S2 in SI), which is much lower than that of isomers VIII (at 768 cm−1) and IX (at 733 cm−1) [28] and matches the frequency of asymmetric vibration of the bridged C–Cl+–C group of the stablest dimethylchloronium cation, which decomposes at elevated temperature [32]. Therefore, the C-Cl bond is weak, and with an increase in temperature, cation VII can lose the HCl molecule.
. The frequency of the C-Cl stretch vibration is seen at 636 cm−1 (Figure S2 in SI), which is much lower than that of isomers VIII (at 768 cm−1) and IX (at 733 cm−1) [28] and matches the frequency of asymmetric vibration of the bridged C–Cl+–C group of the stablest dimethylchloronium cation, which decomposes at elevated temperature [32]. Therefore, the C-Cl bond is weak, and with an increase in temperature, cation VII can lose the HCl molecule.2.3. Comparative Discussion of the IR and NMR Spectroscopic Data
2.3.1. The Chloronium Cation
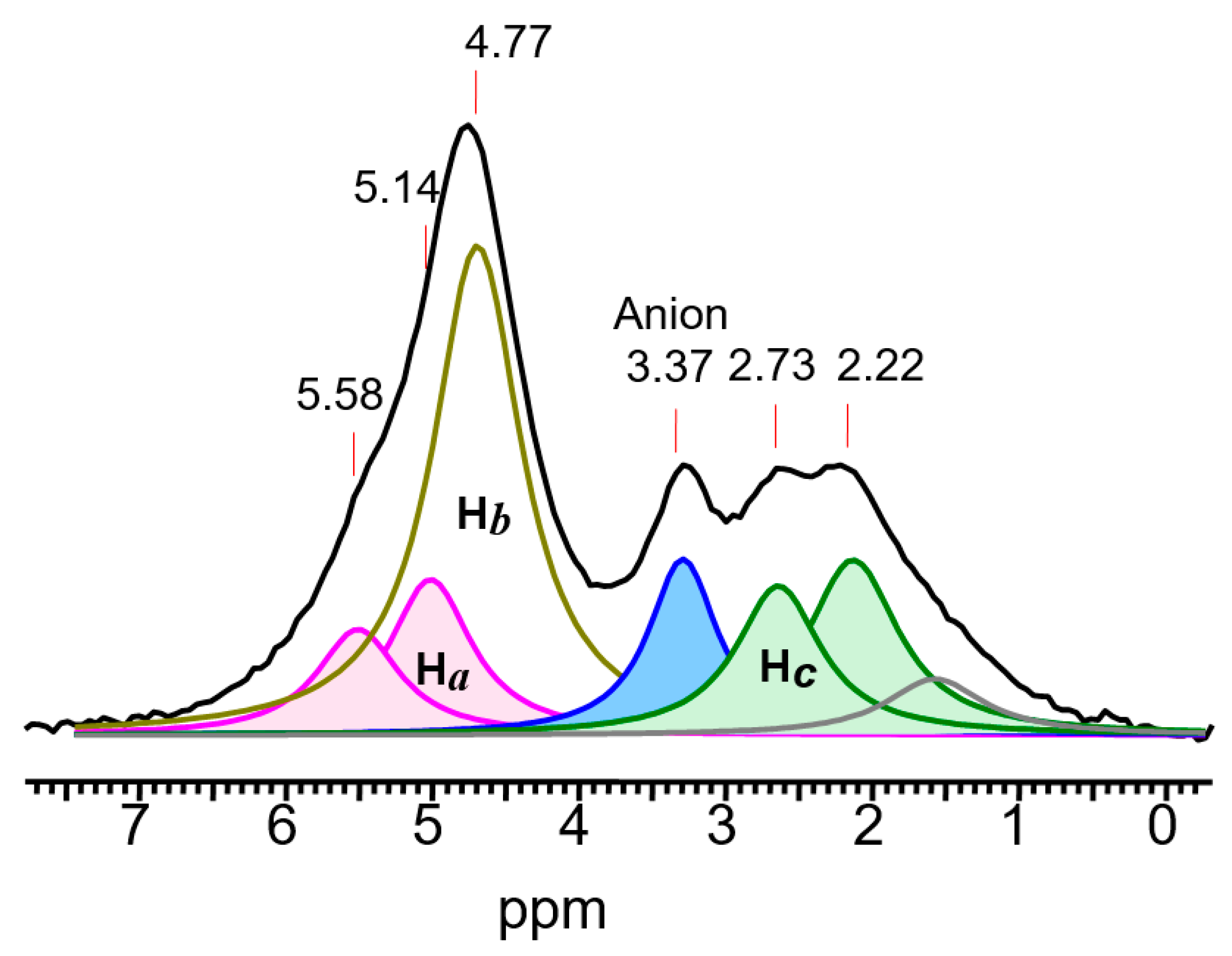
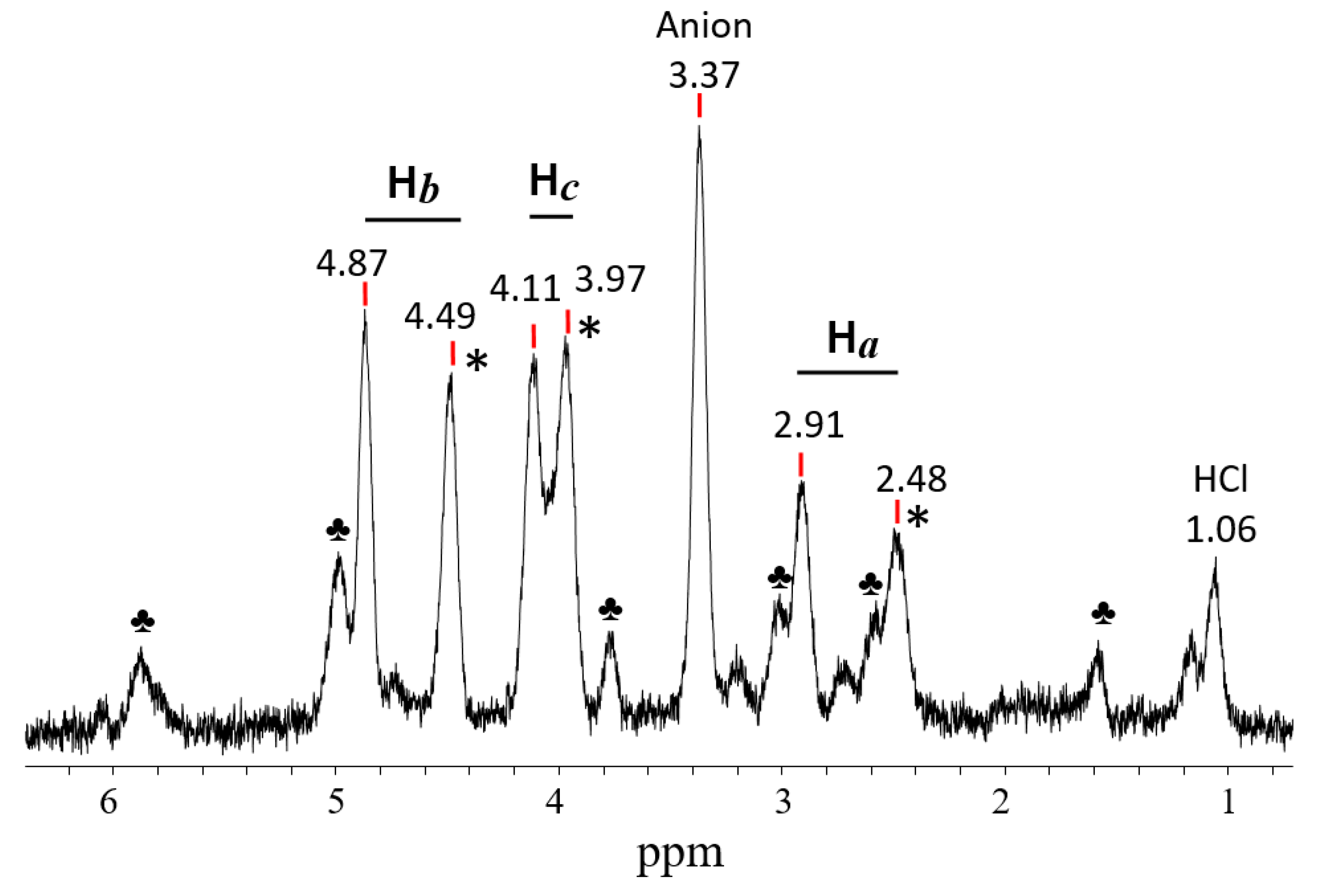
2.3.2. The C4-Carbocations
 . Chemical shifts of its C atoms usually vary depending on the substituents attached to it: from 170 to 190 ppm for C1,3, and 170–150 ppm for C2 atoms, respectively [37], thus pointing to greater charge localization on atoms C1 and C3. If the double bond is not delocalized, as for example, in the
