
Need for a Paradigm Shift in the Treatment of Ischemic Stroke: The Blood-Brain Barrier

 , , ,
, , ,  ,
,  ,
,  ,
,
Abstract
:1. Introduction
2. BBB Structure and Function
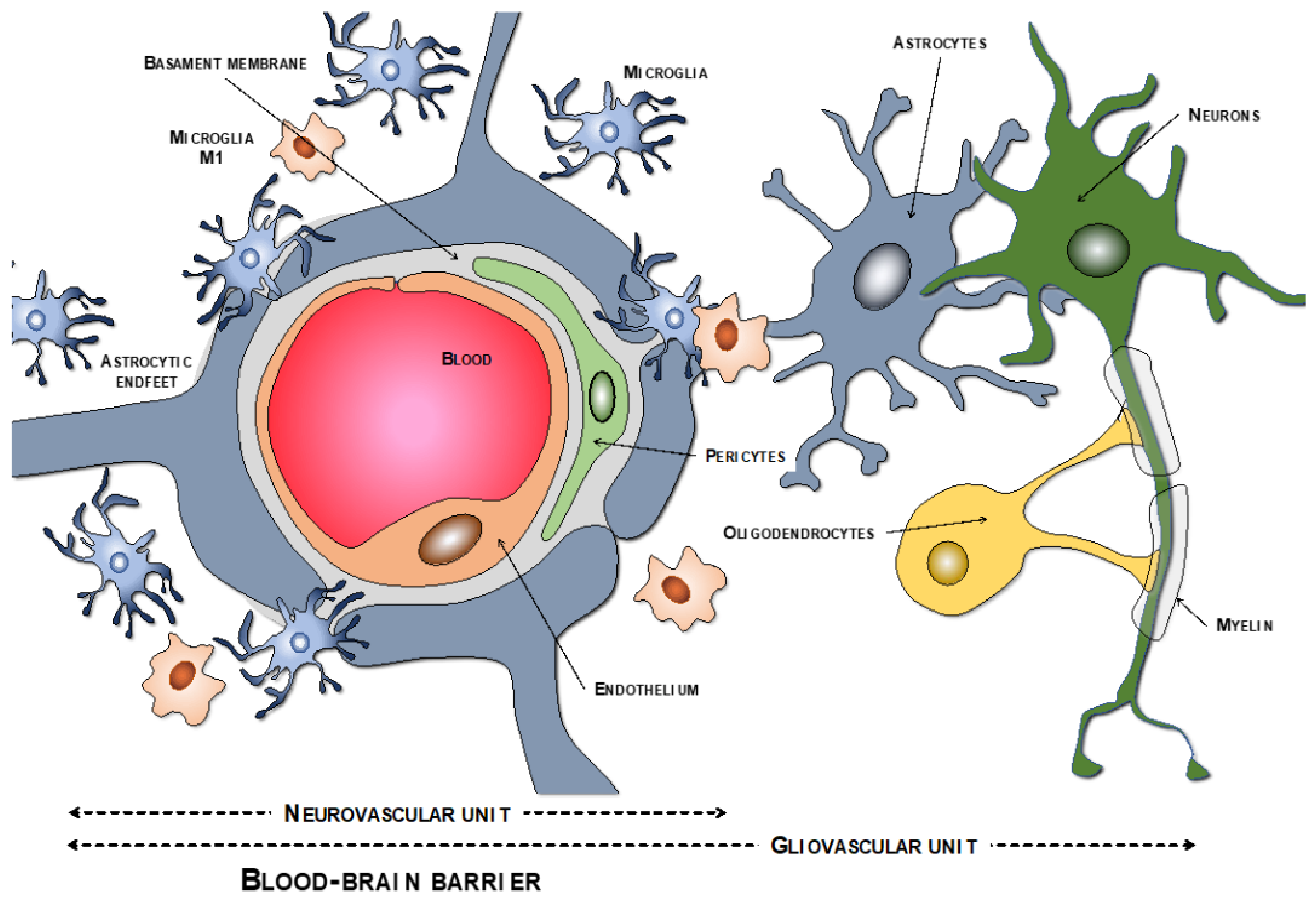
2.1. BBB Molecular Components: The Neurovascular Unit
2.1.1. Endothelial Cells
2.1.2. Tight Junction
2.1.3. Pericytes
2.1.4. Basement Membrane
2.1.5. Astrocytes
2.1.6. Macrophages and Microglia
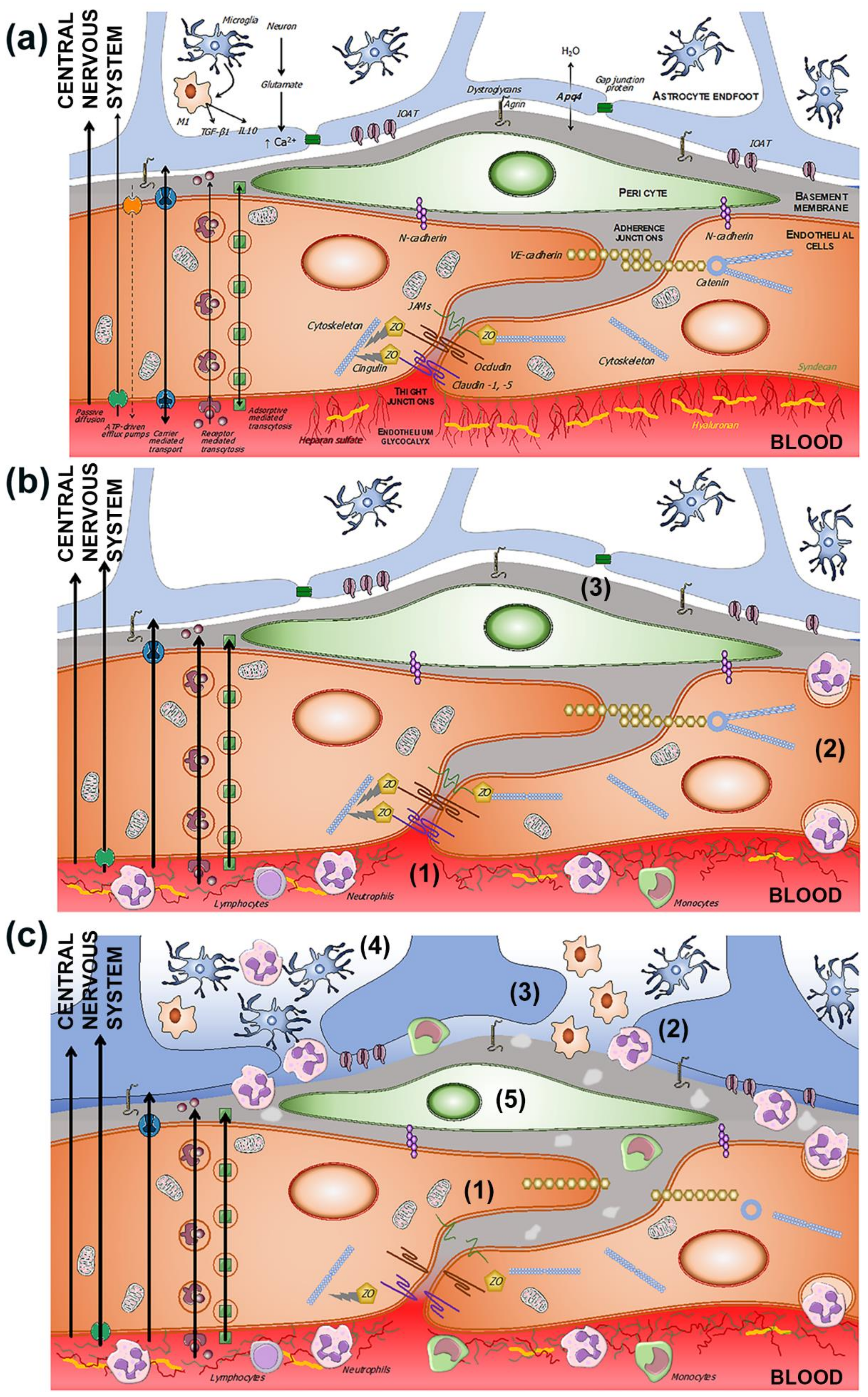
2.2. Transport Pathways across the BBB
2.2.1. Passive Diffusion
2.2.2. Active Efflux
2.2.3. Carrier-Mediated Transport
2.2.4. Receptor-Mediated Transport
3. Evolution of BBB Dysfunction in the Setting of Ischemic Stroke
3.1. Endothelials Cells
3.2. Microglia
3.3. Basement Membrane
3.4. Tight Junction
3.5. BBB Dysfunction and Transport Mechanisms
3.6. Neuroinflammation in BBB Dysfunction
3.6.1. Cytokines
3.6.2. Ischemic Neurons
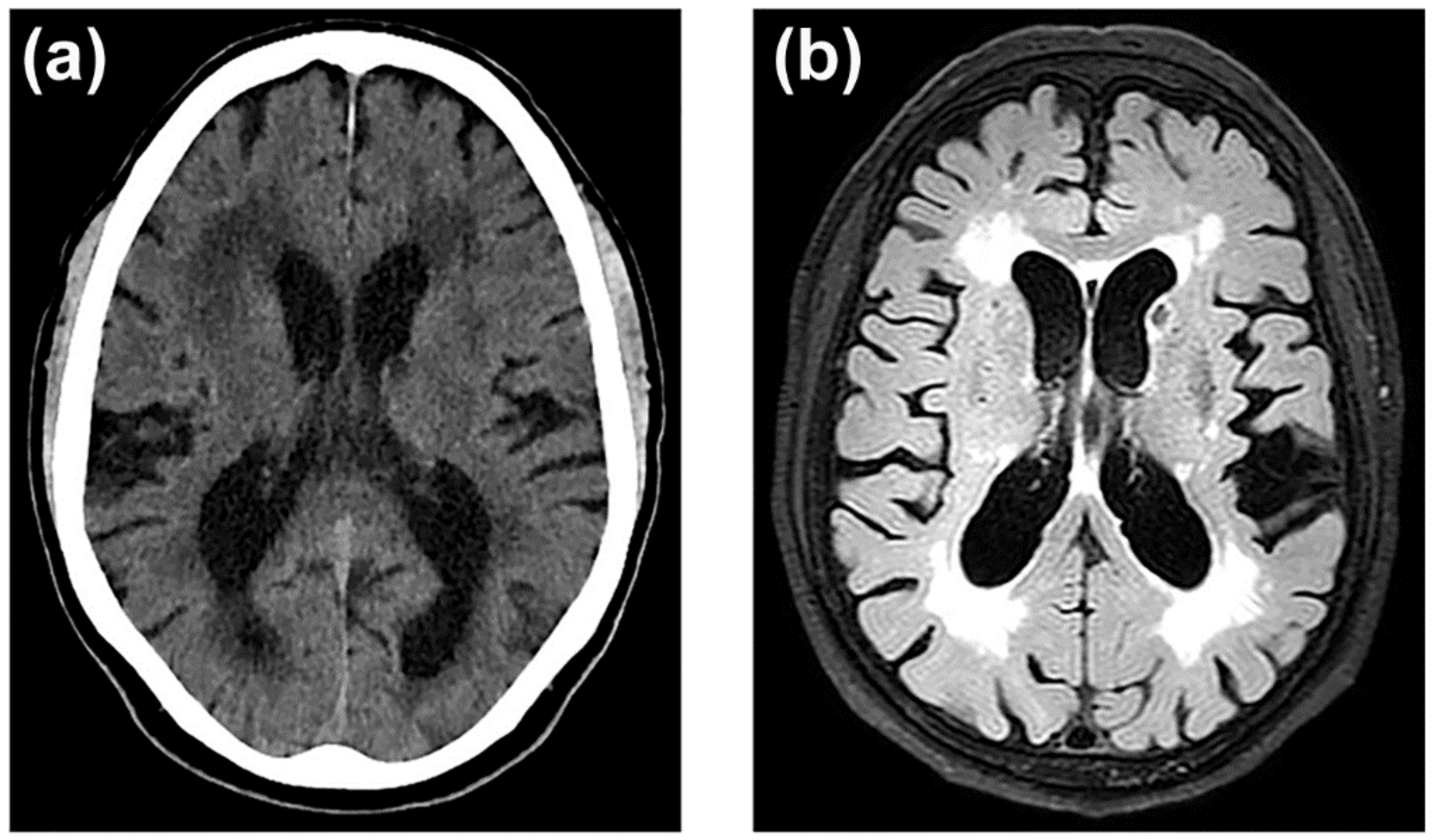
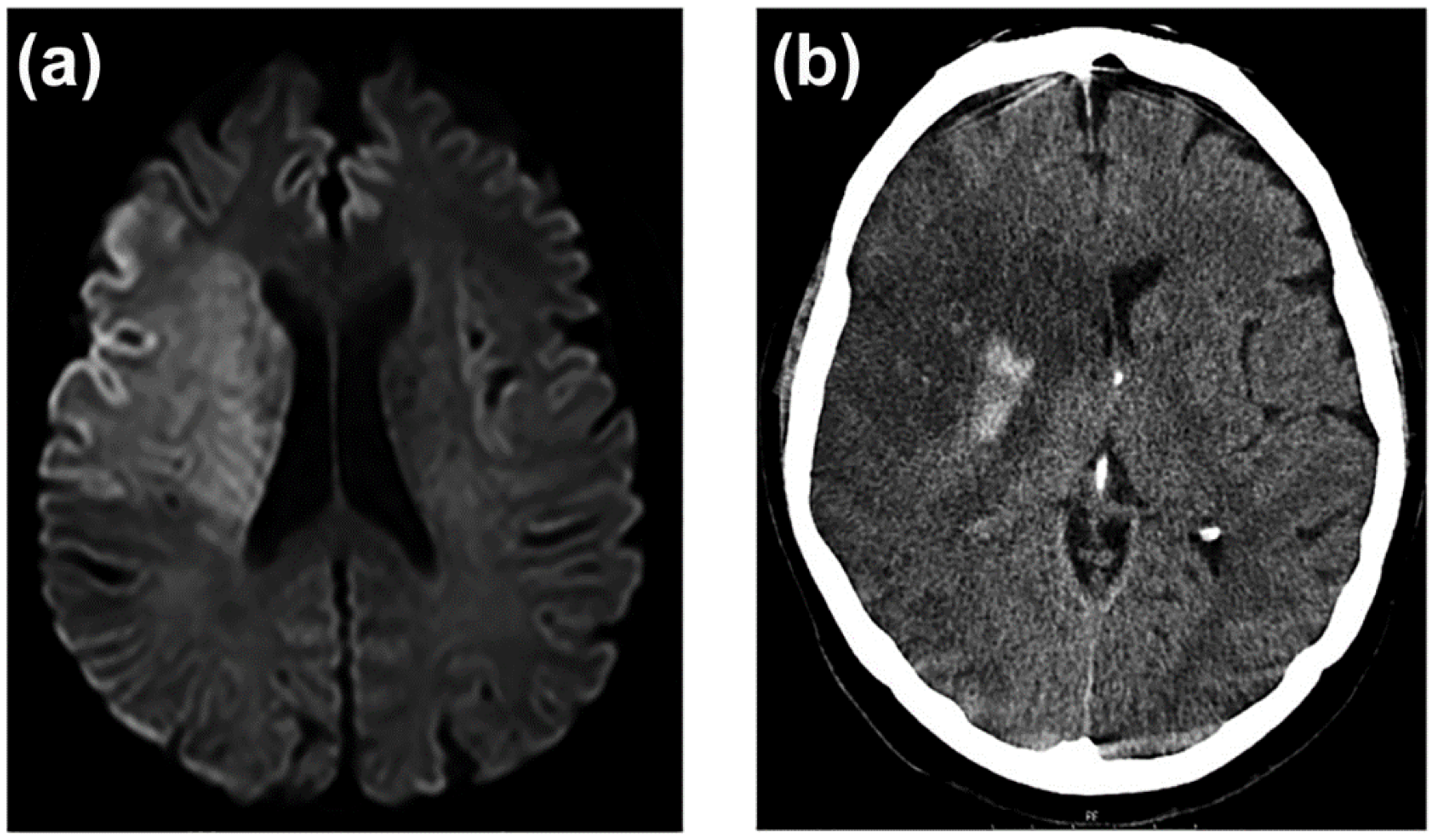
3.7. Neuroimaging of BBB Dysfunction
4. BBB as a Target for Brain Aging and Cerebrovascular Disease Prevention
Nanomedicine and Drug Delivery Systems
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- GBD 2016 Neurology Collaborators. Global, Regional, and National Burden of Neurological Disorders, 1990–2016: A Systematic Analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 459–480. [Google Scholar] [CrossRef] [Green Version]
- Wafa, H.A.; Wolfe, C.D.A.; Emmett, E.; Roth, G.A.; Johnson, C.O.; Wang, Y. Burden of Stroke in Europe. Stroke 2020, 51, 2418–2427. [Google Scholar] [CrossRef] [PubMed]
- Walker, E.R.; McGee, R.E.; Druss, B.G. Mortality in Mental Disorders and Global Disease Burden Implications. JAMA Psychiatry 2015, 72, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Deuschl, G.; Beghi, E.; Fazekas, F.; Varga, T.; Christoforidi, K.A.; Sipido, E.; Bassetti, C.L.; Vos, T.; Feigin, V.L. The Burden of Neurological Diseases in Europe: An Analysis for the Global Burden of Disease Study 2017. Lancet Public Health 2020, 5, e551–e567. [Google Scholar] [CrossRef]
- Feigin, V.L.; Roth, G.A.; Naghavi, M.; Parmar, P.; Krishnamurthi, R.; Chugh, S.; Mensah, G.A.; Norrving, B.; Shiue, I.; Ng, M.; et al. Global Burden of Stroke and Risk Factors in 188 Countries, during 1990-2013: A Systematic Analysis for the Global Burden of Disease Study 2013. Lancet Neurol. 2016, 15, 913–924. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Castro, E.; López-Dequit, I.; Santamaría-Cadavid, M.; Arias-Rivas, S.; Rodríguez-Yáñez, M.; Pumar, J.M.; Hervella, P.; López-Arias, E.; da Silva-Candal, A.; Estany, A.; et al. Trends in Stroke Outcomes in the Last Ten Years in a European Tertiary Hospital. BMC Neurol. 2018, 18, 164. [Google Scholar] [CrossRef]
- Chang, A.Y.; Skirbekk, V.F.; Tyrovolas, S.; Kassebaum, N.J.; Dieleman, J.L. Measuring Population Ageing: An Analysis of the Global Burden of Disease Study 2017. Lancet Public Health 2019, 4, e159–e167. [Google Scholar] [CrossRef] [Green Version]
- Olesen, J.; Gustavsson, A.; Svensson, M.; Wittchen, H.-U.; Jönsson, B.; CDBE2010 study group. European Brain Council the Economic Cost of Brain Disorders in Europe. Eur. J. Neurol. 2012, 19, 155–162. [Google Scholar] [CrossRef]
- Henderson, S.J.; Weitz, J.I.; Kim, P.Y. Fibrinolysis: Strategies to Enhance the Treatment of Acute Ischemic Stroke. J. Thromb. Haemost. 2018, 16, 1932–1940. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, K.; Matsumaru, Y.; Takeuchi, M.; Morimoto, M.; Kanazawa, R.; Takayama, Y.; Kamiya, Y.; Shigeta, K.; Okubo, S.; Hayakawa, M.; et al. Effect of Mechanical Thrombectomy without vs. with Intravenous Thrombolysis on Functional Outcome Among Patients with Acute Ischemic Stroke: The SKIP Randomized Clinical Trial. JAMA 2021, 325, 244–253. [Google Scholar] [CrossRef]
- Zi, W.; Qiu, Z.; Li, F.; Sang, H.; Wu, D.; Luo, W.; Liu, S.; Yuan, J.; Song, J.; Shi, Z.; et al. Effect of Endovascular Treatment Alone vs. Intravenous Alteplase Plus Endovascular Treatment on Functional Independence in Patients with Acute Ischemic Stroke: The DEVT Randomized Clinical Trial. JAMA 2021, 325, 234–243. [Google Scholar] [CrossRef]
- Schrag, M.; Kirshner, H. Management of Intracerebral Hemorrhage: JACC Focus Seminar. J. Am. Coll. Cardiol. 2020, 75, 1819–1831. [Google Scholar] [CrossRef]
- Santschi, V.; Wuerzner, G.; Pais, B.; Chiolero, A.; Schaller, P.; Cloutier, L.; Paradis, G.; Burnier, M. Team-Based Care for Improving Hypertension Management: A Pragmatic Randomized Controlled Trial. Front. Cardiovasc. Med. 2021, 8, 760662. [Google Scholar] [CrossRef]
