
Targeting the Sphingolipid Rheostat in Gliomas


Abstract
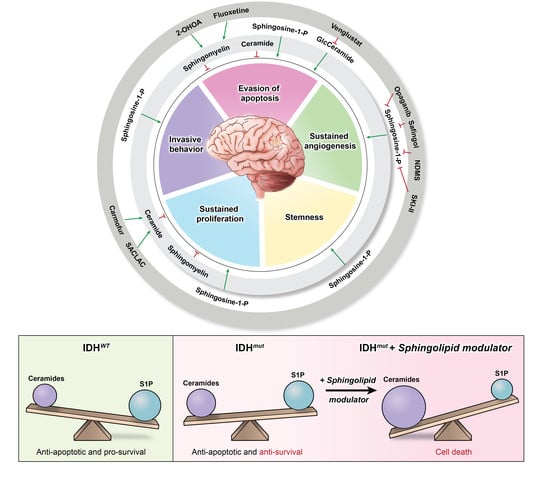
:
1. Introduction
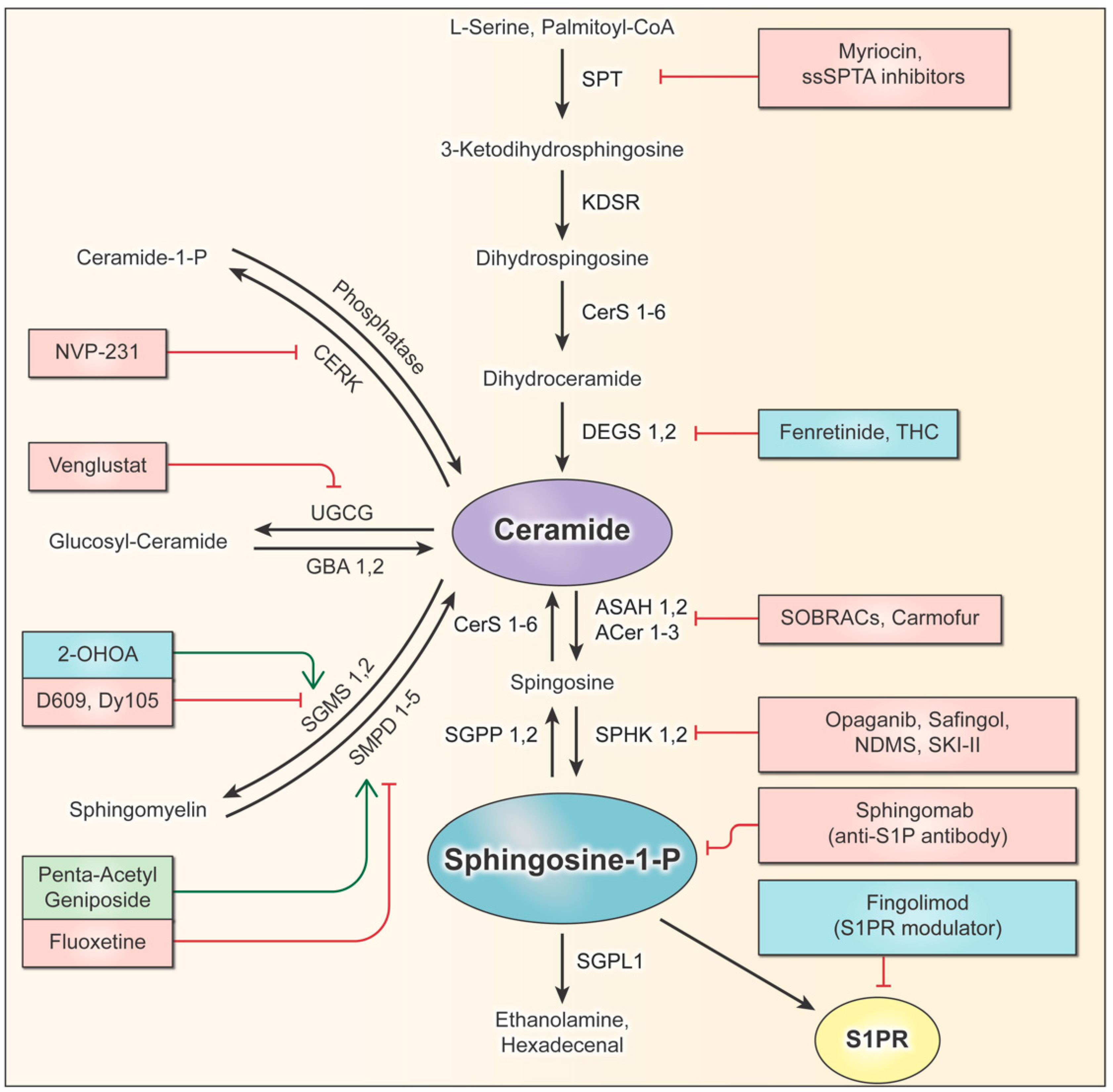
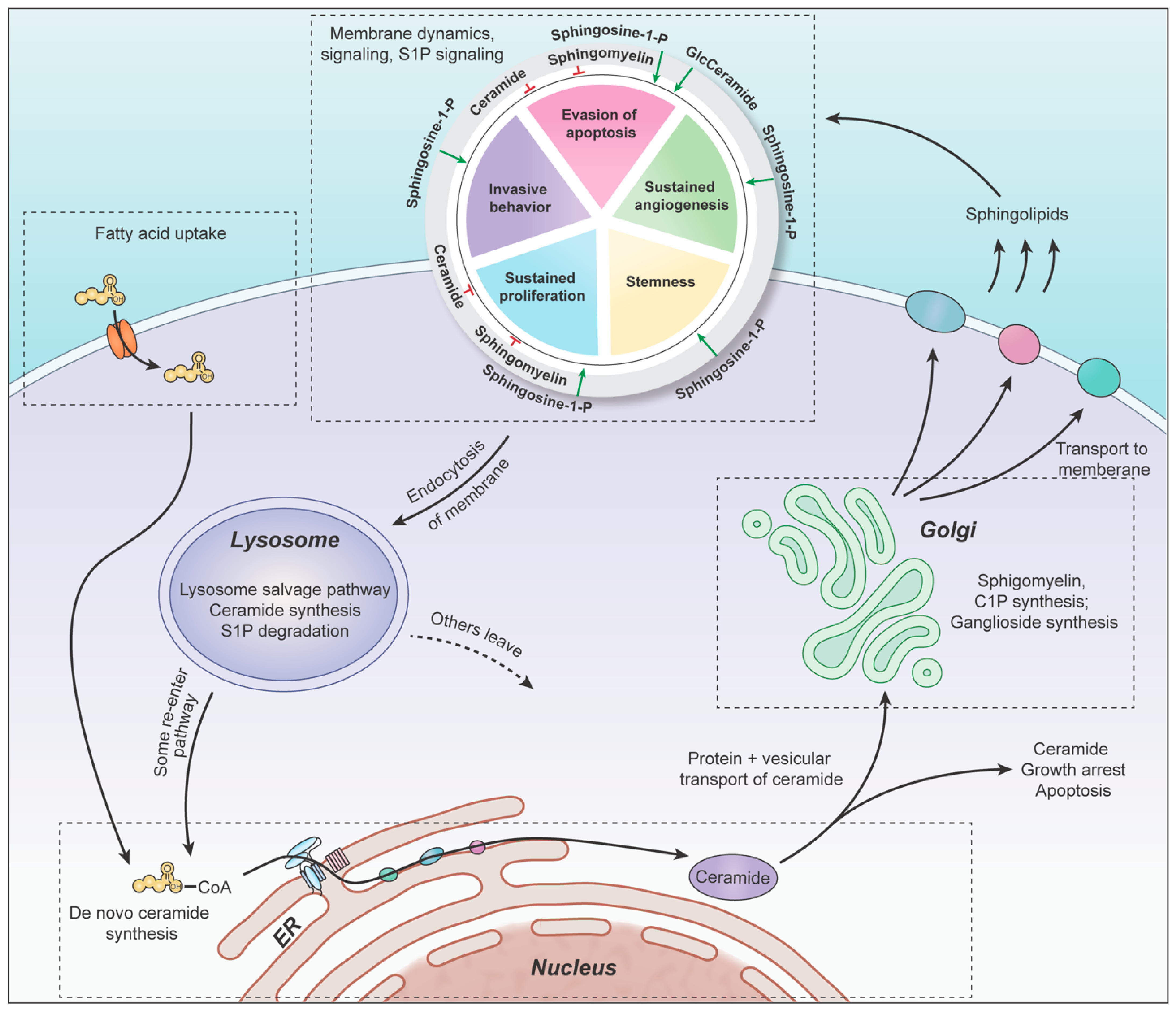
2. Sphingolipid Pathway
3. Palmitic Acid Uptake and Incorporation into the Sphingolipid Pathway
3.1. PA Transport into the Brain
3.2. PA Storage in Lipid Droplets
3.3. PA Activation for Sphingolipid Synthesis
4. Endoplasmic Reticulum: De Novo Synthesis of Ceramide
4.1. First Step of Sphingolipid Synthesis
4.2. Production of Dihydroceramide
4.3. Production of Ceramide
5. Golgi: Complex Sphingolipids Synthesis
5.1. Ceramide Transport to the Golgi
5.2. Production of Sphingomyelin
5.3. Phosphorylation of Ceramide
5.4. Initiation of the Glycosphingolipid Synthesis Pathway
6. Plasma Membrane
6.1. Production of Sphingomyelin at the Membrane
6.2. Degradation of Sphingomyelin at the Membrane
6.3. Production of Sphingosine-1-Phosphate at the Membrane
6.4. Degradation of Ceramide-1-Phosphate
7. Lysosomes: Salvage Pathway
7.1. Lysosomal Degradation of Sphingomyelin
7.2. Lysosomal Degradation of Ceramide
8. Cytoplasm: Exiting the Sphingolipid Pathway
8.1. Phosphorylation of Sphingosine
8.2. Dephosphorylation of Sphingosine-1-Phosphate
8.3. Degradation of Sphingosine
9. Current Advancements in Sphingolipid Targeting
9.1. Drugs Targeting the Sphingolipid Pathway Assessed in Glioma Clinical Trials
9.2. Future Drug Development
9.3. Pre-Clinical Drugs Targeting De Novo Sphingolipid Synthesis
9.4. Pre-Clinical Drugs Targeting Complex Sphingolipid Synthesis
9.5. Pre-Clinical Drugs Targeting Sphingolipid Metabolism at the Membrane
9.6. Pre-Clinical Drugs Targeting Lysosomal Sphingolipid Degradation
9.7. Pre-Clinical Drugs Targeting Sphingosine Kinase
10. Perspectives and Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ostrom, Q.T.; Bauchet, L.; Davis, F.G.; Deltour, I.; Fisher, J.L.; Langer, C.E.; Pekmezci, M.; Schwartzbaum, J.A.; Turner, M.C.; Walsh, K.M.; et al. The Epidemiology of Glioma in Adults: A “State of the Science” Review. Neuro Oncol. 2014, 16, 896–913. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.R.; O’Neill, B.P. Glioblastoma Survival in the United States before and during the Temozolomide Era. J. Neurooncol. 2012, 107, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Bi, J.; Chowdhry, S.; Wu, S.; Zhang, W.; Masui, K.; Mischel, P.S. Altered Cellular Metabolism in Gliomas—An Emerging Landscape of Actionable Co-Dependency Targets. Nat. Rev. Cancer 2020, 20, 57–70. [Google Scholar] [CrossRef]
- Bleeker, F.E.; Lamba, S.; Leenstra, S.; Troost, D.; Hulsebos, T.; Vandertop, W.P.; Frattini, M.; Molinari, F.; Knowles, M.; Cerrato, A.; et al. IDH1 Mutations at Residue p.R132 (IDH1R132) Occur Frequently in High-Grade Gliomas but Not in Other Solid Tumors. Hum. Mutat. 2009, 30, 7–11. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A Summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef]
- Fack, F.; Tardito, S.; Hochart, G.; Oudin, A.; Zheng, L.; Fritah, S.; Golebiewska, A.; Nazarov, P.V.; Bernard, A.; Hau, A.; et al. Altered Metabolic Landscape in IDH-mutant Gliomas Affects Phospholipid, Energy, and Oxidative Stress Pathways. EMBO Mol. Med. 2017, 9, 1681–1695. [Google Scholar] [CrossRef]
- Watanabe, T.; Nobusawa, S.; Kleihues, P.; Ohgaki, H. IDH1 Mutations Are Early Events in the Development of Astrocytomas and Oligodendrogliomas. Am. J. Pathol. 2009, 174, 1149–1153. [Google Scholar] [CrossRef]
- Juratli, T.A.; Kirsch, M.; Robel, K.; Soucek, S.; Geiger, K.; von Kummer, R.; Schackert, G.; Krex, D. IDH Mutations as an Early and Consistent Marker in Low-Grade Astrocytomas WHO Grade II and Their Consecutive Secondary High-Grade Gliomas. J. Neurooncol. 2012, 108, 403–410. [Google Scholar] [CrossRef]
- Rakheja, D.; Medeiros, L.; Bevan, S.; Chen, W. The Emerging Role of D-2-Hydroxyglutarate as an Oncometabolite in Hematolymphoid and Central Nervous System Neoplasms. Front. Oncol. 2013, 3, 169. [Google Scholar] [CrossRef]
- Dowdy, T.; Zhang, L.; Celiku, O.; Movva, S.; Lita, A.; Ruiz-Rodado, V.; Gilbert, M.R.; Larion, M. Sphingolipid Pathway as a Source of Vulnerability in IDH1mut Glioma. Cancers 2020, 12, 2910. [Google Scholar] [CrossRef] [PubMed]
- Abdel Hadi, L.; Di Vito, C.; Marfia, G.; Navone, S.E.; Campanella, R.; Riboni, L. The Role and Function of Sphingolipids in Glioblastoma Multiforme. In Bioactive Sphingolipids in Cancer Biology and Therapy; Hannun, Y.A., Luberto, C., Mao, C., Obeid, L.M., Eds.; Springer International Publishing: Cham, Switzerland, 2015; pp. 259–293. ISBN 978-3-319-20750-6. [Google Scholar]
- Abuhusain, H.J.; Matin, A.; Qiao, Q.; Shen, H.; Kain, N.; Day, B.W.; Stringer, B.W.; Daniels, B.; Laaksonen, M.A.; Teo, C.; et al. A Metabolic Shift Favoring Sphingosine 1-Phosphate at the Expense of Ceramide Controls Glioblastoma Angiogenesis. J. Biol. Chem. 2013, 288, 37355–37364. [Google Scholar] [CrossRef] [PubMed]
- Young, N.; Pearl, D.K.; Van Brocklyn, J.R. Sphingosine-1-Phosphate Regulates Glioblastoma Cell Invasiveness Through The Urokinase Plasminogen Activator System and CCN1/Cyr61. Mol. Cancer Res. 2009, 7, 23–32. [Google Scholar] [CrossRef]
- Marfia, G.; Campanella, R.; Navone, S.E.; Di Vito, C.; Riccitelli, E.; Hadi, L.A.; Bornati, A.; de Rezende, G.; Giussani, P.; Tringali, C.; et al. Autocrine/Paracrine Sphingosine-1-Phosphate Fuels Proliferative and Stemness Qualities of Glioblastoma Stem Cells. Glia 2014, 62, 1968–1981. [Google Scholar] [CrossRef] [PubMed]
- Van Brocklyn, J.R.; Letterle, C.A.; Snyder, P.J.; Prior, T.W. Sphingosine-1-Phosphate Stimulates Human Glioma Cell Proliferation through Gi-Coupled Receptors: Role of ERK MAP Kinase and Phosphatidylinositol 3-Kinase β. Cancer Lett. 2002, 181, 195–204. [Google Scholar] [CrossRef]
- Riccitelli, E.; Giussani, P.; Vito, C.D.; Condomitti, G.; Tringali, C.; Caroli, M.; Galli, R.; Viani, P.; Riboni, L. Extracellular Sphingosine-1-Phosphate: A Novel Actor in Human Glioblastoma Stem Cell Survival. PLoS ONE 2013, 8, e68229. [Google Scholar] [CrossRef] [PubMed]
- Mahajan-Thakur, S.; Bien-Möller, S.; Marx, S.; Schroeder, H.; Rauch, B.H. Sphingosine 1-Phosphate (S1P) Signaling in Glioblastoma Multiforme-A Systematic Review. Int. J. Mol. Sci. 2017, 18, E2448. [Google Scholar] [CrossRef]
- Riboni, L.; Campanella, R.; Bassi, R.; Villani, R.; Gaini, S.M.; Martinelli-Boneschi, F.; Viani, P.; Tettamanti, G. Ceramide Levels Are Inversely Associated with Malignant Progression of Human Glial Tumors. Glia 2002, 39, 105–113. [Google Scholar] [CrossRef]
- Van Brocklyn, J. Sphingolipid Signaling Pathways as Potential Therapeutic Targets in Gliomas. MRMC 2007, 7, 984–990. [Google Scholar] [CrossRef]
- Tea, M.N.; Poonnoose, S.I.; Pitson, S.M. Targeting the Sphingolipid System as a Therapeutic Direction for Glioblastoma. Cancers 2020, 12, 111. [Google Scholar] [CrossRef]