. Chemical shifts of its C atoms usually vary depending on the substituents attached to it: from 170 to 190 ppm for C1,3, and 170–150 ppm for C2 atoms, respectively [37], thus pointing to greater charge localization on atoms C1 and C3. If the double bond is not delocalized, as for example, in the  moiety of the 1-cyclopropylcyclopropyli-dene methyl cation, then the positive charge is located mainly on the C2 atom with a chemical shift of 234 ppm and less on atoms C1 (51.7 ppm) and C3 (21.2 ppm) [38]. In vinyl-type carbocations R′C+=CR″2 stabilized by electron-donating groups R′ and R″, such as
moiety of the 1-cyclopropylcyclopropyli-dene methyl cation, then the positive charge is located mainly on the C2 atom with a chemical shift of 234 ppm and less on atoms C1 (51.7 ppm) and C3 (21.2 ppm) [38]. In vinyl-type carbocations R′C+=CR″2 stabilized by electron-donating groups R′ and R″, such as  , the positive charge on the C=C bond is greatly reduced due to the combined influence of t-Bu and the two β-silyl substituents directly attached to it [39]. In the C13 NMR spectra, the signal from the Cα atom is observed in a weak field at 202.7 ppm and from the Cβ atom at 75.5 ppm, that is, the charge is mainly localized on the Cα atom.
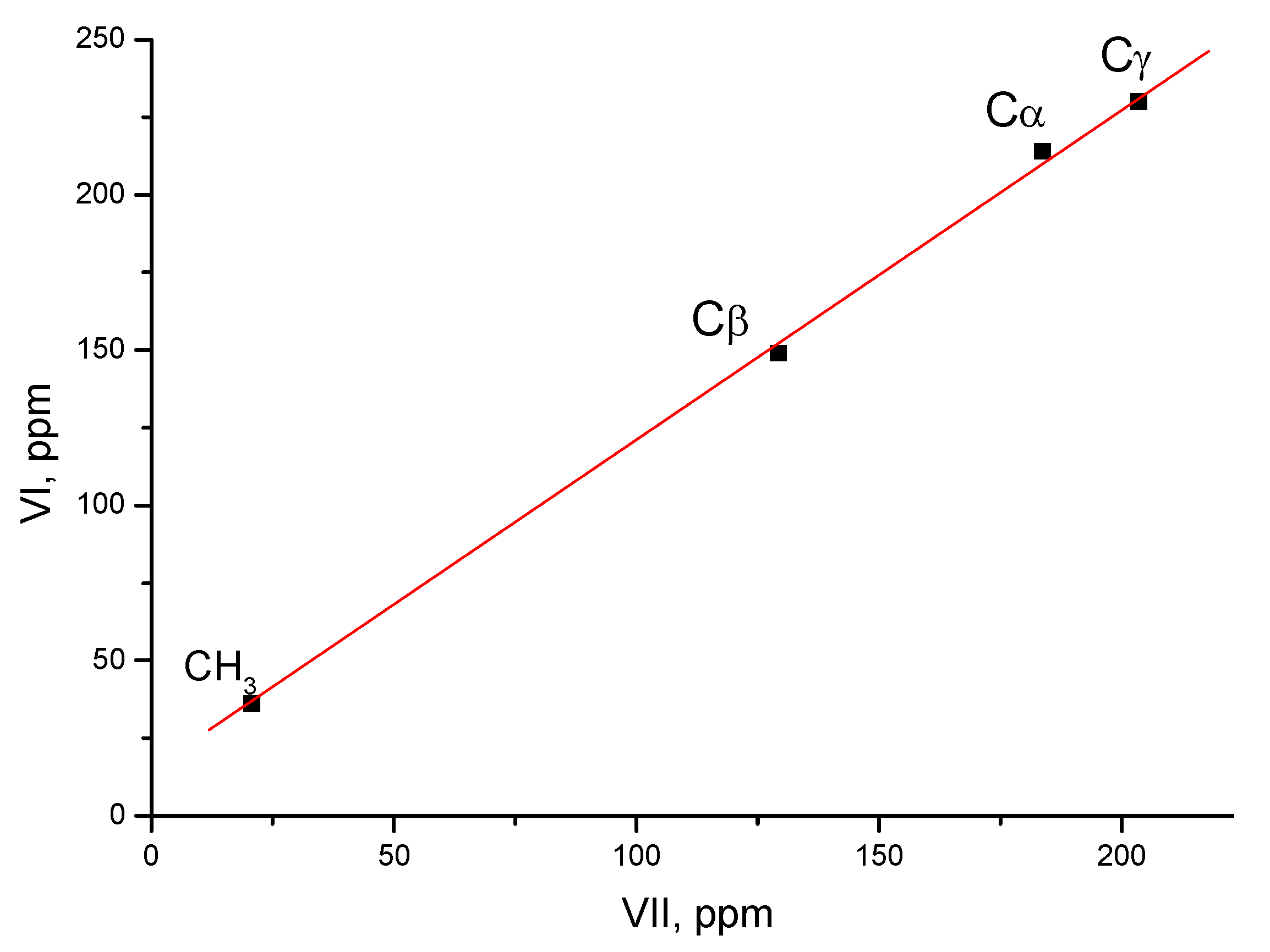
, the positive charge on the C=C bond is greatly reduced due to the combined influence of t-Bu and the two β-silyl substituents directly attached to it [39]. In the C13 NMR spectra, the signal from the Cα atom is observed in a weak field at 202.7 ppm and from the Cβ atom at 75.5 ppm, that is, the charge is mainly