- Sofogianni, A.; Stalikas, N.; Antza, C.; Tziomalos, K. Cardiovascular Risk Prediction Models and Scores in the Era of Personalized Medicine. J. Pers. Med. 2022, 12, 1180. [Google Scholar] [CrossRef]
- Ruff, C.T.; Giugliano, R.P.; Braunwald, E.; Hoffman, E.B.; Deenadayalu, N.; Ezekowitz, M.D.; Camm, A.J.; Weitz, J.I.; Lewis, B.S.; Parkhomenko, A.; et al. Comparison of the Efficacy and Safety of New Oral Anticoagulants with Warfarin in Patients with Atrial Fibrillation: A Meta-Analysis of Randomised Trials. Lancet 2014, 383, 955–962. [Google Scholar] [CrossRef]
- GBD 2019 Stroke Collaborators. Global, Regional, and National Burden of Stroke and Its Risk Factors, 1990–2019: A Systematic Analysis for the Global Burden of Disease Study 2019. Lancet Neurol. 2021, 20, 795–820. [Google Scholar] [CrossRef]
- Meschia, J.F.; Bushnell, C.; Boden-Albala, B.; Braun, L.T.; Bravata, D.M.; Chaturvedi, S.; Creager, M.A.; Eckel, R.H.; Elkind, M.S.V.; Fornage, M.; et al. Guidelines for the Primary Prevention of Stroke: A Statement for Healthcare Professionals from the American Heart Association/American Stroke Association. Stroke 2014, 45, 3754–3832. [Google Scholar] [CrossRef] [Green Version]
- Diener, H.-C.; Hankey, G.J. Primary and Secondary Prevention of Ischemic Stroke and Cerebral Hemorrhage. J. Am. Coll. Cardiol. 2020, 75, 1804–1818. [Google Scholar] [CrossRef]
- Gelbenegger, G.; Postula, M.; Pecen, L.; Halvorsen, S.; Lesiak, M.; Schoergenhofer, C.; Jilma, B.; Hengstenberg, C.; Siller-Matula, J.M. Aspirin for Primary Prevention of Cardiovascular Disease: A Meta-Analysis with a Particular Focus on Subgroups. BMC Med. 2019, 17, 198. [Google Scholar] [CrossRef]
- Peacock, E.; Krousel-Wood, M. Adherence to Antihypertensive Therapy. Med. Clin. N. Am. 2017, 101, 229–245. [Google Scholar] [CrossRef] [Green Version]
- Iancu, M.A.; Mateiciuc, I.-I.; Stanescu, A.-M.A.; Matei, D.; Diaconu, C.C. Therapeutic Compliance of Patients with Arterial Hypertension in Primary Care. Medicina 2020, 56, 631. [Google Scholar] [CrossRef]
- Iulita, M.F.; Girouard, H. Treating Hypertension to Prevent Cognitive Decline and Dementia: Re-Opening the Debate. Adv. Exp. Med. Biol. 2017, 956, 447–473. [Google Scholar] [CrossRef]
- Hughes, D.; Judge, C.; Murphy, R.; Loughlin, E.; Costello, M.; Whiteley, W.; Bosch, J.; O’Donnell, M.J.; Canavan, M. Association of Blood Pressure Lowering with Incident Dementia or Cognitive Impairment: A Systematic Review and Meta-Analysis. JAMA 2020, 323, 1934–1944. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Zhao, Z.; Montagne, A.; Nelson, A.R.; Zlokovic, B.V. Blood-Brain Barrier: From Physiology to Disease and Back. Physiol. Rev. 2019, 99, 21–78. [Google Scholar] [CrossRef]
- Daneman, R.; Prat, A. The Blood-Brain Barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [Green Version]
- Aird, W.C. Phenotypic Heterogeneity of the Endothelium: I. Structure, Function, and Mechanisms. Circ. Res. 2007, 100, 158–173. [Google Scholar] [CrossRef]
- Nation, D.A.; Sweeney, M.D.; Montagne, A.; Sagare, A.P.; D’Orazio, L.M.; Pachicano, M.; Sepehrband, F.; Nelson, A.R.; Buennagel, D.P.; Harrington, M.G.; et al. Blood–Brain Barrier Breakdown Is an Early Biomarker of Human Cognitive Dysfunction. Nat. Med. 2019, 25, 270–276. [Google Scholar] [CrossRef]
- Zhao, Z.; Nelson, A.R.; Betsholtz, C.; Zlokovic, B.V. Establishment and Dysfunction of the Blood-Brain Barrier. Cell 2015, 163, 1064–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadry, H.; Noorani, B.; Cucullo, L. A Blood-Brain Barrier Overview on Structure, Function, Impairment, and Biomarkers of Integrity. Fluids Barriers CNS 2020, 17, 69. [Google Scholar] [CrossRef] [PubMed]
- Escobar, A.; Gómez-González, B. Barrera Hematoencefálica. Neurobiología, Implicaciones Clínicas y Efectos Del Estrés Sobre Su Desarrollo. Rev. Mex. Neurocienc. 2008, 9, 395–405. [Google Scholar]
- Brown, P.D.; Davies, S.L.; Speake, T.; Millar, I.D. Molecular Mechanisms of Cerebrospinal Fluid Production. Neuroscience 2004, 129, 955–968. [Google Scholar] [CrossRef] [Green Version]
- Ufnal, M.; Skrzypecki, J. Blood Borne Hormones in a Cross-Talk between Peripheral and Brain Mechanisms Regulating Blood Pressure, the Role of Circumventricular Organs. Neuropeptides 2014, 48, 65–73. [Google Scholar] [CrossRef]
- Obermeier, B.; Daneman, R.; Ransohoff, R.M. Development, Maintenance and Disruption of the Blood-Brain Barrier. Nat. Med. 2013, 19, 1584–1596. [Google Scholar] [CrossRef]
- Profaci, C.P.; Munji, R.N.; Pulido, R.S.; Daneman, R. The Blood-Brain Barrier in Health and Disease: Important Unanswered Questions. J. Exp. Med. 2020, 217, e20190062. [Google Scholar] [CrossRef]