- Noda, S.; Yoshimura, S.; Sawada, M.; Naganawa, T.; Iwama, T.; Nakashima, S.; Sakai, N. Role of Ceramide During Cisplatin-Induced Apoptosis in C6 Glioma Cells. J. Neurooncol. 2001, 52, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Hara, S.; Nakashima, S.; Kiyono, T.; Sawada, M.; Yoshimura, S.; Iwama, T.; Banno, Y.; Shinoda, J.; Sakai, N. P53-Independent Ceramide Formation in Human Glioma Cells during Gamma-Radiation-Induced Apoptosis. Cell Death Differ. 2004, 11, 853–861. [Google Scholar] [CrossRef] [PubMed]
- Sawada, M.; Kiyono, T.; Nakashima, S.; Shinoda, J.; Naganawa, T.; Hara, S.; Iwama, T.; Sakai, N. Molecular Mechanisms of TNF-α-Induced Ceramide Formation in Human Glioma Cells:P53-Mediated Oxidant Stress-Dependent and -Independent Pathways. Cell Death Differ. 2004, 11, 997–1008. [Google Scholar] [CrossRef]
- Morales, A.; París, R.; Villanueva, A.; Llacuna, L.; García-Ruiz, C.; Fernández-Checa, J.C. Pharmacological Inhibition or Small Interfering RNA Targeting Acid Ceramidase Sensitizes Hepatoma Cells to Chemotherapy and Reduces Tumor Growth in Vivo. Oncogene 2007, 26, 905–916. [Google Scholar] [CrossRef]
- Charruyer, A.; Bell, S.M.; Kawano, M.; Douangpanya, S.; Yen, T.-Y.; Macher, B.A.; Kumagai, K.; Hanada, K.; Holleran, W.M.; Uchida, Y. Decreased Ceramide Transport Protein (CERT) Function Alters Sphingomyelin Production Following UVB Irradiation. J. Biol. Chem. 2008, 283, 16682–16692. [Google Scholar] [CrossRef] [PubMed]
- Dumitru, C.A.; Weller, M.; Gulbins, E. Ceramide Metabolism Determines Glioma Cell Resistance to Chemotherapy. J. Cell Physiol. 2009, 221, 688–695. [Google Scholar] [CrossRef]
- Giussani, P.; Bassi, R.; Anelli, V.; Brioschi, L.; De Zen, F.; Riccitelli, E.; Caroli, M.; Campanella, R.; Gaini, S.M.; Viani, P.; et al. Glucosylceramide Synthase Protects Glioblastoma Cells against Autophagic and Apoptotic Death Induced by Temozolomide and Paclitaxel. Cancer Investig. 2012, 30, 27–37. [Google Scholar] [CrossRef]
- Noack, J.; Choi, J.; Richter, K.; Kopp-Schneider, A.; Régnier-Vigouroux, A. A Sphingosine Kinase Inhibitor Combined with Temozolomide Induces Glioblastoma Cell Death through Accumulation of Dihydrosphingosine and Dihydroceramide, Endoplasmic Reticulum Stress and Autophagy. Cell Death Dis. 2014, 5, e1425. [Google Scholar] [CrossRef]
- Doan, N.B.; Nguyen, H.S.; Al-Gizawiy, M.M.; Mueller, W.M.; Sabbadini, R.A.; Rand, S.D.; Connelly, J.M.; Chitambar, C.R.; Schmainda, K.M.; Mirza, S.P. Acid Ceramidase Confers Radioresistance to Glioblastoma Cells. Oncol. Rep. 2017, 38, 1932–1940. [Google Scholar] [CrossRef]
- Van Brocklyn, J.R.; Williams, J.B. The Control of the Balance between Ceramide and Sphingosine-1-Phosphate by Sphingosine Kinase: Oxidative Stress and the Seesaw of Cell Survival and Death. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2012, 163, 26–36. [Google Scholar] [CrossRef]
- Gault, C.; Obeid, L.; Hannun, Y. An Overview of Sphingolipid Metabolism: From Synthesis to Breakdown. Adv. Exp. Med. Biol. 2010, 688, 1–23. [Google Scholar] [PubMed]
- Carta, G.; Murru, E.; Banni, S.; Manca, C. Palmitic Acid: Physiological Role, Metabolism and Nutritional Implications. Front. Physiol. 2017, 8, 902. [Google Scholar] [CrossRef] [PubMed]
- Shakya, S.; Gromovsky, A.D.; Hale, J.S.; Knudsen, A.M.; Prager, B.; Wallace, L.C.; Penalva, L.O.F.; Brown, H.A.; Kristensen, B.W.; Rich, J.N.; et al. Altered Lipid Metabolism Marks Glioblastoma Stem and Non-Stem Cells in Separate Tumor Niches. Acta Neuropathol. Commun. 2021, 9, 101. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Lupu, R. Fatty Acid Synthase and the Lipogenic Phenotype in Cancer Pathogenesis. Nat. Rev. Cancer 2007, 7, 763–777. [Google Scholar] [CrossRef]
- Dhopeshwarkar, G.A.; Mead, J.F. Uptake and Transport of Fatty Acids into the Brain and the Role of the Blood-Brain Barrier System. Adv. Lipid Res. 1973, 11, 109–142. [Google Scholar] [CrossRef]
- Mitchell, R.W.; On, N.H.; Bigio, M.R.D.; Miller, D.W.; Hatch, G.M. Fatty Acid Transport Protein Expression in Human Brain and Potential Role in Fatty Acid Transport across Human Brain Microvessel Endothelial Cells. J. Neurochem. 2011, 117, 735–746. [Google Scholar] [CrossRef]
- García-Martínez, C.; Marotta, M.; Moore-Carrasco, R.; Guitart, M.; Camps, M.; Busquets, S.; Montell, E.; Gómez-Foix, A.M. Impact on Fatty Acid Metabolism and Differential Localization of FATP1 and FAT/CD36 Proteins Delivered in Cultured Human Muscle Cells. Am. J. Physiol.-Cell Physiol. 2005, 288, C1264–C1272. [Google Scholar] [CrossRef]
- Bowman, R.L.; Wang, Q.; Carro, A.; Verhaak, R.G.W.; Squatrito, M. GlioVis Data Portal for Visualization and Analysis of Brain Tumor Expression Datasets. Neuro Oncol. 2017, 19, 139–141. [Google Scholar] [CrossRef]
- Cheng, A.; Jia, W.; Kawahata, I.; Fukunaga, K. A Novel Fatty Acid-Binding Protein 5 and 7 Inhibitor Ameliorates Oligodendrocyte Injury in Multiple Sclerosis Mouse Models. eBioMedicine 2021, 72, 103582. [Google Scholar] [CrossRef]
- Nath, A.; Li, I.; Roberts, L.R.; Chan, C. Elevated Free Fatty Acid Uptake via CD36 Promotes Epithelial-Mesenchymal Transition in Hepatocellular Carcinoma. Sci. Rep. 2015, 5, 14752. [Google Scholar] [CrossRef]
- Ng, Y.-W.; Say, Y.-H. Palmitic Acid Induces Neurotoxicity and Gliatoxicity in SH-SY5Y Human Neuroblastoma and T98G Human Glioblastoma Cells. PeerJ 2018, 6, e4696. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Shah, N.; Almohaisin, M.I.; Saha, S.; Lu, F. Assessing Fatty Acid-Induced Lipotoxicity and Its Therapeutic Potential in Glioblastoma Using Stimulated Raman Microscopy. Sci. Rep. 2021, 11, 7422. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Geng, F.; Pan, M.; Wu, X.; Zhong, Y.; Wang, C.; Tian, Z.; Cheng, C.; Zhang, R.; Puduvalli, V.; et al. Targeting DGAT1 Ameliorates Glioblastoma by Increasing Fat Catabolism and Oxidative Stress. Cell Metab. 2020, 32, 229–242.e8. [Google Scholar] [CrossRef] [PubMed]
- Berge, K.; Tronstad, K.J.; Bohov, P.; Madsen, L.; Berge, R.K. Impact of Mitochondrial β-Oxidation in Fatty Acid-Mediated Inhibition of Glioma Cell Proliferation. J. Lipid Res. 2003, 44, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.B.; Louie, S.M.; Daniele, J.R.; Tran, Q.; Dillin, A.; Zoncu, R.; Nomura, D.K.; Olzmann, J.A. DGAT1-Dependent Lipid Droplet Biogenesis Protects Mitochondrial Function during Starvation-Induced Autophagy. Dev. Cell 2017, 42, 9–21.e5. [Google Scholar] [CrossRef] [PubMed]
- Listenberger, L.L.; Han, X.; Lewis, S.E.; Cases, S.; Farese, R.V.; Ory, D.S.; Schaffer, J.E. Triglyceride Accumulation Protects against Fatty Acid-Induced Lipotoxicity. Proc. Natl. Acad. Sci. USA 2003, 100, 3077–3082. [Google Scholar] [CrossRef]
- Hernández-Corbacho, M.J.; Obeid, L.M. A Novel Role for DGATs in Cancer. Adv. Biol. Regul. 2019, 72, 89–101. [Google Scholar] [CrossRef]