localized on the Cα atom. moiety, whose carbon atoms possess the chemical shifts Cγ (203 ppm), Cβ (129 ppm) and Cα (183 ppm). They do not match a triatomic moiety with one CC double bond but rather correspond to slightly asymmetric allyl moiety
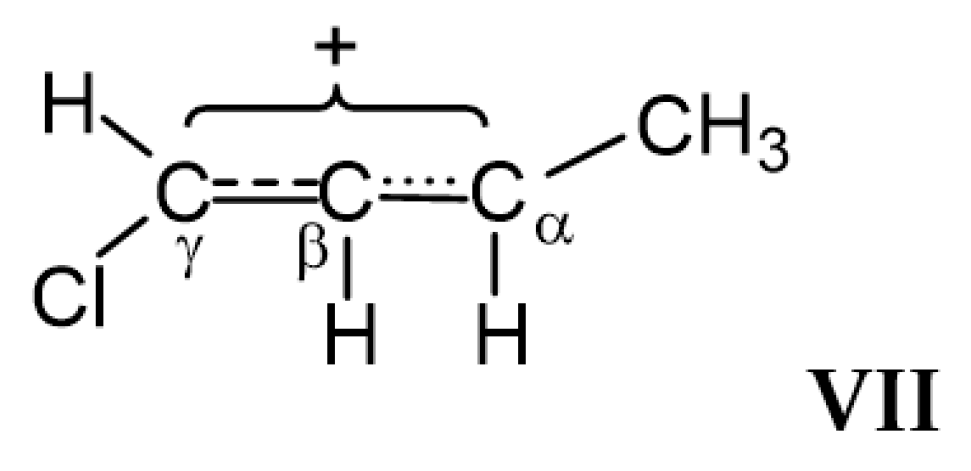
moiety, whose carbon atoms possess the chemical shifts Cγ (203 ppm), Cβ (129 ppm) and Cα (183 ppm). They do not match a triatomic moiety with one CC double bond but rather correspond to slightly asymmetric allyl moiety  . Therefore, it is more correct to represent structure VII as shown in Scheme 8.
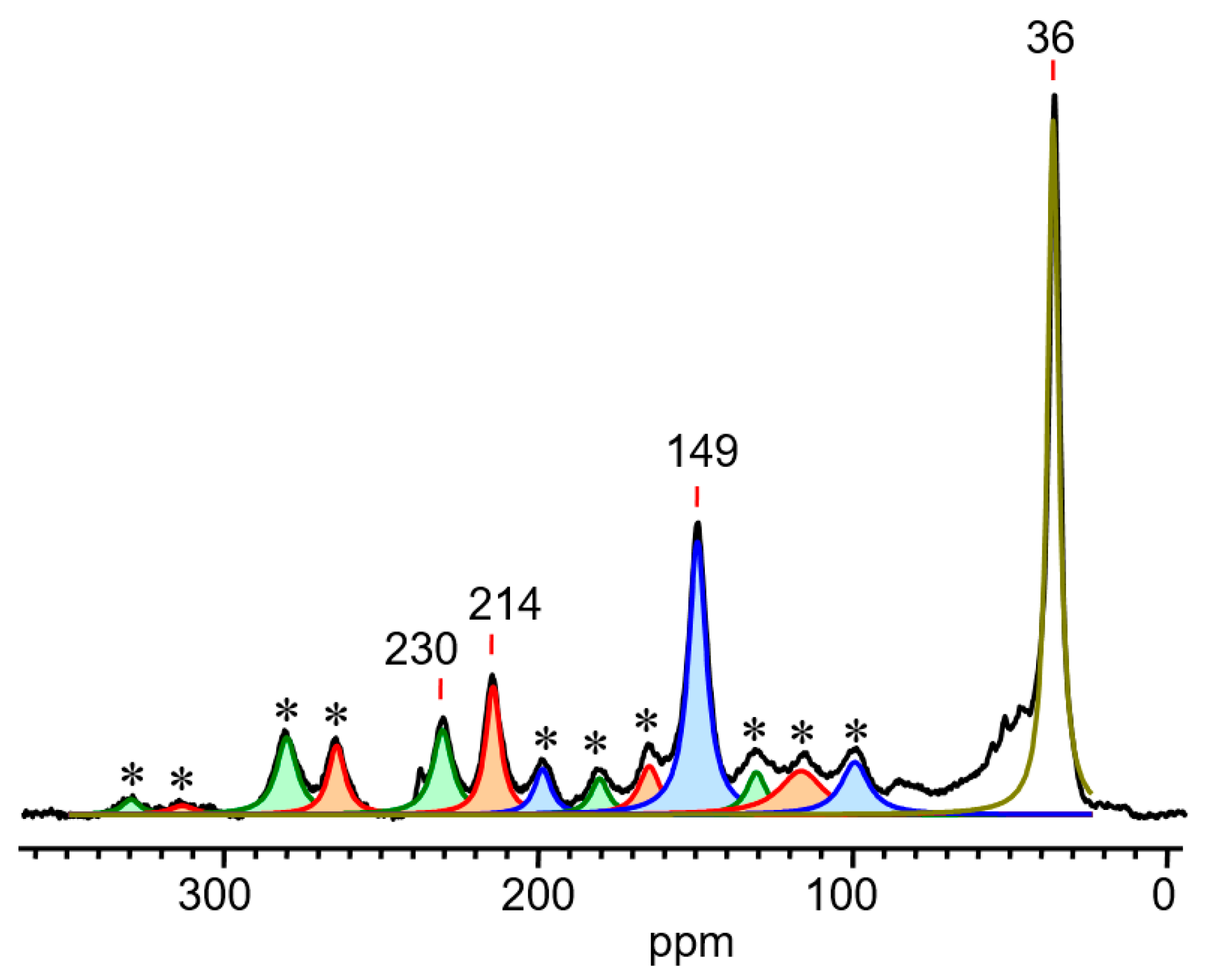
. Therefore, it is more correct to represent structure VII as shown in Scheme 8. moiety (Scheme 6). The chemical shifts of its C atoms (230 (Cγ), 150 (Cβ) and 215 ppm (Cα)) are consistent with those of the
moiety (Scheme 6). The chemical shifts of its C atoms (230 (Cγ), 150 (Cβ) and 215 ppm (Cα)) are consistent with those of the  part of vinyl cation VII (Figure 11). This means that electron density distributions—across the CC bonds of the two moieties, which differ in formal double and triple CC bonds—are of the same type, with a lower positive charge on the Cα atom than on atoms Cβ and Cγ. The observed participation of H atoms of the CH3 group in hyperconjugation with the partially empty 2pz orbital of the Cα atom indicates its sp2 hybridization. That is, formally, the Cα atom must carry a considerable positive charge.