- Alajangi, H.K.; Kaur, M.; Sharma, A.; Rana, S.; Thakur, S.; Chatterjee, M.; Singla, N.; Jaiswal, P.K.; Singh, G.; Barnwal, R.P. Blood–Brain Barrier: Emerging Trends on Transport Models and New-Age Strategies for Therapeutics Intervention against Neurological Disorders. Mol. Brain 2022, 15, 49. [Google Scholar] [CrossRef]
- Banks, W.A.; Reed, M.J.; Logsdon, A.F.; Rhea, E.M.; Erickson, M.A. Healthy Aging and the Blood–Brain Barrier. Nat. Aging 2021, 1, 243–254. [Google Scholar] [CrossRef]
- Hagan, N.; Ben-Zvi, A. The Molecular, Cellular, and Morphological Components of Blood-Brain Barrier Development during Embryogenesis. Semin. Cell Dev. Biol. 2015, 38, 7–15. [Google Scholar] [CrossRef]
- Jin, J.; Fang, F.; Gao, W.; Chen, H.; Wen, J.; Wen, X.; Chen, J. The Structure and Function of the Glycocalyx and Its Connection with Blood-Brain Barrier. Front. Cell. Neurosci. 2021, 15, 739699. [Google Scholar] [CrossRef]
- Zhang, X.; Sun, D.; Song, J.W.; Zullo, J.; Lipphardt, M.; Coneh-Gould, L.; Goligorsky, M.S. Endothelial Cell Dysfunction and Glycocalyx—A Vicious Circle. Matrix Biol. 2018, 71–72, 421–431. [Google Scholar] [CrossRef]
- Cosgun, Z.C.; Fels, B.; Kusche-Vihrog, K. Nanomechanics of the Endothelial Glycocalyx: From Structure to Function. Am. J. Pathol. 2020, 190, 732–741. [Google Scholar] [CrossRef]
- Dogné, S.; Flamion, B. Endothelial Glycocalyx Impairment in Disease: Focus on Hyaluronan Shedding. Am. J. Pathol. 2020, 190, 768–780. [Google Scholar] [CrossRef] [PubMed]
- Tkachenko, E.; Rhodes, J.M.; Simons, M. Syndecans: New Kids on the Signaling Block. Circ. Res. 2005, 96, 488–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheslow, L.; Alvarez, J.I. Glial-Endothelial Crosstalk Regulates Blood-Brain Barrier Function. Curr. Opin. Pharmacol. 2016, 26, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Liebner, S.; Dijkhuizen, R.M.; Reiss, Y.; Plate, K.H.; Agalliu, D.; Constantin, G. Functional Morphology of the Blood-Brain Barrier in Health and Disease. Acta Neuropathol. 2018, 135, 311–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohtsuki, S.; Hirayama, M.; Ito, S.; Uchida, Y.; Tachikawa, M.; Terasaki, T. Quantitative Targeted Proteomics for Understanding the Blood-Brain Barrier: Towards Pharmacoproteomics. Expert Rev. Proteom. 2014, 11, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Lochhead, J.J.; Yang, J.; Ronaldson, P.T.; Davis, T.P. Structure, Function, and Regulation of the Blood-Brain Barrier Tight Junction in Central Nervous System Disorders. Front. Physiol. 2020, 11, 914. [Google Scholar] [CrossRef] [PubMed]
- Citi, S. The Mechanobiology of Tight Junctions. Biophys. Rev. 2019, 11, 783–793. [Google Scholar] [CrossRef]
- Hudson, N.; Campbell, M. Tight Junctions of the Neurovascular Unit. Front. Mol. Neurosci. 2021, 14, 752781. [Google Scholar] [CrossRef]
- Hou, J.; Gomes, A.S.; Paul, D.L.; Goodenough, D.A. Study of Claudin Function by RNA Interference. J. Biol. Chem. 2006, 281, 36117–36123. [Google Scholar] [CrossRef] [Green Version]
- Matter, K.; Balda, M.S. Signalling to and from Tight Junctions. Nat. Rev. Mol. Cell Biol. 2003, 4, 225–236. [Google Scholar] [CrossRef]
- Majesky, M.W. Developmental Basis of Vascular Smooth Muscle Diversity. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1248–1258. [Google Scholar] [CrossRef] [Green Version]
- Campisi, M.; Shin, Y.; Osaki, T.; Hajal, C.; Chiono, V.; Kamm, R.D. 3D Self-Organized Microvascular Model of the Human Blood-Brain Barrier with Endothelial Cells, Pericytes and Astrocytes. Biomaterials 2018, 180, 117–129. [Google Scholar] [CrossRef]
- Armulik, A.; Genové, G.; Betsholtz, C. Pericytes: Developmental, Physiological, and Pathological Perspectives, Problems, and Promises. Dev. Cell. 2011, 21, 193–215. [Google Scholar] [CrossRef] [Green Version]
- Laredo, F.; Plebanski, J.; Tedeschi, A. Pericytes: Problems and Promises for CNS Repair. Front. Cell. Neurosci. 2019, 13, 546. [Google Scholar] [CrossRef]
- Sorokin, L. The Impact of the Extracellular Matrix on Inflammation. Nat. Rev. Immunol. 2010, 10, 712–723. [Google Scholar] [CrossRef]
- Logsdon, A.F.; Rhea, E.M.; Reed, M.; Banks, W.A.; Erickson, M.A. The Neurovascular Extracellular Matrix in Health and Disease. Exp. Biol. Med. 2021, 246, 835–844. [Google Scholar] [CrossRef]
- Xu, L.; Nirwane, A.; Yao, Y. Basement Membrane and Blood–Brain Barrier. Stroke Vasc. Neurol. 2019, 4, 78–82. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, K.; Ikeuchi, T.; Nara, K.; Rhodes, C.S.; Zhang, P.; Chiba, Y.; Kazuno, S.; Miura, Y.; Ago, T.; Arikawa-Hirasawa, E.; et al. Perlecan Regulates Pericyte Dynamics in the Maintenance and Repair of the Blood–Brain Barrier. J. Cell Biol. 2019, 218, 3506–3525. [Google Scholar] [CrossRef] [Green Version]