- Senkal, C.E.; Salama, M.F.; Snider, A.J.; Allopenna, J.J.; Rana, N.A.; Koller, A.; Hannun, Y.A.; Obeid, L.M. Ceramide Is Metabolized to Acylceramide and Stored in Lipid Droplets. Cell Metab. 2017, 25, 686–697. [Google Scholar] [CrossRef]
- Watkins, P.A.; Maiguel, D.; Jia, Z.; Pevsner, J. Evidence for 26 Distinct Acyl-Coenzyme A Synthetase Genes in the Human Genomes. J. Lipid Res. 2007, 48, 2736–2750. [Google Scholar] [CrossRef]
- Ohkuni, A.; Ohno, Y.; Kihara, A. Identification of Acyl-CoA Synthetases Involved in the Mammalian Sphingosine 1-Phosphate Metabolic Pathway. Biochem. Biophys. Res. Commun. 2013, 442, 195–201. [Google Scholar] [CrossRef]
- Marszalek, J.R.; Kitidis, C.; DiRusso, C.C.; Lodish, H.F. Long-Chain Acyl-CoA Synthetase 6 Preferentially Promotes DHA Metabolism. J. Biol. Chem. 2005, 280, 10817–10826. [Google Scholar] [CrossRef]
- Mashek, D.G.; McKenzie, M.A.; Van Horn, C.G.; Coleman, R.A. Rat Long Chain Acyl-CoA Synthetase 5 Increases Fatty Acid Uptake and Partitioning to Cellular Triacylglycerol in McArdle-RH7777 Cells. J. Biol. Chem. 2006, 281, 945–950. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Kawarabayasi, Y.; Kondo, J.; Abe, T.; Nishikawa, K.; Kimura, S.; Hashimoto, T.; Yamamoto, T. Structure and Regulation of Rat Long-Chain Acyl-CoA Synthetase. J. Biol. Chem. 1990, 265, 8681–8685. [Google Scholar] [CrossRef]
- Oikawa, E.; Iijima, H.; Suzuki, T.; Sasano, H.; Sato, H.; Kamataki, A.; Nagura, H.; Kang, M.-J.; Fujino, T.; Suzuki, H.; et al. A Novel Acyl-CoA Synthetase, ACS5, Expressed in Intestinal Epithelial Cells and Proliferating Preadipocytes. J. Biochem. 1998, 124, 679–685. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, Y.; Kumabe, T.; Cho, Y.-Y.; Watanabe, M.; Kawagishi, J.; Yoshimoto, T.; Fujino, T.; Kang, M.-J.; Yamamoto, T.T. Fatty Acid Induced Glioma Cell Growth Is Mediated by the Acyl-CoA Synthetase 5 Gene Located on Chromosome 10q25.1-Q25.2, a Region Frequently Deleted in Malignant Gliomas. Oncogene 2000, 19, 5919–5925. [Google Scholar] [CrossRef] [PubMed]
- Mashima, T.; Sato, S.; Sugimoto, Y.; Tsuruo, T.; Seimiya, H. Promotion of Glioma Cell Survival by Acyl-CoA Synthetase 5 under Extracellular Acidosis Conditions. Oncogene 2009, 28, 9–19. [Google Scholar] [CrossRef]
- Zhou, L.; Wang, Z.; Hu, C.; Zhang, C.; Kovatcheva-Datchary, P.; Yu, D.; Liu, S.; Ren, F.; Wang, X.; Li, Y.; et al. Integrated Metabolomics and Lipidomics Analyses Reveal Metabolic Reprogramming in Human Glioma with IDH1 Mutation. J. Proteome Res. 2019, 18, 960–969. [Google Scholar] [CrossRef] [PubMed]
- Van Horn, C.G.; Caviglia, J.M.; Li, L.O.; Wang, S.; Granger, D.A.; Coleman, R.A. Characterization of Recombinant Long-Chain Rat Acyl-CoA Synthetase Isoforms 3 and 6: Identification of a Novel Variant of Isoform 6. Biochemistry 2005, 44, 1635–1642. [Google Scholar] [CrossRef]
- Cao, Y.; Traer, E.; Zimmerman, G.A.; McIntyre, T.M.; Prescott, S.M. Cloning, Expression, and Chromosomal Localization of Human Long-Chain Fatty Acid-CoA Ligase 4 (FACL4). Genomics 1998, 49, 327–330. [Google Scholar] [CrossRef]
- Lita, A.; Pliss, A.; Kuzmin, A.; Yamasaki, T.; Zhang, L.; Dowdy, T.; Burks, C.; de Val, N.; Celiku, O.; Ruiz-Rodado, V.; et al. IDH1 Mutations Induce Organelle Defects via Dysregulated Phospholipids. Nat. Commun. 2021, 12, 614. [Google Scholar] [CrossRef]
- Cheng, J.; Fan, Y.-Q.; Liu, B.-H.; Zhou, H.; Wang, J.-M.; Chen, Q.-X. ACSL4 Suppresses Glioma Cells Proliferation via Activating Ferroptosis. Oncol. Rep. 2020, 43, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Kolar, E.A.; Shi, X.; Clay, E.M.; Moser, A.B.; Lal, B.; Nirujogi, R.S.; Pandey, A.; Bandaru, V.V.R.; Laterra, J.; Pei, Z.; et al. Very Long-Chain Acyl-CoA Synthetase 3 Mediates Onco-Sphingolipid Metabolism in Malignant Glioma. Med. Res. Arch. 2021, 9, 2433. [Google Scholar] [CrossRef] [PubMed]
- Pei, Z.; Sun, P.; Huang, P.; Lal, B.; Laterra, J.; Watkins, P.A. Acyl-CoA Synthetase VL3 Knockdown Inhibits Human Glioma Cell Proliferation and Tumorigenicity. Cancer Res. 2009, 69, 9175–9182. [Google Scholar] [CrossRef] [PubMed]
- Mandon, E.C.; Ehses, I.; Rother, J.; van Echten, G.; Sandhoff, K. Subcellular Localization and Membrane Topology of Serine Palmitoyltransferase, 3-Dehydrosphinganine Reductase, and Sphinganine N-Acyltransferase in Mouse Liver. J. Biol. Chem. 1992, 267, 11144–11148. [Google Scholar] [CrossRef]
- Tidhar, R.; Futerman, A.H. The Complexity of Sphingolipid Biosynthesis in the Endoplasmic Reticulum. Biochim. Et Biophys. Acta (BBA)-Mol. Cell Res. 2013, 1833, 2511–2518. [Google Scholar] [CrossRef]
- Hanada, K.; Hara, T.; Nishijima, M. Purification of the Serine Palmitoyltransferase Complex Responsible for Sphingoid Base Synthesis by Using Affinity Peptide Chromatography Techniques. J. Biol. Chem. 2000, 275, 8409–8415. [Google Scholar] [CrossRef]
- Kihara, A.; Igarashi, Y. FVT-1 Is a Mammalian 3-Ketodihydrosphingosine Reductase with an Active Site That Faces the Cytosolic Side of the Endoplasmic Reticulum Membrane. J. Biol. Chem. 2004, 279, 49243–49250. [Google Scholar] [CrossRef]
- Mizutani, Y.; Kihara, A.; Igarashi, Y. Mammalian Lass6 and Its Related Family Members Regulate Synthesis of Specific Ceramides. Biochem. J. 2005, 390, 263–271. [Google Scholar] [CrossRef]
- Michel, C.; van Echten-Deckert, G. Conversion of Dihydroceramide to Ceramide Occurs at the Cytosolic Face of the Endoplasmic Reticulum. FEBS Lett. 1997, 416, 153–155. [Google Scholar] [CrossRef]
- Furuya, S. An Essential Role for de Novo Biosynthesis of L-Serine in CNS Development. Asia Pac. J. Clin. Nutr. 2008, 17, 312–315. [Google Scholar]
- Inuzuka, M.; Hayakawa, M.; Ingi, T. Serinc, an Activity-Regulated Protein Family, Incorporates Serine into Membrane Lipid Synthesis. J. Biol. Chem. 2005, 280, 35776–35783. [Google Scholar] [CrossRef]
- Han, G.; Gupta, S.D.; Gable, K.; Niranjanakumari, S.; Moitra, P.; Eichler, F.; Brown, R.H.; Harmon, J.M.; Dunn, T.M. Identification of Small Subunits of Mammalian Serine Palmitoyltransferase That Confer Distinct Acyl-CoA Substrate Specificities. Proc. Natl. Acad. Sci. USA 2009, 106, 8186–8191. [Google Scholar] [CrossRef] [PubMed]
- Bernhart, E.; Damm, S.; Wintersperger, A.; Nusshold, C.; Brunner, A.M.; Plastira, I.; Rechberger, G.; Reicher, H.; Wadsack, C.; Zimmer, A.; et al. Interference with Distinct Steps of Sphingolipid Synthesis and Signaling Attenuates Proliferation of U87MG Glioma Cells. Biochem. Pharmacol. 2015, 96, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Medlock, K.A.; Merrill, A.H. Inhibition of Serine Palmitoyltransferase in Vitro and Long-Chain Base Biosynthesis in Intact Chinese Hamster Ovary Cells by.Beta.-Chloroalanine. Biochemistry 1988, 27, 7079–7084. [Google Scholar] [CrossRef] [PubMed]