part of vinyl cation VII (Figure 11). This means that electron density distributions—across the CC bonds of the two moieties, which differ in formal double and triple CC bonds—are of the same type, with a lower positive charge on the Cα atom than on atoms Cβ and Cγ. The observed participation of H atoms of the CH3 group in hyperconjugation with the partially empty 2pz orbital of the Cα atom indicates its sp2 hybridization. That is, formally, the Cα atom must carry a considerable positive charge.3. Methods and Materials
4. Conclusions
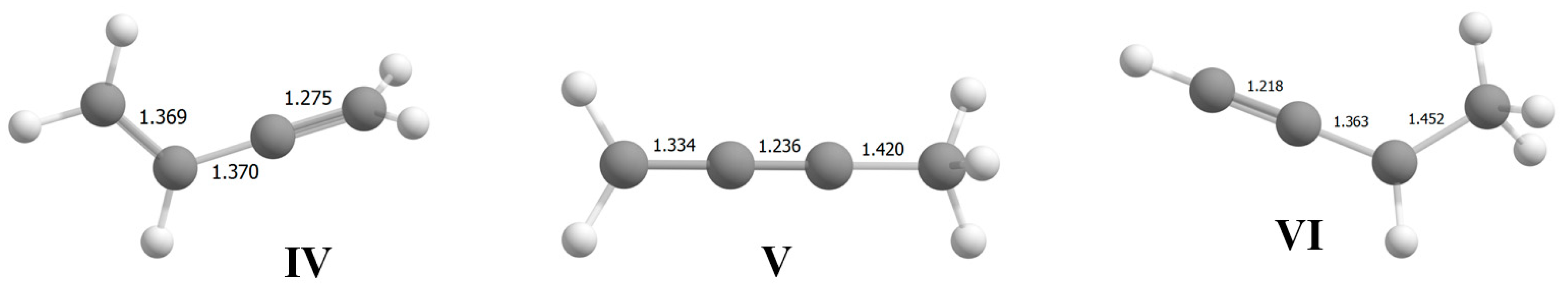
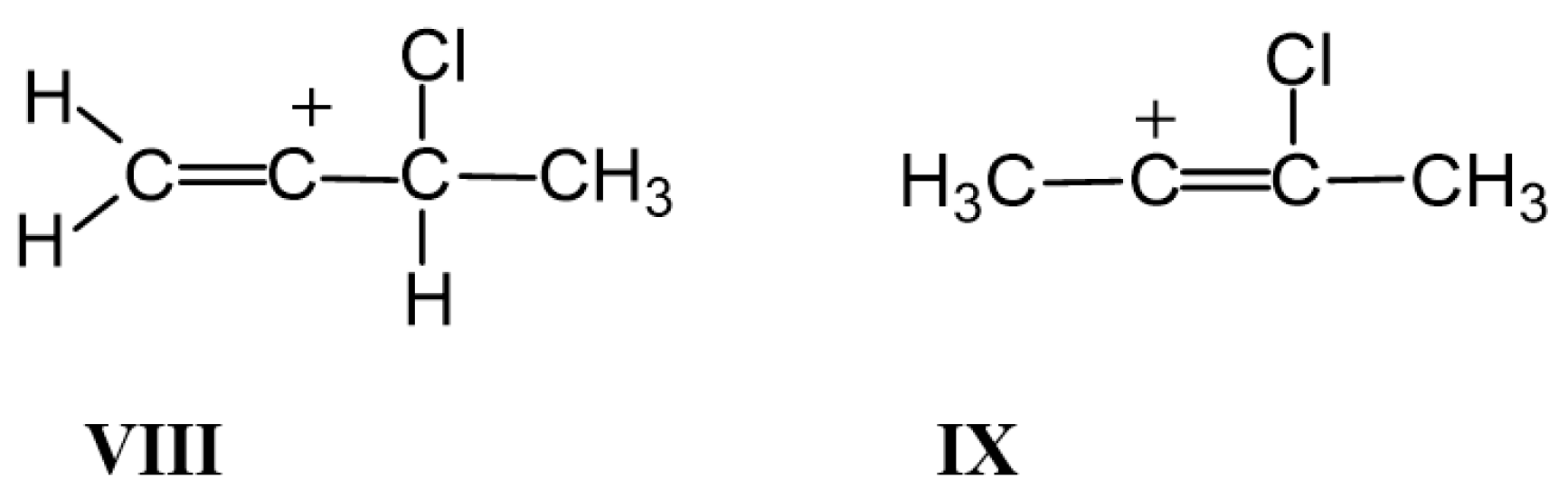
 . When the Cl atom is attached to the Cα/Cβ, atom, isomers
. When the Cl atom is attached to the Cα/Cβ, atom, isomers  and
and  arise, with well-defined and close-to-equivalent C=C double bonds. If the Cl atom is attached to the Cγ atom, then allyl-type isomer VII is formed with considerable alignment of the CC bonds in the
arise, with well-defined and close-to-equivalent C=C double bonds. If the Cl atom is attached to the Cγ atom, then allyl-type isomer VII is formed with considerable alignment of the CC bonds in the  moiety, with the charge dispersed over it. This phenomenon drives strong change in the CC stretch frequencies.