- Muoio, V.; Persson, P.B.; Sendeski, M.M. The Neurovascular Unit–Concept Review. Acta Physiol. 2014, 210, 790–798. [Google Scholar] [CrossRef]
- Wolburg, H.; Noell, S.; Wolburg-Buchholz, K.; Mack, A.; Fallier-Becker, P. Agrin, Aquaporin-4, and Astrocyte Polarity as an Important Feature of the Blood-Brain Barrier. Neuroscientist 2009, 15, 180–193. [Google Scholar] [CrossRef]
- Alvarez, J.I.; Katayama, T.; Prat, A. Glial Influence on the Blood Brain Barrier. Glia 2013, 61, 1939–1958. [Google Scholar] [CrossRef] [Green Version]
- Stokum, J.A.; Kurland, D.B.; Gerzanich, V.; Simard, J.M. Mechanisms of Astrocyte-Mediated Cerebral Edema. Neurochem. Res. 2015, 40, 317–328. [Google Scholar] [CrossRef] [Green Version]
- Pati, R.; Palazzo, C.; Valente, O.; Abbrescia, P.; Messina, R.; Surdo, N.C.; Lefkimmiatis, K.; Signorelli, F.; Nicchia, G.P.; Frigeri, A. The Readthrough Isoform AQP4ex Is Constitutively Phosphorylated in the Perivascular Astrocyte Endfeet of Human Brain. Biomolecules 2022, 12, 633. [Google Scholar] [CrossRef]
- Pan, W. From Blood to Brain through BBB and Astrocytic Signaling. Peptides 2015, 72, 121–127. [Google Scholar] [CrossRef]
- Silva, I.; Silva, J.; Ferreira, R.; Trigo, D. Glymphatic System, AQP4, and Their Implications in Alzheimer’s Disease. Neurol. Res. Pract. 2021, 3, 5. [Google Scholar] [CrossRef]
- Huber, V.J.; Igarashi, H.; Ueki, S.; Kwee, I.L.; Nakada, T. Aquaporin-4 Facilitator TGN-073 Promotes Interstitial Fluid Circulation within the Blood–Brain Barrier. Neuroreport 2018, 29, 697–703. [Google Scholar] [CrossRef]
- Nakada, T.; Kwee, I.; Igarashi, H.; Suzuki, Y. Aquaporin-4 Functionality and Virchow-Robin Space Water Dynamics: Physiological Model for Neurovascular Coupling and Glymphatic Flow. Int. J. Mol. Sci. 2017, 18, 1798. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.-Y.; Yang, Y.; Ju, W.-N.; Wang, X.; Zhang, H.-L. Emerging Roles of Astrocytes in Neuro-Vascular Unit and the Tripartite Synapse with Emphasis on Reactive Gliosis in the Context of Alzheimer’s Disease. Front. Cell. Neurosci. 2018, 12, 193. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Mato, M.; Iglesias-Rey, R.; Vieites-Prado, A.; Dopico-López, A.; Argibay, B.; Fernández-Susavila, H.; da Silva-Candal, A.; Pérez-Díaz, A.; Correa-Paz, C.; Günther, A.; et al. Blood Glutamate EAAT2-Cell Grabbing Therapy in Cerebral Ischemia. eBioMedicine 2019, 39, 118–131. [Google Scholar] [CrossRef] [Green Version]
- Zaghmi, A.; Dopico-López, A.; Pérez-Mato, M.; Iglesias-Rey, R.; Hervella, P.; Greschner, A.A.; Bugallo-Casal, A.; da Silva, A.; Gutiérrez-Fernández, M.; Castillo, J.; et al. Sustained Blood Glutamate Scavenging Enhances Protection in Ischemic Stroke. Commun. Biol. 2020, 3, 729. [Google Scholar] [CrossRef]
- Theparambil, S.M.; Hosford, P.S.; Ruminot, I.; Kopach, O.; Reynolds, J.R.; Sandoval, P.Y.; Rusakov, D.A.; Barros, L.F.; Gourine, A.V. Astrocytes Regulate Brain Extracellular PH via a Neuronal Activity-Dependent Bicarbonate Shuttle. Nat. Commun. 2020, 11, 5073. [Google Scholar] [CrossRef] [PubMed]
- Da Silva-Candal, A.; Pérez-Díaz, A.; Santamaría, M.; Correa-Paz, C.; Rodríguez-Yáñez, M.; Ardá, A.; Pérez-Mato, M.; Iglesias-Rey, R.; Brea, J.; Azuaje, J.; et al. Clinical Validation of Blood/Brain Glutamate Grabbing in Acute Ischemic Stroke. Ann. Neurol. 2018, 84, 260–273. [Google Scholar] [CrossRef] [PubMed]
- Hladky, S.B.; Barrand, M.A. Elimination of Substances from the Brain Parenchyma: Efflux via Perivascular Pathways and via the Blood-Brain Barrier. Fluids Barriers CNS 2018, 15, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polfliet, M.M.; Zwijnenburg, P.J.; van Furth, A.M.; van der Poll, T.; Döpp, E.A.; Renardel de Lavalette, C.; van Kesteren-Hendrikx, E.M.; van Rooijen, N.; Dijkstra, C.D.; van den Berg, T.K. Meningeal and Perivascular Macrophages of the Central Nervous System Play a Protective Role during Bacterial Meningitis. J. Immunol. 2001, 167, 4644–4650. [Google Scholar] [CrossRef] [Green Version]
- Dudvarski Stankovic, N.; Teodorczyk, M.; Ploen, R.; Zipp, F.; Schmidt, M.H.H. Microglia-Blood Vessel Interactions: A Double-Edged Sword in Brain Pathologies. Acta Neuropathol. 2016, 131, 347–363. [Google Scholar] [CrossRef]
- Ronaldson, P.T.; Davis, T.P. Regulation of Blood-Brain Barrier Integrity by Microglia in Health and Disease: A Therapeutic Opportunity. J. Cereb. Blood Flow Metab. 2020, 40, S6–S24. [Google Scholar] [CrossRef]