- Davis, D.; Kannan, M.; Wattenberg, B. Orm/ORMDL Proteins: Gate Guardians and Master Regulators. Adv. Biol. Regul. 2018, 70, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Siow, D.L.; Wattenberg, B.W. Mammalian ORMDL Proteins Mediate the Feedback Response in Ceramide Biosynthesis. J. Biol. Chem. 2012, 287, 40198–40204. [Google Scholar] [CrossRef]
- Siow, D.; Sunkara, M.; Dunn, T.M.; Morris, A.J.; Wattenberg, B. ORMDL/Serine Palmitoyltransferase Stoichiometry Determines Effects of ORMDL3 Expression on Sphingolipid Biosynthesis. J. Lipid Res. 2015, 56, 898–908. [Google Scholar] [CrossRef] [PubMed]
- Davis, D.L.; Gable, K.; Suemitsu, J.; Dunn, T.M.; Wattenberg, B.W. The ORMDL/Orm–Serine Palmitoyltransferase (SPT) Complex Is Directly Regulated by Ceramide: Reconstitution of SPT Regulation in Isolated Membranes. J. Biol. Chem. 2019, 294, 5146–5156. [Google Scholar] [CrossRef]
- Gupta, S.D.; Gable, K.; Alexaki, A.; Chandris, P.; Proia, R.L.; Dunn, T.M.; Harmon, J.M. Expression of the ORMDLS, Modulators of Serine Palmitoyltransferase, Is Regulated by Sphingolipids in Mammalian Cells. J. Biol. Chem. 2015, 290, 90–98. [Google Scholar] [CrossRef]
- Venkataraman, K.; Riebeling, C.; Bodennec, J.; Riezman, H.; Allegood, J.C.; Sullards, M.C.; Merrill, A.H.; Futerman, A.H. Upstream of Growth and Differentiation Factor 1 (Uog1), a Mammalian Homolog of the Yeast Longevity Assurance Gene 1 (LAG1), RegulatesN-Stearoyl-Sphinganine (C18-(Dihydro)Ceramide) Synthesis in a Fumonisin B1-Independent Manner in Mammalian Cells. J. Biol. Chem. 2002, 277, 35642–35649. [Google Scholar] [CrossRef]
- Merrill, A.H.; van Echten, G.; Wang, E.; Sandhoff, K. Fumonisin B1 Inhibits Sphingosine (Sphinganine) N-Acyltransferase and de Novo Sphingolipid Biosynthesis in Cultured Neurons in Situ. J. Biol. Chem. 1993, 268, 27299–27306. [Google Scholar] [CrossRef]
- Laviad, E.L.; Albee, L.; Pankova-Kholmyansky, I.; Epstein, S.; Park, H.; Merrill, A.H.; Futerman, A.H. Characterization of Ceramide Synthase 2: TISSUE DISTRIBUTION, SUBSTRATE SPECIFICITY, AND INHIBITION BY SPHINGOSINE 1-PHOSPHATE. J. Biol. Chem. 2008, 283, 5677–5684. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, Y.; Kihara, A.; Igarashi, Y. LASS3 (Longevity Assurance Homologue 3) Is a Mainly Testis-Specific (Dihydro)Ceramide Synthase with Relatively Broad Substrate Specificity. Biochem. J. 2006, 398, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Kota, V.; Dhople, V.M.; Fullbright, G.; Smythe, N.M.; Szulc, Z.M.; Bielawska, A.; Hama, H. 2′-Hydroxy C16-Ceramide Induces Apoptosis-Associated Proteomic Changes in C6 Glioma Cells. J. Proteome Res. 2013, 12, 4366–4375. [Google Scholar] [CrossRef]
- Riebeling, C.; Allegood, J.C.; Wang, E.; Merrill, A.H.; Futerman, A.H. Two Mammalian Longevity Assurance Gene (LAG1) Family Members, Trh1 and Trh4, Regulate Dihydroceramide Synthesis Using Different Fatty Acyl-CoA Donors. J. Biol. Chem. 2003, 278, 43452–43459. [Google Scholar] [CrossRef]
- Morell, P.; Radin, N.S. Specificity in Ceramide Biosynthesis from Long Chain Bases and Various Fatty Acyl Coenzyme A’s by Brain Microsomes. J. Biol. Chem. 1970, 245, 342–350. [Google Scholar] [CrossRef]
- Wang, Z.; Wen, L.; Zhu, F.; Wang, Y.; Xie, Q.; Chen, Z.; Li, Y. Overexpression of Ceramide Synthase 1 Increases C18-Ceramide and Leads to Lethal Autophagy in Human Glioma. Oncotarget 2017, 8, 104022–104036. [Google Scholar] [CrossRef]
- Yu, R.K.; Nakatani, Y.; Yanagisawa, M. The Role of Glycosphingolipid Metabolism in the Developing Brain. J. Lipid Res. 2009, 50, S440–S445. [Google Scholar] [CrossRef]
- Fabris, D.; Rožman, M.; Sajko, T.; Vukelić, Ž. Aberrant Ganglioside Composition in Glioblastoma Multiforme and Peritumoral Tissue: A Mass Spectrometry Characterization. Biochimie 2017, 137, 56–68. [Google Scholar] [CrossRef]
- Yu, D.; Xuan, Q.; Zhang, C.; Hu, C.; Li, Y.; Zhao, X.; Liu, S.; Ren, F.; Zhang, Y.; Zhou, L.; et al. Metabolic Alterations Related to Glioma Grading Based on Metabolomics and Lipidomics Analyses. Metabolites 2020, 10, 478. [Google Scholar] [CrossRef]
- Geeraert, L.; Mannaerts, G.P.; van Veldhoven, P.P. Conversion of Dihydroceramide into Ceramide: Involvement of a Desaturase. Biochem. J. 1997, 327 Pt 1, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Bielawska, A.; Crane, H.M.; Liotta, D.; Obeid, L.M.; Hannun, Y.A. Selectivity of Ceramide-Mediated Biology. Lack of Activity of Erythro-Dihydroceramide. J. Biol. Chem. 1993, 268, 26226–26232. [Google Scholar] [CrossRef]
- Idkowiak-Baldys, J.; Apraiz, A.; Li, L.; Rahmaniyan, M.; Clarke, C.J.; Kraveka, J.M.; Asumendi, A.; Hannun, Y.A. Dihydroceramide Desaturase Activity Is Modulated by Oxidative Stress. Biochem. J. 2010, 427, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Michel, C.; van Echten-Deckert, G.; Rother, J.; Sandhoff, K.; Wang, E.; Merrill, A.H. Characterization of Ceramide Synthesis. A Dihydroceramide Desaturase Introduces the 4,5-Trans-Double Bond of Sphingosine at the Level of Dihydroceramide. J. Biol. Chem. 1997, 272, 22432–22437. [Google Scholar] [CrossRef] [PubMed]
- Savile, C.K.; Fabriàs, G.; Buist, P.H. Dihydroceramide Δ 4 Desaturase Initiates Substrate Oxidation at C-4. J. Am. Chem. Soc. 2001, 123, 4382–4385. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, Y.; Kihara, A.; Igarashi, Y. Identification of the Human Sphingolipid C4-Hydroxylase, HDES2, and Its up-Regulation during Keratinocyte Differentiation. FEBS Lett. 2004, 563, 93–97. [Google Scholar] [CrossRef]
- Hwang, O.; Kim, G.; Jang, Y.J.; Kim, S.W.; Choi, G.; Choi, H.J.; Jeon, S.Y.; Lee, D.G.; Lee, J.D. Synthetic Phytoceramides Induce Apoptosis with Higher Potency than Ceramides. Mol. Pharmacol. 2001, 59, 1249–1255. [Google Scholar] [CrossRef]
- Pant, D.C.; Dorboz, I.; Schluter, A.; Fourcade, S.; Launay, N.; Joya, J.; Aguilera-Albesa, S.; Yoldi, M.E.; Casasnovas, C.; Willis, M.J.; et al. Loss of the Sphingolipid Desaturase DEGS1 Causes Hypomyelinating Leukodystrophy. J. Clin. Investig. 2019, 129, 1240–1256. [Google Scholar] [CrossRef]
- Hanada, K.; Kumagai, K.; Yasuda, S.; Miura, Y.; Kawano, M.; Fukasawa, M.; Nishijima, M. Molecular Machinery for Non-Vesicular Trafficking of Ceramide. Nature 2003, 426, 803–809. [Google Scholar] [CrossRef]
- Giussani, P.; Colleoni, T.; Brioschi, L.; Bassi, R.; Hanada, K.; Tettamanti, G.; Riboni, L.; Viani, P. Ceramide Traffic in C6 Glioma Cells: Evidence for CERT-Dependent and Independent Transport from ER to the Golgi Apparatus. Biochim. Biophys. Acta 2008, 1781, 40–51. [Google Scholar] [CrossRef]
- Kumagai, K.; Yasuda, S.; Okemoto, K.; Nishijima, M.; Kobayashi, S.; Hanada, K. CERT Mediates Intermembrane Transfer of Various Molecular Species of Ceramides. J. Biol. Chem. 2005, 280, 6488–6495. [Google Scholar] [CrossRef] [PubMed]
- Lamour, N.F.; Stahelin, R.V.; Wijesinghe, D.S.; Maceyka, M.; Wang, E.; Allegood, J.C.; Merrill, A.H.; Cho, W.; Chalfant, C.E. Ceramide Kinase Uses Ceramide Provided by Ceramide Transport Protein: Localization to Organelles of Eicosanoid Synthesis. J. Lipid Res. 2007, 48, 1293–1304. [Google Scholar] [CrossRef] [PubMed]