moiety, with the charge dispersed over it. This phenomenon drives strong change in the CC stretch frequencies. moiety is evidenced by the higher frequency of the
moiety is evidenced by the higher frequency of the  stretch at 1563 cm−1 than that of the double C=C bond in vinyl cations (1490 cm−1) [26,27]. Despite the asymmetry, the positive charge is effectively distributed over both CC bonds: the involvement of the hydrogen atoms of the CH3 group in hyperconjugation with the 2pz orbital of the Cα atom means that it has sp2 hybridization and a major positive charge is concentrated on it. Meanwhile, it follows from the MAS NMR spectra that a comparable charge is also localized on the Cγ atom.
stretch at 1563 cm−1 than that of the double C=C bond in vinyl cations (1490 cm−1) [26,27]. Despite the asymmetry, the positive charge is effectively distributed over both CC bonds: the involvement of the hydrogen atoms of the CH3 group in hyperconjugation with the 2pz orbital of the Cα atom means that it has sp2 hybridization and a major positive charge is concentrated on it. Meanwhile, it follows from the MAS NMR spectra that a comparable charge is also localized on the Cγ atom.Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Olah, G.A.; Baker, E.B.; Evans, J.C.; Tolgyesi, W.S.; McIntyre, J.S.; Bastien, I.J. Stable Carbonium Ions. V.1a Alkylcarbonium Hexafluoroantimonates. J. Am. Chem. Soc. 1964, 86, 1360–1373. [Google Scholar] [CrossRef]
- Olah, G.A.; Bollinger, J.M.; Cupas, C.A.; Lukas, J. Stable carbonium ions. XXXIV. 1-Methylcyclopentyl cation. J. Am. Chem. Soc. 1967, 89, 2692–2694. [Google Scholar] [CrossRef]
- Olah, G.A.; DeMember, J.R.; Commeyras, A.; Bribes, J.L. Stable carbonium ions. LXXXV. Laser Raman and infrared spectroscopic study of alkylcarbonium ions. J. Am. Chem. Soc. 1971, 93, 459–463. [Google Scholar] [CrossRef]
- Olah, G.A. Stable Carbonium Ions in Solution. Science 1970, 168, 1298–1311. [Google Scholar] [CrossRef] [PubMed]
- Brouwer, D.M.; Hogeveen, H. Electrophilic Substitutions at Alkanes and in Alkylcarbonium Ions. Prog. Phys. Org. Chem. 1972, 9, 179–240. [Google Scholar]
- Stoyanov, E.S.; Gomes, G.P. tert-Butyl Carbocation in Condensed Phases: Stabilization via Hyperconjugation, Polarization, and Hydrogen Bonding. J. Phys. Chem. A 2015, 119, 8619–8629. [Google Scholar] [CrossRef]
- Stoyanov, E.S.; Nizovtsev, A.S. Stabilization of carbocations CH3+, C2H5+, i-C3H7+, tert-Bu+, and cyclo-pentyl+ in solid phases: Experimental data versus calculations. Phys. Chem. Chem. Phys. 2017, 19, 7270–7279. [Google Scholar] [CrossRef]
- Stoyanov, E.S. Stabilization of Saturated Carbocations in Condensed Phases. J. Phys. Chem. A 2017, 121, 9638–9644. [Google Scholar] [CrossRef]
- Koptyug, V.A. Contemporary Problems in Carbonium Ion Chemistry III: Arenium Ions-Structure and Reactivity; Rees, C., Ed.; Springer: Berlin/Heidelberg, Germany, 1984; pp. 1–227. [Google Scholar]
- Reed, C.A.; Kim, K.-C.; Stoyanov, E.S.; Stasko, D.; Tham, F.S.; Mueller, L.J.; Boyd, P.D.W. Isolating Benzenium Ion Salts. J. Am. Chem. Soc. 2003, 125, 1796–1804. [Google Scholar] [CrossRef]
- Duncan, M.A. Infrared Laser Spectroscopy of Mass-Selected Carbocations. J. Phys. Chem. A 2012, 116, 11477–11491. [Google Scholar]