- Akhmetzyanova, E.; Kletenkov, K.; Mukhamedshina, Y.; Rizvanov, A. Different Approaches to Modulation of Microglia Phenotypes After Spinal Cord Injury. Front. Syst. Neurosci. 2019, 13, 37. [Google Scholar] [CrossRef] [Green Version]
- Subramaniam, S.R.; Federoff, H.J. Targeting Microglial Activation States as a Therapeutic Avenue in Parkinson’s Disease. Front. Aging Neurosci. 2017, 9, 176. [Google Scholar] [CrossRef]
- Rutkowska, A.; Shimshek, D.R.; Sailer, A.W.; Dev, K.K. EBI2 Regulates Pro-Inflammatory Signalling and Cytokine Release in Astrocytes. Neuropharmacology 2018, 133, 121–128. [Google Scholar] [CrossRef]
- Rustenhoven, J.; Jansson, D.; Smyth, L.C.; Dragunow, M. Brain Pericytes as Mediators of Neuroinflammation. Trends Pharmacol. Sci. 2017, 38, 291–304. [Google Scholar] [CrossRef]
- Abdullahi, W.; Davis, T.P.; Ronaldson, P.T. Functional Expression of P-Glycoprotein and Organic Anion Transporting Polypeptides at the Blood-Brain Barrier: Understanding Transport Mechanisms for Improved CNS Drug Delivery? AAPS J. 2017, 19, 931–939. [Google Scholar] [CrossRef] [Green Version]
- Roberts, L.M.; Black, D.S.; Raman, C.; Woodford, K.; Zhou, M.; Haggerty, J.E.; Yan, A.T.; Cwirla, S.E.; Grindstaff, K.K. Subcellular Localization of Transporters along the Rat Blood-Brain Barrier and Blood-Cerebral-Spinal Fluid Barrier by in Vivo Biotinylation. Neuroscience 2008, 155, 423–438. [Google Scholar] [CrossRef]
- Sanchez-Covarrubias, L.; Slosky, L.M.; Thompson, B.J.; Davis, T.P.; Ronaldson, P.T. Transporters at CNS Barrier Sites: Obstacles or Opportunities for Drug Delivery? Curr. Pharm. Des. 2014, 20, 1422–1449. [Google Scholar] [CrossRef] [Green Version]
- More, V.R.; Campos, C.R.; Evans, R.A.; Oliver, K.D.; Chan, G.N.; Miller, D.S.; Cannon, R.E. PPAR-α, a Lipid-Sensing Transcription Factor, Regulates Blood–Brain Barrier Efflux Transporter Expression. J. Cereb. Blood Flow Metab. 2017, 37, 1199–1212. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.; Yee, S.W.; Kim, R.B.; Giacomini, K.M. SLC Transporters as Therapeutic Targets: Emerging Opportunities. Nat. Rev. Drug Discov. 2015, 14, 543–560. [Google Scholar] [CrossRef] [Green Version]
- Pardridge, W.M. Blood-Brain Barrier Endogenous Transporters as Therapeutic Targets: A New Model for Small Molecule CNS Drug Discovery. Expert Opin. Ther. Targets 2015, 19, 1059–1072. [Google Scholar] [CrossRef]
- Abdullahi, W.; Tripathi, D.; Ronaldson, P.T. Blood-Brain Barrier Dysfunction in Ischemic Stroke: Targeting Tight Junctions and Transporters for Vascular Protection. Am. J. Physiol. Cell Physiol. 2018, 315, C343–C356. [Google Scholar] [CrossRef]
- Andreone, B.J.; Chow, B.W.; Tata, A.; Lacoste, B.; Ben-Zvi, A.; Bullock, K.; Deik, A.A.; Ginty, D.D.; Clish, C.B.; Gu, C. Blood-Brain Barrier Permeability Is Regulated by Lipid Transport-Dependent Suppression of Caveolae-Mediated Transcytosis. Neuron 2017, 94, 581–594. [Google Scholar] [CrossRef] [Green Version]
- Bray, N. Biologics: Transferrin’ Bispecific Antibodies across the Blood-Brain Barrier. Nat. Rev. Drug Discov. 2015, 14, 14–15. [Google Scholar] [CrossRef]
- Ghosh, D.; Peng, X.; Leal, J.; Mohanty, R.P. Peptides as Drug Delivery Vehicles across Biological Barriers. J. Pharm. Investig. 2018, 48, 89–111. [Google Scholar] [CrossRef]
- Zhao, Z.; Zlokovic, B.V. Blood-Brain Barrier: A Dual Life of MFSD2A? Neuron 2014, 82, 728–730. [Google Scholar] [CrossRef] [Green Version]
- Cui, Y.; Wang, Y.; Song, X.; Ning, H.; Zhang, Y.; Teng, Y.; Wang, J.; Yang, X. Brain Endothelial PTEN/AKT/NEDD4-2/MFSD2A Axis Regulates Blood-Brain Barrier Permeability. Cell Rep. 2021, 36, 109327. [Google Scholar] [CrossRef]
- Zhou, M.; Shi, S.X.; Liu, N.; Jiang, Y.; Karim, M.S.; Vodovoz, S.J.; Wang, X.; Zhang, B.; Dumont, A.S. Caveolae-Mediated Endothelial Transcytosis across the Blood-Brain Barrier in Acute Ischemic Stroke. J. Clin. Med. 2021, 10, 3795. [Google Scholar] [CrossRef]
- Abassi, Z.; Armaly, Z.; Heyman, S.N. Glycocalyx Degradation in Ischemia-Reperfusion Injury. Am. J. Pathol. 2020, 190, 752–767. [Google Scholar] [CrossRef]
- Yang, R.; Chen, M.; Zheng, J.; Li, X.; Zhang, X. The Role of Heparin and Glycocalyx in Blood-Brain Barrier Dysfunction. Front. Immunol. 2021, 12, 754141. [Google Scholar] [CrossRef]
- Tarbell, J.M.; Pahakis, M.Y. Mechanotransduction and the Glycocalyx. J. Intern. Med. 2006, 259, 339–350. [Google Scholar] [CrossRef]
- Zhu, J.; Li, X.; Yin, J.; Hu, Y.; Gu, Y.; Pan, S. Glycocalyx Degradation Leads to Blood-Brain Barrier Dysfunction and Brain Edema after Asphyxia Cardiac Arrest in Rats. J. Cereb. Blood Flow Metab. 2018, 38, 1979–1992. [Google Scholar] [CrossRef]