- Boath, A.; Graf, C.; Lidome, E.; Ullrich, T.; Nussbaumer, P.; Bornancin, F. Regulation and Traffic of Ceramide 1-Phosphate Produced by Ceramide Kinase: Comparative Analysis to Glucosylceramide and Sphingomyelin. J. Biol. Chem. 2008, 283, 8517–8526. [Google Scholar] [CrossRef] [PubMed]
- Yamaji, T.; Horie, A.; Tachida, Y.; Sakuma, C.; Suzuki, Y.; Kushi, Y.; Hanada, K. Role of Intracellular Lipid Logistics in the Preferential Usage of Very Long Chain-Ceramides in Glucosylceramide. Int. J. Mol. Sci. 2016, 17, 1761. [Google Scholar] [CrossRef] [PubMed]
- Chandran, S.; Machamer, C.E. Inactivation of Ceramide Transfer Protein during Pro-Apoptotic Stress by Golgi Disassembly and Caspase Cleavage. Biochem. J. 2012, 442, 391–401. [Google Scholar] [CrossRef]
- Nakao, N.; Ueno, M.; Sakai, S.; Egawa, D.; Hanzawa, H.; Kawasaki, S.; Kumagai, K.; Suzuki, M.; Kobayashi, S.; Hanada, K. Natural Ligand-Nonmimetic Inhibitors of the Lipid-Transfer Protein CERT. Commun. Chem. 2019, 2, 1–11. [Google Scholar] [CrossRef]
- Sadeghlar, F.; Sandhoff, K.; van Echten-Deckert, G. Cell Type Specific Localization of Sphingomyelin Biosynthesis. FEBS Lett. 2000, 478, 9–12. [Google Scholar] [CrossRef]
- Huitema, K.; van den Dikkenberg, J.; Brouwers, J.F.H.M.; Holthuis, J.C.M. Identification of a Family of Animal Sphingomyelin Synthases. EMBO J. 2004, 23, 33–44. [Google Scholar] [CrossRef]
- Riboni, L.; Viani, P.; Bassi, R.; Giussani, P.; Tettamanti, G. Basic Fibroblast Growth Factor-Induced Proliferation of Primary Astrocytes: EVIDENCE FOR THE INVOLVEMENT OF SPHINGOMYELIN BIOSYNTHESIS. J. Biol. Chem. 2001, 276, 12797–12804. [Google Scholar] [CrossRef]
- Barceló-Coblijn, G.; Martin, M.; de Almeida, R.; Noguera-Salvà, M.; Marcilla-Etxenike, A.; Guardiola Serrano, F.; Lüth, A.; Kleuser, B.; Halver, J.; Escriba, P. Sphingomyelin and Sphingomyelin Synthase (SMS) in the Malignant Transformation of Glioma Cells and in 2-Hydroxyoleic Acid Therapy. Proc. Natl. Acad. Sci. USA 2011, 108, 19569–19574. [Google Scholar] [CrossRef]
- Terés, S.; Lladó, V.; Higuera, M.; Barceló-Coblijn, G.; Martin, M.L.; Noguera-Salvà, M.A.; Marcilla-Etxenike, A.; García-Verdugo, J.M.; Soriano-Navarro, M.; Saus, C.; et al. 2-Hydroxyoleate, a Nontoxic Membrane Binding Anticancer Drug, Induces Glioma Cell Differentiation and Autophagy. Proc. Natl. Acad. Sci. USA 2012, 109, 8489–8494. [Google Scholar] [CrossRef] [PubMed]
- Bajjalieh, S.M.; Martin, T.F.; Floor, E. Synaptic Vesicle Ceramide Kinase: A Calcium-Stimulated Lipid Kinase That Co-Purifies with Brain Synaptic Vesicles. J. Biol. Chem. 1989, 264, 14354–14360. [Google Scholar] [CrossRef]
- Simanshu, D.K.; Kamlekar, R.K.; Wijesinghe, D.S.; Zou, X.; Zhai, X.; Mishra, S.K.; Molotkovsky, J.G.; Malinina, L.; Hinchcliffe, E.H.; Chalfant, C.E.; et al. Nonvesicular Trafficking by a Ceramide-1-Phosphate Transfer Protein Regulates Eicosanoids. Nature 2013, 500, 463–467. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Muñoz, A.; Frago, L.M.; Alvarez, L.; Varela-Nieto, I. Stimulation of DNA Synthesis by Natural Ceramide 1-Phosphate. Biochem. J. 1997, 325, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, S.; Sakiyama, H.; Suzuki, G.; Hidari, K.I.; Hirabayashi, Y. Expression Cloning of a CDNA for Human Ceramide Glucosyltransferase That Catalyzes the First Glycosylation Step of Glycosphingolipid Synthesis. Proc. Natl. Acad. Sci. USA 1996, 93, 4638–4643. [Google Scholar] [CrossRef]
- Richardson, C.L.; Baker, S.R.; Morré, D.J.; Keenan, T.W. Glycosphingolipid Synthesis and Tumorigenesis. A Role for the Golgi Apparatus in the Origin of Specific Receptor Molecules of the Mammalian Cell Surface. Biochim. Biophys. Acta 1975, 417, 175–186. [Google Scholar] [CrossRef]
- Zhuo, D.; Li, X.; Guan, F. Biological Roles of Aberrantly Expressed Glycosphingolipids and Related Enzymes in Human Cancer Development and Progression. Front. Physiol. 2018, 9, 466. [Google Scholar] [CrossRef]
- Yeh, S.-C.; Wang, P.-Y.; Lou, Y.-W.; Khoo, K.-H.; Hsiao, M.; Hsu, T.-L.; Wong, C.-H. Glycolipid GD3 and GD3 Synthase Are Key Drivers for Glioblastoma Stem Cells and Tumorigenicity. Proc. Natl. Acad. Sci. USA 2016, 113, 5592–5597. [Google Scholar] [CrossRef]
- Bi, J.; Khan, A.; Tang, J.; Armando, A.M.; Wu, S.; Zhang, W.; Gimple, R.C.; Reed, A.; Jing, H.; Koga, T.; et al. Targeting Glioblastoma Signaling and Metabolism with a Re-Purposed Brain-Penetrant Drug. Cell Rep. 2021, 37, 109957. [Google Scholar] [CrossRef]
- Fernández-García, P.; Rosselló, C.A.; Rodríguez-Lorca, R.; Beteta-Göbel, R.; Fernández-Díaz, J.; Lladó, V.; Busquets, X.; Escribá, P.V. The Opposing Contribution of SMS1 and SMS2 to Glioma Progression and Their Value in the Therapeutic Response to 2OHOA. Cancers 2019, 11, 88. [Google Scholar] [CrossRef]
- Hofmann, K.; Tomiuk, S.; Wolff, G.; Stoffel, W. Cloning and Characterization of the Mammalian Brain-Specific, Mg2+-Dependent Neutral Sphingomyelinase. Proc. Natl. Acad. Sci. USA 2000, 97, 5895–5900. [Google Scholar] [CrossRef] [PubMed]
- Tabatadze, N.; Savonenko, A.; Song, H.; Bandaru, V.V.R.; Chu, M.; Haughey, N.J. Inhibition of Neutral Sphingomyelinase-2 Perturbs Brain Sphingolipid Balance and Spatial Memory in Mice. J. Neurosci. Res. 2010, 88, 2940–2951. [Google Scholar] [CrossRef] [PubMed]
- Tani, M.; Hannun, Y.A. Neutral Sphingomyelinase 2 Is Palmitoylated on Multiple Cysteine Residues. Role of Palmitoylation in Subcellular Localization. J. Biol. Chem. 2007, 282, 10047–10056. [Google Scholar] [CrossRef] [PubMed]
- Tani, M.; Igarashi, Y.; Ito, M. Involvement of Neutral Ceramidase in Ceramide Metabolism at the Plasma Membrane and in Extracellular Milieu. J. Biol. Chem. 2005, 280, 36592–36600. [Google Scholar] [CrossRef] [PubMed]
- Shinghal, R.; Scheller, R.H.; Bajjalieh, S.M. Ceramide 1-Phosphate Phosphatase Activity in Brain. J. Neurochem. 1993, 61, 2279–2285. [Google Scholar] [CrossRef]
- Wang, X.; Rahman, Z.; Sun, P.; Meuillet, E.; George, D.; Bremer, E.G.; Al-Qamari, A.; Paller, A.S. Ganglioside Modulates Ligand Binding to the Epidermal Growth Factor Receptor. J. Investig. Dermatol. 2001, 116, 69–76. [Google Scholar] [CrossRef]
- Schissel, S.L.; Schuchman, E.H.; Williams, K.J.; Tabas, I. Zn2+-Stimulated Sphingomyelinase Is Secreted by Many Cell Types and Is a Product of the Acid Sphingomyelinase Gene. J. Biol. Chem. 1996, 271, 18431–18436. [Google Scholar] [CrossRef]
- Zeidan, Y.H.; Hannun, Y.A. The Acid Sphingomyelinase/Ceramide Pathway: Biomedical Significance and Mechanisms of Regulation. CMM 2010, 10, 454–466. [Google Scholar] [CrossRef]
- Sawada, M.; Nakashima, S.; Kiyono, T.; Yamada, J.; Hara, S.; Nakagawa, M.; Shinoda, J.; Sakai, N. Acid Sphingomyelinase Activation Requires Caspase-8 but Not P53 nor Reactive Oxygen Species during Fas-Induced Apoptosis in Human Glioma Cells. Exp. Cell Res. 2002, 273, 157–168. [Google Scholar] [CrossRef]