- Ricks, A.M.; Douberly, G.E.; Schleyer, P.V.R.; Duncan, M.A. Infrared spectroscopy of gas phase C3H3+ ions: The cyclopropenyl and propargyl cations. J. Chem. Phys. 2010, 132, 051101. [Google Scholar] [CrossRef] [PubMed]
- Douberly, G.E.; Risks, A.M.; Schleyer, P.V.R.; Duncan, M.A. Infrared spectroscopy of gas phase C3H5+: The allyl and 2-propenyl cations. J. Chem. Phys. 2008, 128, 021102. [Google Scholar] [CrossRef] [PubMed]
- Vogel, P. Carbocation Chemistry; Elsevier: Amsterdam, The Netherlands, 1985; p. 173. [Google Scholar]
- Buzek, P.; Schleyer, P.R.; Vančik, H.; Mihalic, Z.; Gauss, J. Generation of the Parent Allyl Cation in a Superacid Cryogenic Matrix. Angew. Chem. Int. Ed. 1994, 33, 448–451. [Google Scholar] [CrossRef]
- Mišić, V.; Piech, K.; Bally, T. Carbocations Generated under Stable Conditions by Ionization of Matrix-Isolated Radicals: The Allyl and Benzyl Cations. J. Am. Chem. Soc. 2013, 135, 8625–8631. [Google Scholar] [CrossRef]
- Siehl, H.-U.; Müller, T.; Gauss, J.; Buzek, P.; Schleyer, P.V.R. Cyclopropylcyclopropylidenemethyl Cation: A Unique Stabilized Vinyl Cation Characterized by NMR Spectroscopy and Quantum Chemical ab Initio Calculations. J. Am. Chem. Soc. 1994, 116, 6384–6387. [Google Scholar] [CrossRef]
- Siehl, H.-U.; Müller, T.; Gauss, J. NMR Spectroscopic and quantum chemical characterization of the (E)- and (Z)-isomers of the penta-1,3-dienyl-2-cation. J. Phys. Org. Chem. 2003, 16, 577–581. [Google Scholar] [CrossRef]
- Olah, G.A.; Staral, J.S.; Liang., G. Novel aromatic systems. I. Homocyclopropenyl cation, the simplest 2.pi. homoaromatic system. J. Am. Chem. Soc. 1974, 96, 6233–6235. [Google Scholar] [CrossRef]
- Mayr, H.; Olah, G.A. Stable carbocations. 202. Ring closure reactions of allyl to cyclopropylcarbinyl cations. J. Am. Chem. Soc. 1977, 99, 510–513. [Google Scholar] [CrossRef]
- Olah, G.A.; Spear, R.J. Stable carbocations. CLXXX. Carbon-13 and proton nuclear magnetic resonance spectroscopic study of phenyl-, methyl-, and cyclopropyl-substituted alkenyl (allyl) cations. Further studies of the trend of charge distribution and the relative delocalization afforded by phenyl, methyl, and cyclopropyl. J. Am. Chem. Soc. 1975, 97, 1539–1546. [Google Scholar]
- Siehl, H.-U. Dicoordinated Carbocations; Rappoport, Z., Stang, P., Eds.; Wiley: Chichester, UK; New York, NY, USA, 1997; pp. 189–236. [Google Scholar]
- Siehl, H.-U. Stable Carbocation Chemistry; Prakash, G.K.S., Schleyer, P.V.R., Eds.; Wiley: New York, NY, USA, 1997; pp. 165–196. [Google Scholar]
- Siehl, H.-U.; Kaufmann, F.-P.; Apeloig, Y.; Braude, V.; Danovich, D.; Berndt, A.; Stamatis, N. The First Persistent β-Silyl-Substituted Vinyl Cation. Angew. Chem. 1991, 30, 1479–1482. [Google Scholar] [CrossRef]
- Müller, T.; Margraf, D.; Syha, Y.; Nasiri, H.R.; Kaiser, C.; Maier, R.; Boltre, B.; Juhasz, M.; Reed, C.A. Recent Developments in Carbocation and Onium Ion Chemistry; Kenneth, K.L., Ed.; American Chemical Society: Chicago, IL, USA, 2007; Chapter 3; pp. 51–67. [Google Scholar]
- Stoyanov, E.S.; Bagryanskaya, I.Y.; Stoyanova, I.V. Unsaturated vinyl-type carbocation [(CH3)2C=CH]+ in its carborane salts. ACS Omega 2021, 6, 15834–15843. [Google Scholar] [CrossRef] [PubMed]
- Stoyanov, E.S.; Bagryanskaya, I.Y.; Stoyanova, I.V. Isomers of the Allyl Carbocation C3H5+ in Solid Salts: Infrared Spectra and Structures. ACS Omega 2021, 6, 23691–23699. [Google Scholar] [CrossRef] [PubMed]