- Chen, R.; Zhang, X.; Gu, L.; Zhu, H.; Zhong, Y.; Ye, Y.; Xiong, X.; Jian, Z. New Insight into Neutrophils: A Potential Therapeutic Target for Cerebral Ischemia. Front. Immunol. 2021, 12, 692061. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.-M.; Zhang, C.-L.; Chen, A.-Q.; Wang, H.-L.; Zhou, Y.-F.; Li, Y.-N.; Hu, B. Immune Cells in the BBB Disruption After Acute Ischemic Stroke: Targets for Immune Therapy? Front. Immunol. 2021, 12, 678744. [Google Scholar] [CrossRef]
- Vallés, J.; Lago, A.; Santos, M.T.; Latorre, A.M.; Tembl, J.I.; Salom, J.B.; Nieves, C.; Moscardó, A. Neutrophil Extracellular Traps Are Increased in Patients with Acute Ischemic Stroke: Prognostic Significance. Thromb. Haemost. 2017, 117, 1919–1929. [Google Scholar] [CrossRef]
- Van Golen, R.F.; Reiniers, M.J.; Vrisekoop, N.; Zuurbier, C.J.; Olthof, P.B.; van Rheenen, J.; van Gulik, T.M.; Parsons, B.J.; Heger, M. The Mechanisms and Physiological Relevance of Glycocalyx Degradation in Hepatic Ischemia/Reperfusion Injury. Antioxid. Redox Signal. 2014, 21, 1098–1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nahirney, P.C.; Reeson, P.; Brown, C.E. Ultrastructural Analysis of Blood-Brain Barrier Breakdown in the Peri-Infarct Zone in Young Adult and Aged Mice. J. Cereb. Blood Flow Metab. 2016, 36, 413–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Jin, X.; Liu, K.J.; Liu, W. Matrix Metalloproteinase-2-Mediated Occludin Degradation and Caveolin-1-Mediated Claudin-5 Redistribution Contribute to Blood-Brain Barrier Damage in Early Ischemic Stroke Stage. J. Neurosci. 2012, 32, 3044–3057. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.C.; Davis, T.P. Hypoxia/Aglycemia Alters Expression of Occludin and Actin in Brain Endothelial Cells. Biochem. Biophys. Res. Commun. 2005, 327, 1114–1123. [Google Scholar] [CrossRef]
- Li, H.; Gao, A.; Feng, D.; Wang, Y.; Zhang, L.; Cui, Y.; Li, B.; Wang, Z.; Chen, G. Evaluation of the Protective Potential of Brain Microvascular Endothelial Cell Autophagy on Blood–Brain Barrier Integrity During Experimental Cerebral Ischemia–Reperfusion Injury. Transl. Stroke Res. 2014, 5, 618–626. [Google Scholar] [CrossRef]
- Liu, P.; Zhang, R.; Liu, D.; Wang, J.; Yuan, C.; Zhao, X.; Li, Y.; Ji, X.; Chi, T.; Zou, L. Time-Course Investigation of Blood–Brain Barrier Permeability and Tight Junction Protein Changes in a Rat Model of Permanent Focal Ischemia. J. Physiol. Sci. 2018, 68, 121–127. [Google Scholar] [CrossRef] [Green Version]
- Haley, M.J.; Lawrence, C.B. The Blood-Brain Barrier after Stroke: Structural Studies and the Role of Transcytotic Vesicles. J. Cereb. Blood Flow Metab. 2017, 37, 456–470. [Google Scholar] [CrossRef] [Green Version]
- Yang, A.C.; Stevens, M.Y.; Chen, M.B.; Lee, D.P.; Stähli, D.; Gate, D.; Contrepois, K.; Chen, W.; Iram, T.; Zhang, L.; et al. Physiological Blood-Brain Transport Is Impaired with Age by a Shift in Transcytosis. Nature 2020, 583, 425–430. [Google Scholar] [CrossRef]
- Yang, Y.; Kimura-Ohba, S.; Thompson, J.F.; Salayandia, V.M.; Cossé, M.; Raz, L.; Jalal, F.Y.; Rosenberg, G.A. Vascular Tight Junction Disruption and Angiogenesis in Spontaneously Hypertensive Rat with Neuroinflammatory White Matter Injury. Neurobiol. Dis. 2018, 114, 95–110. [Google Scholar] [CrossRef]
- Krueger, M.; Härtig, W.; Reichenbach, A.; Bechmann, I.; Michalski, D. Blood-Brain Barrier Breakdown after Embolic Stroke in Rats Occurs without Ultrastructural Evidence for Disrupting Tight Junctions. PLoS ONE 2013, 8, e56419. [Google Scholar] [CrossRef] [Green Version]
- Andjelkovic, A.V.; Xiang, J.; Stamatovic, S.M.; Hua, Y.; Xi, G.; Wang, M.M.; Keep, R.F. Endothelial Targets in Stroke: Translating Animal Models to Human. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 2240–2247. [Google Scholar] [CrossRef]
- Page, S.; Munsell, A.; Al-Ahmad, A.J. Cerebral Hypoxia/Ischemia Selectively Disrupts Tight Junctions Complexes in Stem Cell-Derived Human Brain Microvascular Endothelial Cells. Fluids Barriers CNS 2016, 13, 16. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, S.F.; Granger, D.N. Blood Cells and Endothelial Barrier Function. Tissue Barriers 2015, 3, e978720. [Google Scholar] [CrossRef] [Green Version]
- Keaney, J.; Campbell, M. The Dynamic Blood-Brain Barrier. FEBS J. 2015, 282, 4067–4079. [Google Scholar] [CrossRef]
- Winkler, L.; Blasig, R.; Breitkreuz-Korff, O.; Berndt, P.; Dithmer, S.; Helms, H.C.; Puchkov, D.; Devraj, K.; Kaya, M.; Qin, Z.; et al. Tight Junctions in the Blood-Brain Barrier Promote Edema Formation and Infarct Size in Stroke–Ambivalent Effects of Sealing Proteins. J. Cereb. Blood Flow Metab. 2021, 41, 132–145. [Google Scholar] [CrossRef]