- Stephan, M.; Edelmann, B.; Winoto-Morbach, S.; Janssen, O.; Bertsch, U.; Perrotta, C.; Schütze, S.; Fritsch, J. Role of Caspases in CD95-Induced Biphasic Activation of Acid Sphingomyelinase. Oncotarget 2017, 8, 20067–20085. [Google Scholar] [CrossRef]
- Grammatikos, G.; Teichgräber, V.; Carpinteiro, A.; Trarbach, T.; Weller, M.; Hengge, U.R.; Gulbins, E. Overexpression of Acid Sphingomyelinase Sensitizes Glioma Cells to Chemotherapy. Antioxid. Redox Signal. 2007, 9, 1449–1456. [Google Scholar] [CrossRef] [PubMed]
- Gramatzki, D.; Herrmann, C.; Happold, C.; Becker, K.A.; Gulbins, E.; Weller, M.; Tabatabai, G. Glioma Cell Death Induced by Irradiation or Alkylating Agent Chemotherapy Is Independent of the Intrinsic Ceramide Pathway. PLoS ONE 2013, 8, e63527. [Google Scholar] [CrossRef] [PubMed]
- Hannun, Y.A.; Loomis, C.R.; Merrill, A.H.; Bell, R.M. Sphingosine Inhibition of Protein Kinase C Activity and of Phorbol Dibutyrate Binding in Vitro and in Human Platelets. J. Biol. Chem. 1986, 261, 12604–12609. [Google Scholar] [CrossRef]
- Mullmann, T.J.; Siegel, M.I.; Egan, R.W.; Billah, M.M. Sphingosine Inhibits Phosphatidate Phosphohydrolase in Human Neutrophils by a Protein Kinase C-Independent Mechanism. J. Biol. Chem. 1991, 266, 2013–2016. [Google Scholar] [CrossRef]
- Ahn, E.H.; Chang, C.-C.; Schroeder, J.J. Evaluation of Sphinganine and Sphingosine as Human Breast Cancer Chemotherapeutic and Chemopreventive Agents. Exp. Biol. Med. 2006, 231, 1664–1672. [Google Scholar] [CrossRef]
- Saied, E.M.; Arenz, C. Inhibitors of Ceramidases. Chem. Phys. Lipids 2016, 197, 60–68. [Google Scholar] [CrossRef]
- Strub, G.M.; Maceyka, M.; Hait, N.C.; Milstien, S.; Spiegel, S. Extracellular and Intracellular Actions of Sphingosine-1-Phosphate. Adv. Exp. Med. Biol. 2010, 688, 141–155. [Google Scholar] [CrossRef]
- Ponnusamy, S.; Meyers-Needham, M.; Senkal, C.E.; Saddoughi, S.A.; Sentelle, D.; Selvam, S.P.; Salas, A.; Ogretmen, B. Sphingolipids and Cancer: Ceramide and Sphingosine-1-Phosphate in the Regulation of Cell Death and Drug Resistance. Future Oncol. 2010, 6, 1603–1624. [Google Scholar] [CrossRef]
- De Ceuster, P.; Mannaerts, G.P.; Van Veldhoven, P.P. Identification and Subcellular Localization of Sphinganine-Phosphatases in Rat Liver. Biochem. J. 1995, 311, 139–146. [Google Scholar] [CrossRef]
- Le Stunff, H.; Galve-Roperh, I.; Peterson, C.; Milstien, S.; Spiegel, S. Sphingosine-1-Phosphate Phosphohydrolase in Regulation of Sphingolipid Metabolism and Apoptosis. J. Cell Biol. 2002, 158, 1039–1049. [Google Scholar] [CrossRef]
- Maceyka, M.; Sankala, H.; Hait, N.C.; Le Stunff, H.; Liu, H.; Toman, R.; Collier, C.; Zhang, M.; Satin, L.S.; Merrill, A.H.; et al. SphK1 and SphK2, Sphingosine Kinase Isoenzymes with Opposing Functions in Sphingolipid Metabolism. J. Biol. Chem. 2005, 280, 37118–37129. [Google Scholar] [CrossRef] [PubMed]
- Karunakaran, I.; Alam, S.; Jayagopi, S.; Frohberger, S.J.; Hansen, J.N.; Kuehlwein, J.; Hölbling, B.V.; Schumak, B.; Hübner, M.P.; Gräler, M.H.; et al. Neural Sphingosine 1-Phosphate Accumulation Activates Microglia and Links Impaired Autophagy and Inflammation. Glia 2019, 67, 1859–1872. [Google Scholar] [CrossRef]
- Reiss, U.; Oskouian, B.; Zhou, J.; Gupta, V.; Sooriyakumaran, P.; Kelly, S.; Wang, E.; Merrill, A.H.; Saba, J.D. Sphingosine-Phosphate Lyase Enhances Stress-Induced Ceramide Generation and Apoptosis. J. Biol. Chem. 2004, 279, 1281–1290. [Google Scholar] [CrossRef] [PubMed]
- Oridate, N.; Suzuki, S.; Higuchi, M.; Mitchell, M.F.; Hong, W.K.; Lotan, R. ARTICLES Involvement of Reactive Oxygen Species in N-(4-Hydroxyphenyl)Retinamide-Induced Apoptosis in Cervical Carcinoma. JNCI J. Natl. Cancer Inst. 1997, 89, 1191–1198. [Google Scholar] [CrossRef] [PubMed]
- Puduvalli, V.K.; Saito, Y.; Xu, R.; Kouraklis, G.P.; Levin, V.A.; Kyritsis, A.P. Fenretinide Activates Caspases and Induces Apoptosis in Gliomas. Clin. Cancer Res. 1999, 5, 2230–2235. [Google Scholar] [PubMed]
- Saitoh, Y.; Goto, T.; Puduvalli, V.K.; Murakami, M.; Kochi, M.; Levin, V.A.; Kyritsis, A.P.; Ushio, Y. Induction of Apoptosis by N-(4-Hydroxyphenyl)Retinamide in Glioma Cells. Int. J. Oncol. 1999, 15, 499–1003. [Google Scholar] [CrossRef]
- Puduvalli, V.K.; Yung, W.K.A.; Hess, K.R.; Kuhn, J.G.; Groves, M.D.; Levin, V.A.; Zwiebel, J.; Chang, S.M.; Cloughesy, T.F.; Junck, L.; et al. Phase II Study of Fenretinide (NSC 374551) in Adults With Recurrent Malignant Gliomas: A North American Brain Tumor Consortium Study. J. Clin. Oncol. 2004, 22, 4282–4289. [Google Scholar] [CrossRef]
- del Pulgar, T.G.; Velasco, G.; Sánchez, C.; Haro, A.; Guzmán, M. De Novo-Synthesized Ceramide Is Involved in Cannabinoid-Induced Apoptosis. Biochem. J. 2002, 363, 183–188. [Google Scholar] [CrossRef]
- Hernández-Tiedra, S.; Fabriàs, G.; Dávila, D.; Salanueva, Í.J.; Casas, J.; Montes, L.R.; Antón, Z.; García-Taboada, E.; Salazar-Roa, M.; Lorente, M.; et al. Dihydroceramide Accumulation Mediates Cytotoxic Autophagy of Cancer Cells via Autolysosome Destabilization. Autophagy 2016, 12, 2213–2229. [Google Scholar] [CrossRef]
- Velasco, G.; Carracedo, A.; Blázquez, C.; Lorente, M.; Aguado, T.; Haro, A.; Sánchez, C.; Galve-Roperh, I.; Guzmán, M. Cannabinoids and Gliomas. Mol. Neurobiol. 2007, 36, 60–67. [Google Scholar] [CrossRef]
- Paugh, S.W.; Payne, S.G.; Barbour, S.E.; Milstien, S.; Spiegel, S. The Immunosuppressant FTY720 Is Phosphorylated by Sphingosine Kinase Type 2. FEBS Lett. 2003, 554, 189–193. [Google Scholar] [CrossRef]
- Horga, A.; Montalban, X. FTY720 (Fingolimod) for Relapsing Multiple Sclerosis. Expert Rev. Neurother. 2008, 8, 699–714. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.L.; Liebisch, G.; Lehneis, S.; Schmitz, G.; Alonso-Sande, M.; Bestard-Escalas, J.; Lopez, D.H.; García-Verdugo, J.M.; Soriano-Navarro, M.; Busquets, X.; et al. Sustained Activation of Sphingomyelin Synthase by 2-Hydroxyoleic Acid Induces Sphingolipidosis in Tumor Cells1[S]. J. Lipid Res. 2013, 54, 1457–1465. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-S.; Choi, K.-M.; Lee, S.; Sin, D.-M.; Yoo, K.-S.; Lim, Y.; Lee, Y.-M.; Hong, J.-T.; Yun, Y.-P.; Yoo, H.-S. Myriocin, a Serine Palmitoyltransferase Inhibitor, Suppresses Tumor Growth in a Murine Melanoma Model by Inhibiting de Novo Sphingolipid Synthesis. Cancer Biol. Ther. 2012, 13, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Adachi, R.; Asano, Y.; Ogawa, K.; Oonishi, M.; Tanaka, Y.; Kawamoto, T. Pharmacological Characterization of Synthetic Serine Palmitoyltransferase Inhibitors by Biochemical and Cellular Analyses. Biochem. Biophys. Res. Commun. 2018, 497, 1171–1176. [Google Scholar] [CrossRef]