- Stoyanov, E.S.; Bagryanskaya, I.Y.; Stoyanova, I.V. An IR-spectroscopic and X-ray-structural study of vinyl-type carbocations in their carborane salts. ACS Omega 2022, 7, 27560–27572. [Google Scholar] [CrossRef]
- Cunje, A.; Rodriquez, C.F.; Lien, M.H.; Hopkinson, A.C. The C4H5+ Potential Energy Surface. Structure, Relative Energies, and Enthalpies of Formation of Isomers of C4H5+. J. Org. Chem. 1996, 61, 5212–5220. [Google Scholar] [CrossRef]
- Reed, C. Carborane acids. New “strong yet gentle” acids for organic and inorganic chemistry. Chem. Commun. 2005, 13, 1669–1677. [Google Scholar] [CrossRef]
- Stoyanov, E.S. Chemical Properties of Dialkyl Halonium Ions (R2Hal+) and Their Neutral Analogues, Methyl Carboranes, CH3−(CHB11Hal11), Where Hal = F., Cl. J. Phys. Chem. A 2017, 121, 2918–2923. [Google Scholar] [CrossRef] [PubMed]
- Stoyanov, E.S.; Stoyanova, I.V.; Tham, F.S.; Reed, C.A. Dialkyl Chloronium Ions. J. Am. Chem. Soc. 2010, 132, 4062–4063. [Google Scholar] [CrossRef] [PubMed]
- Whipple, E.B.; Steward, W.E.; Reddy, G.S.; Goldstein, J.H. NMR Spectra of Vinyl Chloride and the Chloroethylenes. J. Chem. Phys. 1961, 34, 2136–2138. [Google Scholar] [CrossRef]
- Breitmair, E.; Volter, W. Carbon-13 NMR Spectroscopy, 2nd ed.; Gordon & Breach Science: New York, NY, USA, 1978; p. 149. [Google Scholar]
- Narita, S.; Ichinohe, S.; Enomoto, S. Infrared Spectra of Vinyl Chloride and Vinyl Chloride-d3. J. Chem. Phys. 1959, 31, 1151–1157. [Google Scholar] [CrossRef]
- Olah, G.A.; Donovan, D.J. Stable carbocations. 208. Carbon-13 nuclear magnetic resonance spectroscopic study of alkyl cations. The constancy of carbon-13 nuclear magnetic resonance methyl substituent effects and their application in the study of equilibrating carbocations and the mechanism of some rearrangements. J. Am. Chem. Soc. 1977, 99, 5026–5037. [Google Scholar]
- Olah, G.A.; Staral, J.S.; Spear, R.J.; Liang, G. Novel aromatic systems. II. Cyclobutenyl cations and the question of their homoaromaticity. Preparation and study of the homocyclopropenium ion, the simplest homoaromatic system. J. Am. Chem. Soc. 1975, 97, 5489–5497. [Google Scholar] [CrossRef]
- Stanton, J.F.; Gauss, J.; Siehl, H.-U. CCSD(T) calculation of NMR chemical shifts: Consistency of calculated and measured 13C chemical shifts in the 1-cyclopropylcyclopropylidenemethyl cation. Chem. Phys. Lett. 1996, 262, 183–186. [Google Scholar] [CrossRef]
- Muller, T.; Juhasz, M.; Reed, C.A. The X-ray Structure of a Vinyl Cation. Angew. Chem. Int. Ed. 2004, 43, 1543–1546. [Google Scholar] [CrossRef] [PubMed]
- Juhasz, M.; Hoffmann, S.; Stoyanov, E.S.; Kim, K.; Reed, C.A. The strongest isolable acid. Angew. Chem. Int. Ed. 2004, 43, 5352–5355. [Google Scholar] [CrossRef] [PubMed]
- Nava, M.; Stoyanova, I.V.; Cummings, S.; Stoyanov, E.S.; Reed, C.A. The Strongest Brønsted Acid: Protonation of Alkanes by H(CHB11F11) at Room Temperature. Angew. Chem. Int. Ed. 2014, 53, 1131–1134. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Kesharwani, M.K.; Brauer, B.; Martin, J.M.L. Frequency and Zero-Point Vibrational Energy Scale Factors for Double-Hybrid Density Functionals (and Other Selected Methods): Can Anharmonic Force Fields Be Avoided? J. Phys. Chem. A 2015, 119, 1701–1714. [Google Scholar] [CrossRef]






