- Reed, M.J.; Damodarasamy, M.; Banks, W.A. The Extracellular Matrix of the Blood-Brain Barrier: Structural and Functional Roles in Health, Aging, and Alzheimer’s Disease. Tissue Barriers 2019, 7, 1651157. [Google Scholar] [CrossRef]
- Han, L.; Jiang, C. Evolution of Blood-Brain Barrier in Brain Diseases and Related Systemic Nanoscale Brain-Targeting Drug Delivery Strategies. Acta Pharm. Sin. B 2021, 11, 2306–2325. [Google Scholar] [CrossRef]
- Knowland, D.; Arac, A.; Sekiguchi, K.J.; Hsu, M.; Lutz, S.E.; Perrino, J.; Steinberg, G.K.; Barres, B.A.; Nimmerjahn, A.; Agalliu, D. Stepwise Recruitment of Transcellular and Paracellular Pathways Underlies Blood-Brain Barrier Breakdown in Stroke. Neuron 2014, 82, 603–617. [Google Scholar] [CrossRef] [Green Version]
- Betz, A.L.; Keep, R.F.; Beer, M.E.; Ren, X.D. Blood-Brain Barrier Permeability and Brain Concentration of Sodium, Potassium, and Chloride during Focal Ischemia. J. Cereb. Blood Flow Metab. 1994, 14, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Strbian, D.; Durukan, A.; Pitkonen, M.; Marinkovic, I.; Tatlisumak, E.; Pedrono, E.; Abo-Ramadan, U.; Tatlisumak, T. The Blood-Brain Barrier Is Continuously Open for Several Weeks Following Transient Focal Cerebral Ischemia. Neuroscience 2008, 153, 175–181. [Google Scholar] [CrossRef]
- Al-Ahmady, Z.S.; Jasim, D.; Ahmad, S.S.; Wong, R.; Haley, M.; Coutts, G.; Schiessl, I.; Allan, S.M.; Kostarelos, K. Selective Liposomal Transport through Blood Brain Barrier Disruption in Ischemic Stroke Reveals Two Distinct Therapeutic Opportunities. ACS Nano 2019, 13, 12470–12486. [Google Scholar] [CrossRef]
- Fukuda, S.; Fini, C.A.; Mabuchi, T.; Koziol, J.A.; Eggleston, L.L.; del Zoppo, G.J. Focal Cerebral Ischemia Induces Active Proteases That Degrade Microvascular Matrix. Stroke 2004, 35, 998–1004. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Rosenberg, G.A. Blood-Brain Barrier Breakdown in Acute and Chronic Cerebrovascular Disease. Stroke 2011, 42, 3323–3328. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.-R.; Navalitloha, Y.; Singhal, S.; Mehta, J.; Piao, C.-S.; Guo, W.-P.; Kessler, J.A.; Groothuis, D.R. Hematopoietic Growth Factors Pass through the Blood-Brain Barrier in Intact Rats. Exp. Neurol. 2007, 204, 569–573. [Google Scholar] [CrossRef] [Green Version]
- Krizbai, I.A.; Bauer, H.; Bresgen, N.; Eckl, P.M.; Farkas, A.; Szatmári, E.; Traweger, A.; Wejksza, K.; Bauer, H.-C. Effect of Oxidative Stress on the Junctional Proteins of Cultured Cerebral Endothelial Cells. Cell. Mol. Neurobiol. 2005, 25, 129–139. [Google Scholar] [CrossRef]
- Castillo, J.; Rodríguez, J.R.; Corredera, E.; Alvarex, J.M.; Purmar, J.M.; Noya, M. White Matter High-Signal Areas on MRI Associated with Chronic Hypoxia. Eur. J. Neurol. 1996, 3, 533–538. [Google Scholar] [CrossRef]
- Knox, E.G.; Aburto, M.R.; Clarke, G.; Cryan, J.F.; O’Driscoll, C.M. The Blood-Brain Barrier in Aging and Neurodegeneration. Mol. Psychiatry 2022, 27, 2659–2673. [Google Scholar] [CrossRef]
- Castillo, J.; Dávalos, A.; Naveiro, J.; Noya, M. Neuroexcitatory Amino Acids and Their Relation to Infarct Size and Neurological Deficit in Ischemic Stroke. Stroke 1996, 27, 1060–1065. [Google Scholar] [CrossRef] [PubMed]
- Castillo, J.; Dávalos, A.; Noya, M. Progression of Ischaemic Stroke and Excitotoxic Aminoacids. Lancet 1997, 349, 79–83. [Google Scholar] [CrossRef]
- Dávalos, A.; Castillo, J.; Serena, J.; Noya, M. Duration of Glutamate Release after Acute Ischemic Stroke. Stroke 1997, 28, 708–710. [Google Scholar] [CrossRef]
- Puig, N.; Dávalos, A.; Adan, J.; Piulats, J.; Martínez, J.M.; Castillo, J. Serum Amino Acid Levels after Permanent Middle Cerebral Artery Occlusion in the Rat. Cerebrovasc. Dis. 2000, 10, 449–454. [Google Scholar] [CrossRef] [PubMed]
- Castillo, J.; Rodríguez, I. Biochemical Changes and Inflammatory Response as Markers for Brain Ischaemia: Molecular Markers of Diagnostic Utility and Prognosis in Human Clinical Practice. Cerebrovasc. Dis. 2004, 17, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Blanco, M.; Rodríguez-Yáñez, M.; Sobrino, T.; Leira, R.; Castillo, J. Platelets, Inflammation, and Atherothrombotic Neurovascular Disease: The Role of Endothelial Dysfunction. Cerebrovasc. Dis. 2005, 2, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Hawkins, K.E.; Doré, S.; Candelario-Jalil, E. Neuroinflammatory Mechanisms of Blood-Brain Barrier Damage in Ischemic Stroke. Am. J. Physiol. Cell Physiol. 2019, 316, C135–C153. [Google Scholar] [CrossRef]