- Yaguchi, M.; Shibata, S.; Satomi, Y.; Hirayama, M.; Adachi, R.; Asano, Y.; Kojima, T.; Hirata, Y.; Mizutani, A.; Kiba, A.; et al. Antitumor Activity of a Novel and Orally Available Inhibitor of Serine Palmitoyltransferase. Biochem. Biophys. Res. Commun. 2017, 484, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Kojima, T.; Asano, Y.; Kurasawa, O.; Hirata, Y.; Iwamura, N.; Wong, T.-T.; Saito, B.; Tanaka, Y.; Arai, R.; Yonemori, K.; et al. Discovery of Novel Serine Palmitoyltransferase Inhibitors as Cancer Therapeutic Agents. Bioorganic Med. Chem. 2018, 26, 2452–2465. [Google Scholar] [CrossRef]
- Carracedo, A.; Lorente, M.; Egia, A.; Blázquez, C.; García, S.; Giroux, V.; Malicet, C.; Villuendas, R.; Gironella, M.; González-Feria, L.; et al. The Stress-Regulated Protein P8 Mediates Cannabinoid-Induced Apoptosis of Tumor Cells. Cancer Cell 2006, 9, 301–312. [Google Scholar] [CrossRef]
- French, K.J.; Upson, J.J.; Keller, S.N.; Zhuang, Y.; Yun, J.K.; Smith, C.D. Antitumor Activity of Sphingosine Kinase Inhibitors. J. Pharmacol. Exp. Ther. 2006, 318, 596–603. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, F.; Casasampere, M.; Sanllehí, P.; Casas, J.; Bujons, J.; Fabrias, G. Inhibition of Dihydroceramide Desaturase Activity by the Sphingosine Kinase Inhibitor SKI II [S]. J. Lipid Res. 2014, 55, 1711–1720. [Google Scholar] [CrossRef]
- Graf, C.; Klumpp, M.; Habig, M.; Rovina, P.; Billich, A.; Baumruker, T.; Oberhauser, B.; Bornancin, F. Targeting Ceramide Metabolism with a Potent and Specific Ceramide Kinase Inhibitor. Mol. Pharmacol. 2008, 74, 925–932. [Google Scholar] [CrossRef] [PubMed]
- Pastukhov, O.; Schwalm, S.; Zangemeister-Wittke, U.; Fabbro, D.; Bornancin, F.; Japtok, L.; Kleuser, B.; Pfeilschifter, J.; Huwiler, A. The Ceramide Kinase Inhibitor NVP-231 Inhibits Breast and Lung Cancer Cell Proliferation by Inducing M Phase Arrest and Subsequent Cell Death. Br. J. Pharmacol. 2014, 171, 5829–5844. [Google Scholar] [CrossRef] [PubMed]
- Schwalm, S.; Erhardt, M.; Römer, I.; Pfeilschifter, J.; Zangemeister-Wittke, U.; Huwiler, A. Ceramide Kinase Is Upregulated in Metastatic Breast Cancer Cells and Contributes to Migration and Invasion by Activation of PI 3-Kinase and Akt. Int. J. Mol. Sci. 2020, 21, 1396. [Google Scholar] [CrossRef] [PubMed]
- Peterschmitt, M.J.; Crawford, N.P.S.; Gaemers, S.J.M.; Ji, A.J.; Sharma, J.; Pham, T.T. Pharmacokinetics, Pharmacodynamics, Safety, and Tolerability of Oral Venglustat in Healthy Volunteers. Clin. Pharmacol. Drug Dev. 2021, 10, 86–98. [Google Scholar] [CrossRef]
- Ashe, K.M.; Budman, E.; Bangari, D.S.; Siegel, C.S.; Nietupski, J.B.; Wang, B.; Desnick, R.J.; Scheule, R.K.; Leonard, J.P.; Cheng, S.H.; et al. Efficacy of Enzyme and Substrate Reduction Therapy with a Novel Antagonist of Glucosylceramide Synthase for Fabry Disease. Mol. Med. 2015, 21, 389–399. [Google Scholar] [CrossRef]
- Marshall, J.; Sun, Y.; Bangari, D.S.; Budman, E.; Park, H.; Nietupski, J.B.; Allaire, A.; Cromwell, M.A.; Wang, B.; Grabowski, G.A.; et al. CNS-Accessible Inhibitor of Glucosylceramide Synthase for Substrate Reduction Therapy of Neuronopathic Gaucher Disease. Mol. Ther. 2016, 24, 1019–1029. [Google Scholar] [CrossRef]
- Sardi, S.P.; Viel, C.; Clarke, J.; Treleaven, C.M.; Richards, A.M.; Park, H.; Olszewski, M.A.; Dodge, J.C.; Marshall, J.; Makino, E.; et al. Glucosylceramide Synthase Inhibition Alleviates Aberrations in Synucleinopathy Models. Proc. Natl. Acad. Sci. USA 2017, 114, 2699–2704. [Google Scholar] [CrossRef]
- Radin, N.S. Rationales for Cancer Chemotherapy with PDMP, a Specific Inhibitor of Glucosylceramide Synthase. Mol. Chem. Neuropathol. 1994, 21, 111–127. [Google Scholar] [CrossRef]
- Sawada, M.; Nakashima, S.; Banno, Y.; Yamakawa, H.; Hayashi, K.; Takenaka, K.; Nishimura, Y.; Sakai, N.; Nozawa, Y. Ordering of Ceramide Formation, Caspase Activation, and Bax/Bcl-2 Expression during Etoposide-Induced Apoptosis in C6 Glioma Cells. Cell Death Differ. 2000, 7, 761–772. [Google Scholar] [CrossRef]
- Chatterjee, S.; Alsaeedi, N.; Hou, J.; Bandaru, V.V.R.; Wu, L.; Halushka, M.K.; Pili, R.; Ndikuyeze, G.; Haughey, N.J. Use of a Glycolipid Inhibitor to Ameliorate Renal Cancer in a Mouse Model. PLoS ONE 2013, 8, e63726. [Google Scholar] [CrossRef]
- Hartwig, P.; Höglinger, D. The Glucosylceramide Synthase Inhibitor PDMP Causes Lysosomal Lipid Accumulation and MTOR Inactivation. Int. J. Mol. Sci. 2021, 22, 7065. [Google Scholar] [CrossRef] [PubMed]
- Fujii, T.; Tanaka, Y.; Oki, H.; Sato, S.; Shibata, S.; Maru, T.; Tanaka, Y.; Tanaka, M.; Onishi, T. A New Brain-Penetrant Glucosylceramide Synthase Inhibitor as Potential Therapeutics for Gaucher Disease. J. Neurochem. 2021, 159, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Adibhatla, R.M.; Hatcher, J.F.; Gusain, A. Tricyclodecan-9-Yl-Xanthogenate (D609) Mechanism of Actions: A Mini-Review of Literature. Neurochem. Res. 2012, 37, 671–679. [Google Scholar] [CrossRef] [PubMed]
- Mercurio, L.; Cecchetti, S.; Ricci, A.; Pacella, A.; Cigliana, G.; Bozzuto, G.; Podo, F.; Iorio, E.; Carpinelli, G. Phosphatidylcholine-Specific Phospholipase C Inhibition down- Regulates CXCR4 Expression and Interferes with Proliferation, Invasion and Glycolysis in Glioma Cells. PLoS ONE 2017, 12, e0176108. [Google Scholar] [CrossRef]
- Deng, X.; Lin, F.; Zhang, Y.; Li, Y.; Zhou, L.; Lou, B.; Li, Y.; Dong, J.; Ding, T.; Jiang, X.; et al. Identification of Small Molecule Sphingomyelin Synthase Inhibitors. Eur. J. Med. Chem. 2014, 73, 1–7. [Google Scholar] [CrossRef]
- Peng, C.-H.; Huang, C.-N.; Hsu, S.-P.; Wang, C.-J. Penta-Acetyl Geniposide Induce Apoptosis in C6 Glioma Cells by Modulating the Activation of Neutral Sphingomyelinase-Induced P75 Nerve Growth Factor Receptor and Protein Kinase Cδ Pathway. Mol. Pharmacol. 2006, 70, 997–1004. [Google Scholar] [CrossRef]
- Wang, C.J.; Chu, C.Y.; Tseng, T.H.; Lin, J.K. Penta-Acetyl Geniposide Inhibits the Growth and Development of C-6 Glioma Cells in Rats. Cancer Lett. 1993, 70, 113–118. [Google Scholar] [CrossRef]
- Chang, Y.-C.; Tseng, T.-H.; Lee, M.-J.; Hsu, J.-D.; Wang, C.-J. Induction of Apoptosis by Penta-Acetyl Geniposide in Rat C6 Glioma Cells. Chem. -Biol. Interact. 2002, 141, 243–257. [Google Scholar] [CrossRef]
- Huang, H.-P.; Shih, Y.-W.; Wu, C.-H.; Lai, P.-J.; Hung, C.-N.; Wang, C.-J. Inhibitory Effect of Penta-Acetyl Geniposide on C6 Glioma Cells Metastasis by Inhibiting Matrix Metalloproteinase-2 Expression Involved in Both the PI3K and ERK Signaling Pathways. Chem.-Biol. Interact. 2009, 181, 8–14. [Google Scholar] [CrossRef]
- Cattaneo, M.G.; Vanetti, C.; Samarani, M.; Aureli, M.; Bassi, R.; Sonnino, S.; Giussani, P. Cross-Talk between Sphingosine-1-Phosphate and EGFR Signaling Pathways Enhances Human Glioblastoma Cell Invasiveness. FEBS Lett. 2018, 592, 949–961. [Google Scholar] [CrossRef]