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
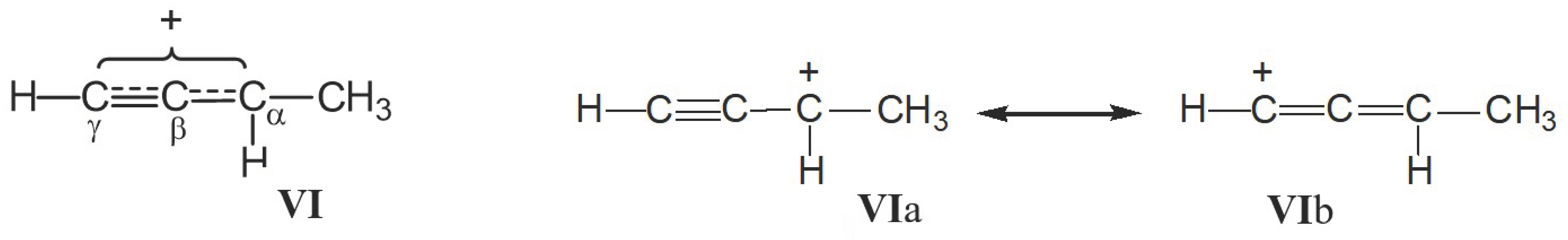{kind=link}
{kind=link}
{kind=link}
| Atom | Cation I | CH2CHCl [33,34] | Atom | Cation I | CH2CHCl [33,34] | |
|---|---|---|---|---|---|---|
| Solid | Solution | Solid | ||||
| Ha | 5.58 5.14 * | 4.87 4.49 * | 6.13 | Cα | 83.2 78.6 * | 124.9 |
| Hb | 4.77 | 4.11 3.97 * | 5.23 | Cβ | 60.6 43.9 * | 116 |
| Hc | 2.73 2.22 * | 2.91 2.49 * | 5.39 | |||
| Sample | C-H Stretches | C-H Bending | CC Stretch | CH Bending | CCCl Bending | CCl+C Stretches | |||
|---|---|---|---|---|---|---|---|---|---|
| protio | 3055 3044 | 2987 2974 2961 2938 | 1418 * | 1324 | 1269 | 1238 | 914 | 740 | 628 594 |
| deuterio | 2303 2295 | 2244 2218 2191 2171 | ** | ** | 960 | 1208 | 694 | 748 | 635 582 |
| ratio | 1.327 1.326 | 1.331 1.341 1.351 1.353 | - | - | 1.323 | 1.024 | 1.320 | 0.989 | 0.989 1.020 |
| Isomer | Stretch Vibrations of C-Cl+-C Group, cm−1 (calc a/exp) | |
|---|---|---|
| C4H6Cl+ (exp) | 628 | 594 |
| I (calc) | 568 (0.91) | 538 (0.91) |
| II (calc) | 494 (0.79) | 385 (0.65) |
| III (calc) | 478 (0.76) | 460 (0.77) |
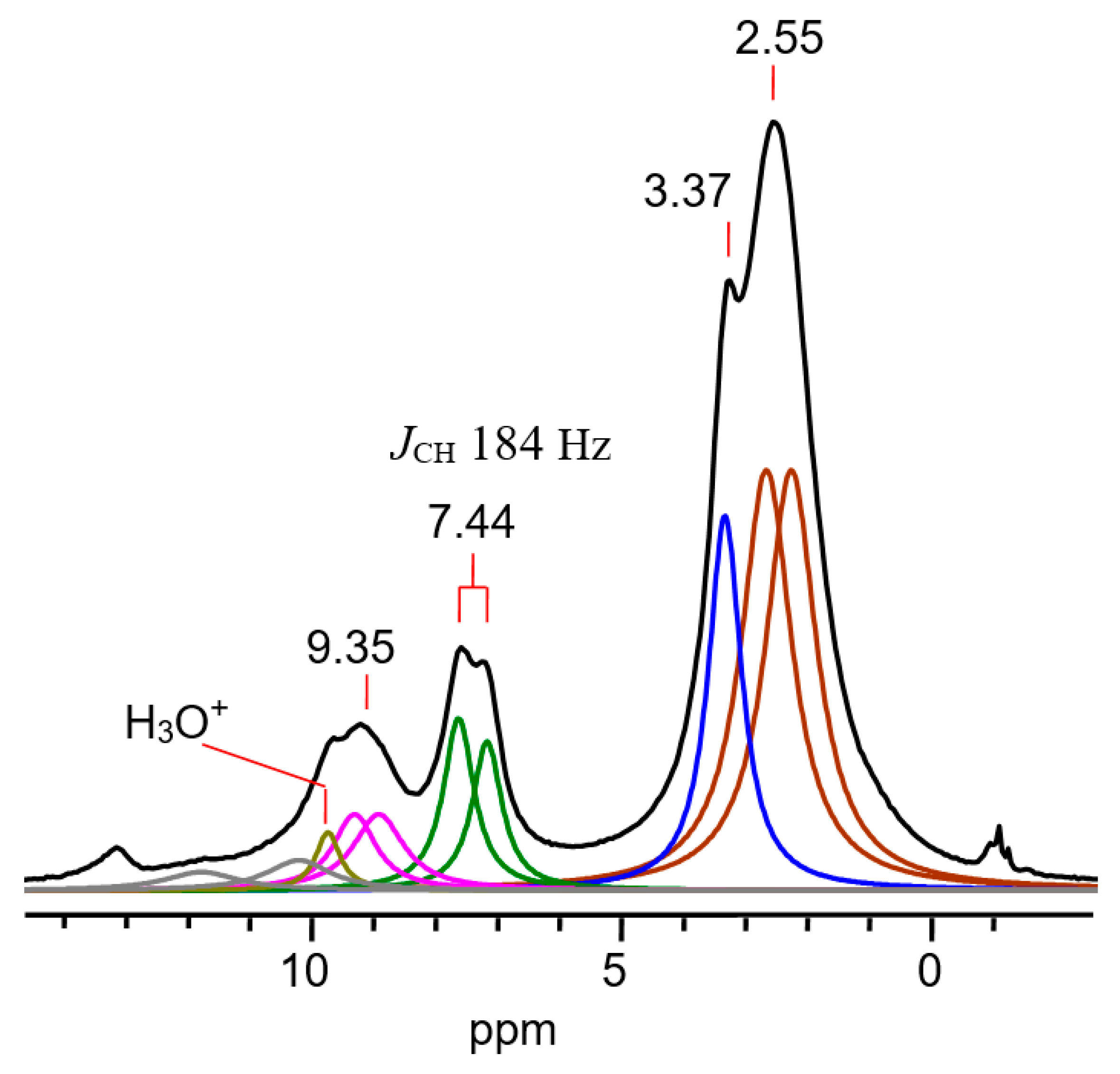
| Nucleus | CH3 | CαH | CβH | CγH (Cl) |
|---|---|---|---|---|
| 1H δ | 2.69 | 8.42 | 7.04 | 9.58 |
| 1JCH | 130 | 158 | 168 | 183 |
| 3JHH | 7 | b | 7 | b |
| 13C δ | 20.8 | 183.6 | 129.2 | 203.6 |
| 1JCC | 39 | 60 | 60 | 60 |
| Nucleus | CH3 | CαH | Cβ | CγH (1JCH in Hz) |
|---|---|---|---|---|
| 1H | 2.25 | 9.35 | - | 7.44 (184) |
| 13C | 36 | 215 | 150 | 231 |
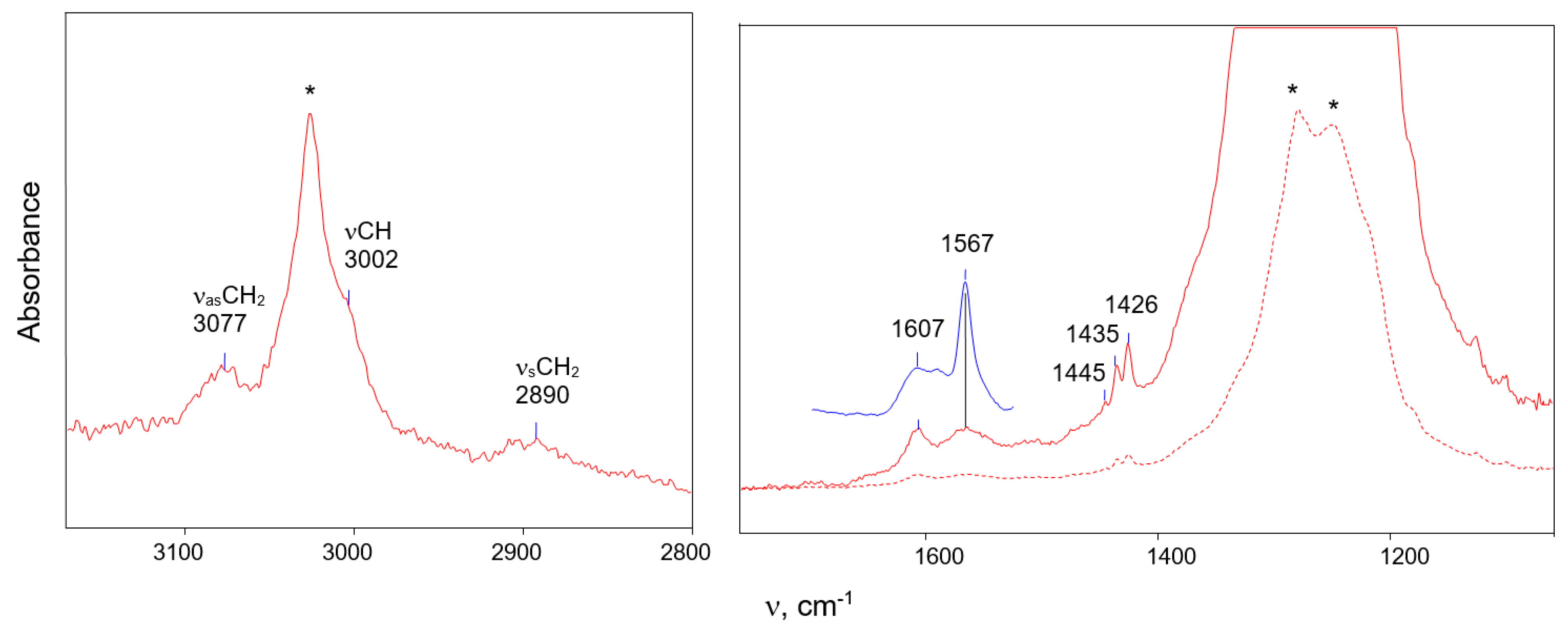
| C4H5+ | C4D5+ | H/D Ratio | Assignment * |
|---|---|---|---|
| 3058 | 2284 | 1.339 | νCαH (CαD) and |
| 3039 | 2251 | 1.350 | νCβH (CβD) |
| 2942 | 2129 | 1.381 | νasCH3 (CD3) |
| 2891 | - | - | 2νCC overtone |
| 2871 | 2077 | 1.380 | νasCH3 (CD3) |
| 2829 | 2055 | 1.377 | νsCH3 (CD3) |
| 1563 | 1521 1497 | 1.028 1.044 | νC≡C |
| 1447 | 1368 | 1.057 | νCC |
| 1327 | 997 | 1.341 | δCH (CD) |
| 1303 | 971 | 1.341 | δCH (CD) |
| 964 | 719 | 1.354 | δCH (CD) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stoyanov, E.S.; Stoyanova, I.V. The Chloronium Cation [(C2H3)2Cl+] and Unsaturated C4-Carbocations with C=C and C≡C Bonds in Their Solid Salts and in Solutions: An H1/C13 NMR and Infrared Spectroscopic Study. Int. J. Mol. Sci. 2022, 23, 9111. https://doi.org/10.3390/ijms23169111
Stoyanov ES, Stoyanova IV. The Chloronium Cation [(C2H3)2Cl+] and Unsaturated C4-Carbocations with C=C and C≡C Bonds in Their Solid Salts and in Solutions: An H1/C13 NMR and Infrared Spectroscopic Study. International Journal of Molecular Sciences. 2022; 23(16):9111. https://doi.org/10.3390/ijms23169111
Chicago/Turabian StyleStoyanov, Evgenii S., and Irina V. Stoyanova. 2022. "The Chloronium Cation [(C2H3)2Cl+] and Unsaturated C4-Carbocations with C=C and C≡C Bonds in Their Solid Salts and in Solutions: An H1/C13 NMR and Infrared Spectroscopic Study" International Journal of Molecular Sciences 23, no. 16: 9111. https://doi.org/10.3390/ijms23169111
APA StyleStoyanov, E. S., & Stoyanova, I. V. (2022). The Chloronium Cation [(C2H3)2Cl+] and Unsaturated C4-Carbocations with C=C and C≡C Bonds in Their Solid Salts and in Solutions: An H1/C13 NMR and Infrared Spectroscopic Study. International Journal of Molecular Sciences, 23(16), 9111. https://doi.org/10.3390/ijms23169111