- Caudill, J.S.; Brown, P.D.; Cerhan, J.H.; Rummans, T.A. Selective Serotonin Reuptake Inhibitors, Glioblastoma Multiforme, and Impact on Toxicities and Overall Survival: The Mayo Clinic Experience. Am. J. Clin. Oncol. 2011, 34, 385–387. [Google Scholar] [CrossRef] [PubMed]
- Otto-Meyer, S.; DeFaccio, R.; Dussold, C.; Ladomersky, E.; Zhai, L.; Lauing, K.L.; Bollu, L.R.; Amidei, C.; Lukas, R.V.; Scholtens, D.M.; et al. A Retrospective Survival Analysis of Glioblastoma Patients Treated with Selective Serotonin Reuptake Inhibitors. Brain Behav. Immun.-Health 2020, 2, 100025. [Google Scholar] [CrossRef]
- Schuchman, E.H. Acid Ceramidase and the Treatment of Ceramide Diseases: The Expanding Role of Enzyme Replacement Therapy. Biochim. Biophys. Acta 2016, 1862, 1459–1471. [Google Scholar] [CrossRef] [PubMed]
- Realini, N.; Solorzano, C.; Pagliuca, C.; Pizzirani, D.; Armirotti, A.; Luciani, R.; Costi, M.P.; Bandiera, T.; Piomelli, D. Discovery of Highly Potent Acid Ceramidase Inhibitors with in Vitro Tumor Chemosensitizing Activity. Sci. Rep. 2013, 3, 1035. [Google Scholar] [CrossRef]
- Dementiev, A.; Joachimiak, A.; Nguyen, H.; Gorelik, A.; Illes, K.; Shabani, S.; Gelsomino, M.; Ahn, E.-Y.E.; Nagar, B.; Doan, N. Molecular Mechanism of Inhibition of Acid Ceramidase by Carmofur. J. Med. Chem. 2019, 62, 987–992. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, C.; Jones, A.; Ziebro, J.; Gordon, E.; Libby, C.; Williford, S.; Allegood, J.; Cooper, S.; Ramanadham, S.; Doan, N.; et al. DDRE-06. TARGETING THE SPHINGOLIPID BALANCE VIA ACID CERAMIDASE INHIBITION TO DECREASE GROWTH OF TMZ-RESISTANT GLIOBLASTOMA AND BLOCK MIGRATION. Neuro-Oncology 2021, 23, vi75. [Google Scholar] [CrossRef]
- Kutlu, H.M.; Sezer, C.V.; Kuş, G.; Çömlekçi, E.; İzgördü, H. A 5-Fluorouracil Derivative: Carmofur as a New Potent Agent for Inhibition of Human Prostate and Breast Cancer Cell Lines. Iran J. Sci. Technol. Trans. Sci. 2022, 46, 13–20. [Google Scholar] [CrossRef]
- Camacho, L.; Meca-Cortés, Ó.; Abad, J.L.; García, S.; Rubio, N.; Díaz, A.; Celià-Terrassa, T.; Cingolani, F.; Bermudo, R.; Fernández, P.L.; et al. Acid Ceramidase as a Therapeutic Target in Metastatic Prostate Cancer. J. Lipid Res. 2013, 54, 1207–1220. [Google Scholar] [CrossRef]
- Pearson, J.M.; Tan, S.-F.; Sharma, A.; Annageldiyev, C.; Fox, T.E.; Abad, J.L.; Fabrias, G.; Desai, D.; Amin, S.; Wang, H.-G.; et al. Ceramide Analog SACLAC Modulates Sphingolipid Levels and Mcl-1 Splicing to Induce Apoptosis in Acute Myeloid Leukemia. Mol. Cancer Res. 2020, 18, 352–363. [Google Scholar] [CrossRef]
- Van Brocklyn, J.R.; Jackson, C.A.; Pearl, D.K.; Kotur, M.S.; Snyder, P.J.; Prior, T.W. Sphingosine Kinase-1 Expression Correlates with Poor Survival of Patients with Glioblastoma Multiforme: Roles of Sphingosine Kinase Isoforms in Growth of Glioblastoma Cell Lines. J. Neuropathol. Exp. Neurol. 2005, 64, 695–705. [Google Scholar] [CrossRef]
- Britten, C.D.; Garrett-Mayer, E.; Chin, S.H.; Shirai, K.; Ogretmen, B.; Bentz, T.A.; Brisendine, A.; Anderton, K.; Cusack, S.L.; Maines, L.W.; et al. A Phase I Study of ABC294640, a First-in-Class Sphingosine Kinase-2 Inhibitor, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2017, 23, 4642–4650. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, J.; Hwang, G.T.; Lee, T.; Kim, S.W.; Oh, Y.S.; Kwon, Y.; Hong, S.W.; Kim, S.; Seop Moon, H.; Baek, D.J.; et al. Synthesis and Biological Evaluation of BODIPY-PF-543. Molecules 2019, 24, 4408. [Google Scholar] [CrossRef] [PubMed]
- Visentin, B.; Vekich, J.A.; Sibbald, B.J.; Cavalli, A.L.; Moreno, K.M.; Matteo, R.G.; Garland, W.A.; Lu, Y.; Yu, S.; Hall, H.S.; et al. Validation of an Anti-Sphingosine-1-Phosphate Antibody as a Potential Therapeutic in Reducing Growth, Invasion, and Angiogenesis in Multiple Tumor Lineages. Cancer Cell 2006, 9, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Larion, M.; Dowdy, T.; Ruiz-Rodado, V.; Meyer, M.W.; Song, H.; Zhang, W.; Davis, D.; Gilbert, M.R.; Lita, A. Detection of Metabolic Changes Induced via Drug Treatments in Live Cancer Cells and Tissue Using Raman Imaging Microscopy. Biosensors 2018, 9, E5. [Google Scholar] [CrossRef]
- Lita, A.; Kuzmin, A.N.; Pliss, A.; Baev, A.; Rzhevskii, A.; Gilbert, M.R.; Larion, M.; Prasad, P.N. Towards Single Organelle Lipidomics in Live Cells. Anal. Chem. 2019, 91, 11380–11387. [Google Scholar] [CrossRef]





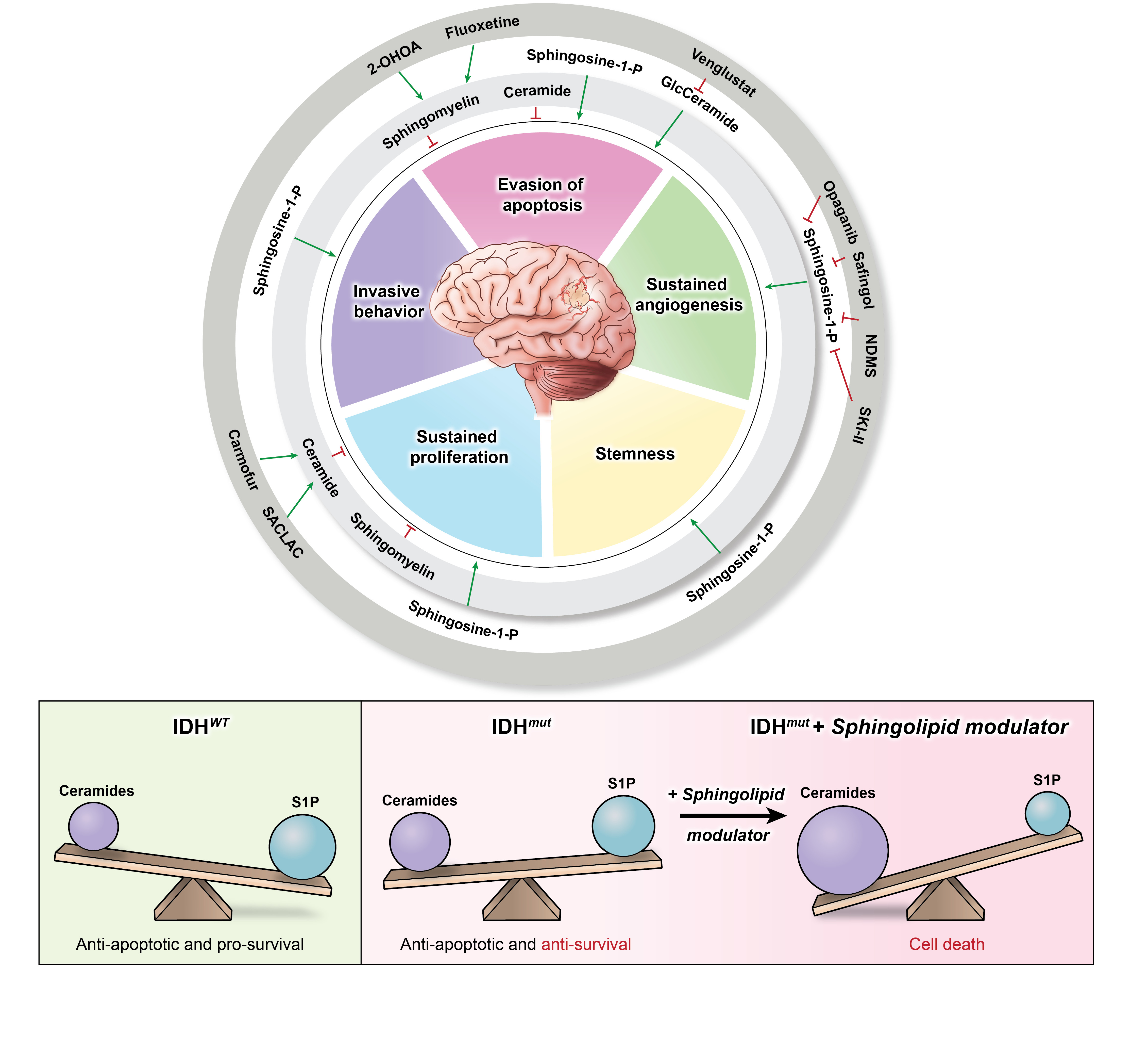{kind=link}
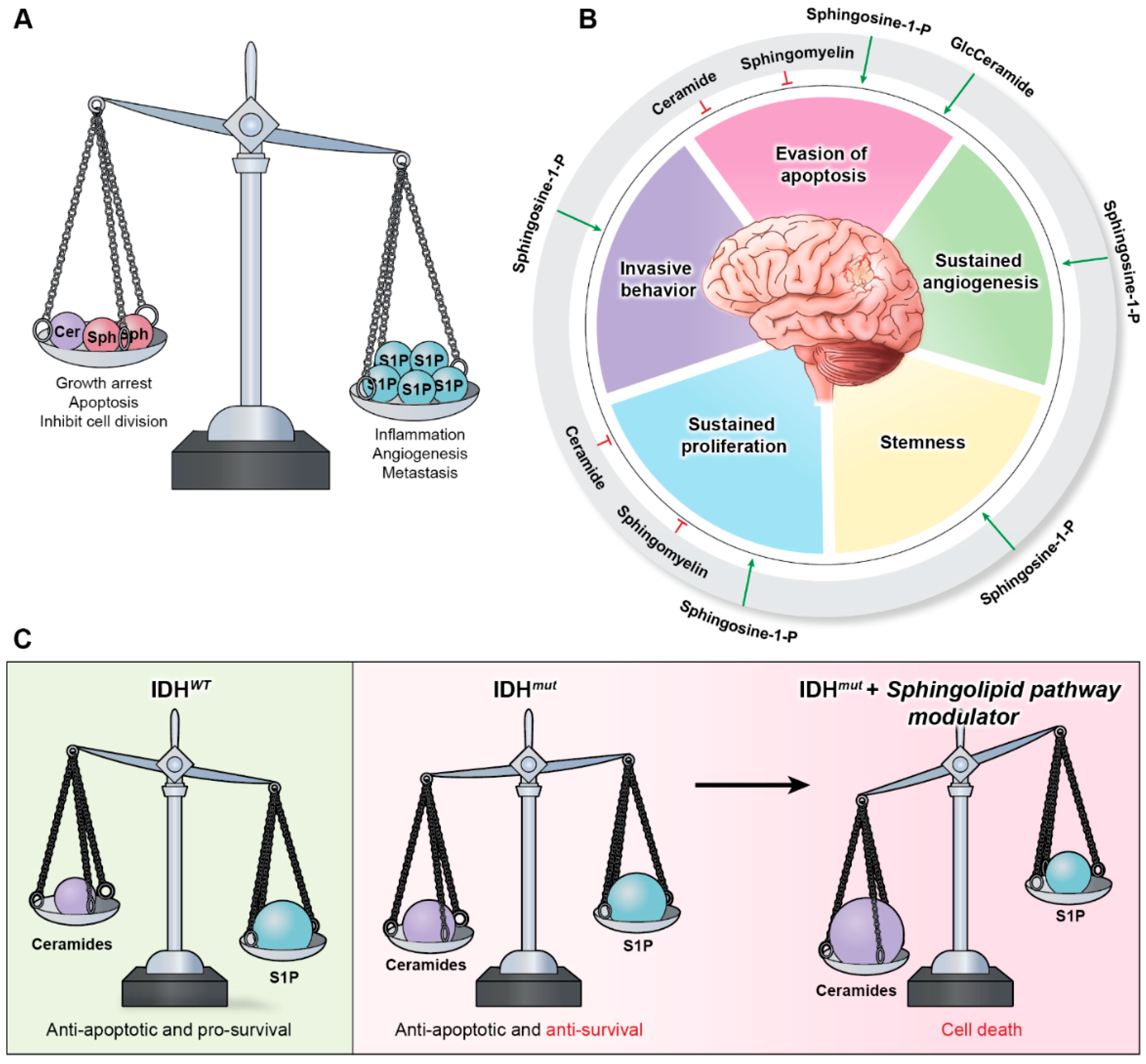{kind=link}
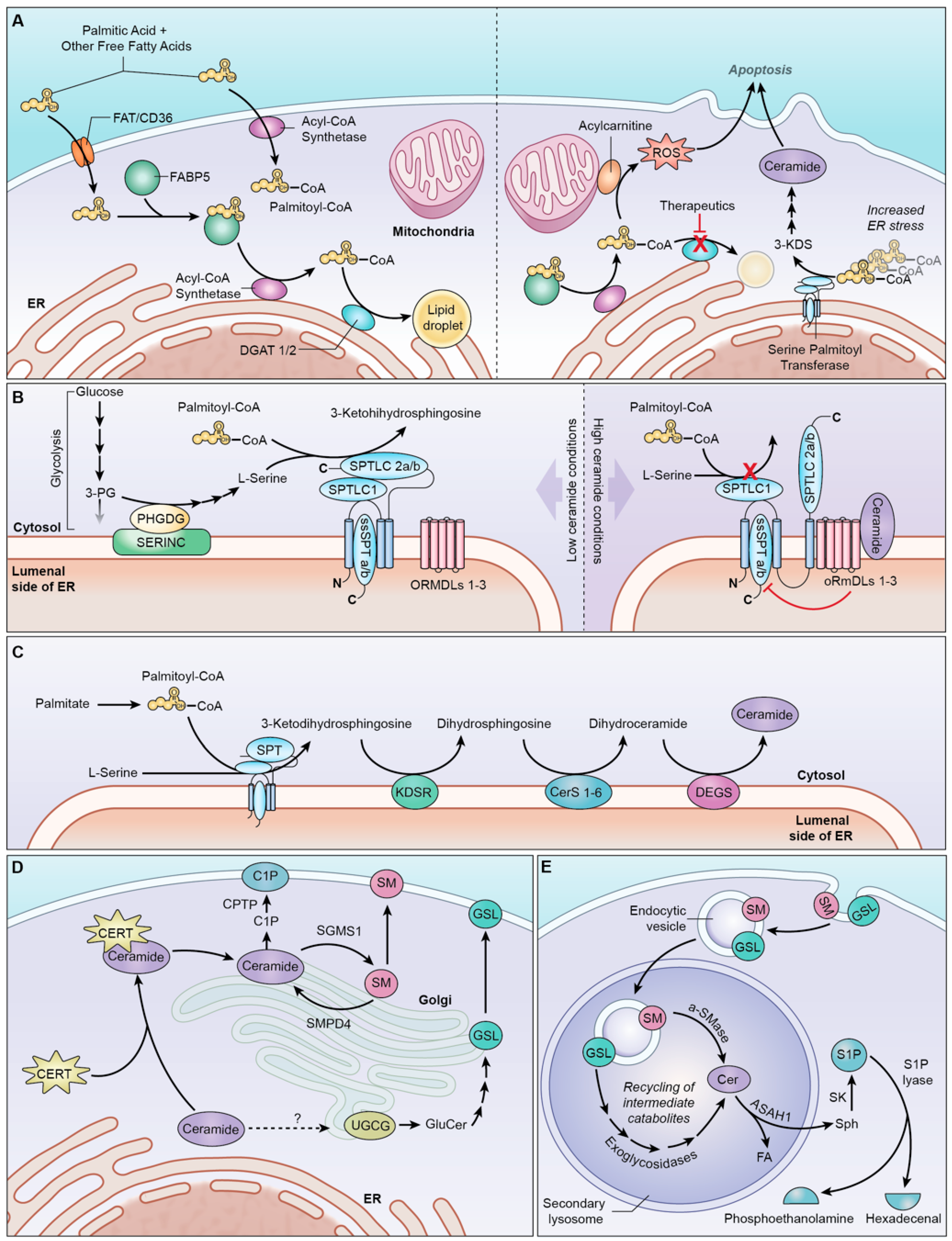{kind=link}
{kind=link}
{kind=link}
| Drug | Target | Study Type | Status | Tumor Type |
|---|---|---|---|---|
| Fenretinide | DEGS1 inhibitor | Phase II (NCT00006080) | Fenretinide was inactive against recurrent glioma at the dosage used | Malignant glioma |
| Fenretinide | DEGS1 inhibitor | Phase I NCT00003191) | Data support phase-II trial in neuroblastoma | Neuroblastoma |
| Fenretinide lipid matrix | DEGS1 inhibitor | Phase I (NCT00295919) | Phase-II trial recommended | Neuroblastoma |
| Cannabis and Temozolomide | DEGS1 inhibitor | Phase I (NCT03246113) | Terminated | Glioblastoma |
| THC + CBD combo and Temozolomide | DEGS1 inhibitor | Phase I–II (NCT03529448) | Ongoing | Glioblastoma |
| Sativex and Temozolomide | DEGS1 inhibitor | Phase I–II (NCT01812616) | Acceptable safety and tolerability, survival differences observed | Glioblastoma |
| Dronabinol | DEGS1 inhibitor | Phase I (NCT00314808) | Potential use as anti-emetic to improve quality of life | Primary Gliomas |
| 2-OHOA and Temozolomide | SGMS1 activator | Phase I (NCT03867123) | Results not publicly available yet | Glioblastoma |
| 2-OHOA | SGMS1 activator | Phase I–II (NCT01792310) | 2-OHOA is well tolerated, antitumor activity warrants further trials | Gliomas and other solid tumors |
| 2-OHOA and Temozolomide | SGMS1 activator | Phase II-III (NCT04250922) | Ongoing | Glioblastoma |
| 2-OHOA | SGMS1 activator | Phase I–II (NCT04299191) | Ongoing | High grade glioma |
| Fingolimod | S1PR modulator | Phase I (NCT02490930) | Results not publicly available | High grade glioma |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zaibaq, F.; Dowdy, T.; Larion, M. Targeting the Sphingolipid Rheostat in Gliomas. Int. J. Mol. Sci. 2022, 23, 9255. https://doi.org/10.3390/ijms23169255
Zaibaq F, Dowdy T, Larion M. Targeting the Sphingolipid Rheostat in Gliomas. International Journal of Molecular Sciences. 2022; 23(16):9255. https://doi.org/10.3390/ijms23169255
Chicago/Turabian StyleZaibaq, Faris, Tyrone Dowdy, and Mioara Larion. 2022. "Targeting the Sphingolipid Rheostat in Gliomas" International Journal of Molecular Sciences 23, no. 16: 9255. https://doi.org/10.3390/ijms23169255
APA StyleZaibaq, F., Dowdy, T., & Larion, M. (2022). Targeting the Sphingolipid Rheostat in Gliomas. International Journal of Molecular Sciences, 23(16), 9255. https://doi.org/10.3390/ijms23169255