- Brea, D.; Sobrino, T.; Ramos-Cabrer, P.; Castillo, J. Inflammatory and Neuroimmunomodulatory Changes in Acute Cerebral Ischemia. Cerebrovasc. Dis. 2009, 27, 48–64. [Google Scholar] [CrossRef]
- Blamire, A.M.; Anthony, D.C.; Rajagopalan, B.; Sibson, N.R.; Perry, V.H.; Styles, P. Interleukin-1beta-Induced Changes in Blood-Brain Barrier Permeability, Apparent Diffusion Coefficient, and Cerebral Blood Volume in the Rat Brain: A Magnetic Resonance Study. J. Neurosci. 2000, 20, 8153–8159. [Google Scholar] [CrossRef] [Green Version]
- Caso, J.R.; Moro, M.A.; Lorenzo, P.; Lizasoain, I.; Leza, J.C. Involvement of IL-1beta in Acute Stress-Induced Worsening of Cerebral Ischaemia in Rats. Eur. Neuropsychopharmacol. 2007, 17, 600–607. [Google Scholar] [CrossRef]
- Vila, N.; Castillo, J.; Dávalos, A.; Chamorro, A. Proinflammatory Cytokines and Early Neurological Worsening in Ischemic Stroke. Stroke 2000, 31, 2325–2329. [Google Scholar] [CrossRef] [Green Version]
- Castellanos, M.; Castillo, J.; García, M.M.; Leira, R.; Serena, J.; Chamorro, A.; Dávalos, A. Inflammation-Mediated Damage in Progressing Lacunar Infarctions: A Potential Therapeutic Target. Stroke 2002, 33, 982–987. [Google Scholar] [CrossRef] [Green Version]
- Castillo, J.; Moro, M.A.; Blanco, M.; Leira, R.; Serena, J.; Lizasoain, I.; Dávalos, A. The Release of Tumor Necrosis Factor-Alpha Is Associated with Ischemic Tolerance in Human Stroke. Ann. Neurol. 2003, 54, 811–819. [Google Scholar] [CrossRef]
- Bustamante, A.; Sobrino, T.; Giralt, D.; García-Berrocoso, T.; Llombart, V.; Ugarriza, I.; Espadaler, M.; Rodríguez, N.; Sudlow, C.; Castellanos, M.; et al. Prognostic Value of Blood Interleukin-6 in the Prediction of Functional Outcome after Stroke: A Systematic Review and Meta-Analysis. J. Neuroimmunol. 2014, 274, 215–224. [Google Scholar] [CrossRef]
- Castillo, J.; Alvarez-Sabín, J.; Martínez-Vila, E.; Montaner, J.; Sobrino, T.; Vivancos, J.; MITICO Study Investigators. Inflammation Markers and Prediction of Post-Stroke Vascular Disease Recurrence: The MITICO Study. J. Neurol. 2009, 256, 217–224. [Google Scholar] [CrossRef]
- Rodríguez-Yáñez, M.; Castillo, J. Role of Inflammatory Markers in Brain Ischemia. Curr. Opin. Neurol. 2008, 21, 353–357. [Google Scholar] [CrossRef]
- Roquer, J.; Segura, T.; Serena, J.; Castillo, J. Endothelial Dysfunction, Vascular Disease and Stroke: The ARTICO Study. Cerebrovasc. Dis. 2009, 1, 25–37. [Google Scholar] [CrossRef]
- Vila, N.; Castillo, J.; Dávalos, A.; Esteve, A.; Planas, A.M.; Chamorro, A. Levels of Anti-Inflammatory Cytokines and Neurological Worsening in Acute Ischemic Stroke. Stroke 2003, 34, 671–675. [Google Scholar] [CrossRef] [Green Version]
- Kraus, J.; Ling, A.K.; Hamm, S.; Voigt, K.; Oschmann, P.; Engelhardt, B. Interferon-Beta Stabilizes Barrier Characteristics of Brain Endothelial Cells in Vitro. Ann. Neurol. 2004, 56, 192–205. [Google Scholar] [CrossRef]
- Luo, J. TGF-β as a Key Modulator of Astrocyte Reactivity: Disease Relevance and Therapeutic Implications. Biomedicines 2022, 10, 1206. [Google Scholar] [CrossRef]
- Burkly, L.C.; Michaelson, J.S.; Hahm, K.; Jakubowski, A.; Zheng, T.S. TWEAKing Tissue Remodeling by a Multifunctional Cytokine: Role of TWEAK/Fn14 Pathway in Health and Disease. Cytokine 2007, 40, 1–16. [Google Scholar] [CrossRef]
- Campbell, S.; Michaelson, J.; Burkly, L.; Putterman, C. The Role of TWEAK/Fn14 in the Pathogenesis of Inflammation and Systemic Autoimmunity. Front. Biosci. 2004, 9, 2273–2284. [Google Scholar] [CrossRef] [Green Version]
- Stephan, D.; Sbai, O.; Wen, J.; Couraud, P.-O.; Putterman, C.; Khrestchatisky, M.; Desplat-Jégo, S. TWEAK/Fn14 Pathway Modulates Properties of a Human Microvascular Endothelial Cell Model of Blood Brain Barrier. J. Neuroinflamm. 2013, 10, 9. [Google Scholar] [CrossRef] [Green Version]
- Nagy, D.; Ennis, K.A.; Wei, R.; Su, S.C.; Hinckley, C.A.; Gu, R.-F.; Gao, B.; Massol, R.H.; Ehrenfels, C.; Jandreski, L.; et al. Developmental Synaptic Regulator, TWEAK/Fn14 Signaling, Is a Determinant of Synaptic Function in Models of Stroke and Neurodegeneration. Proc. Natl. Acad. Sci. USA 2021, 118, e2001679118. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Kim, J.; Ko, J.; Lee, E.J.; Koh, H.J.; Yoon, J.S. Tumor Necrosis Factor-like Weak Inducer of Apoptosis Induces Inflammation in Graves’ Orbital Fibroblasts. PLoS ONE 2018, 13, e0209583. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Makonchuk, D.Y.; Li, H.; Mittal, A.; Kumar, A. TNF-like Weak Inducer of Apoptosis (TWEAK) Activates Proinflammatory Signaling Pathways and Gene Expression through the Activation of TGF-Beta-Activated Kinase 1. J. Immunol. 2009, 182, 2439–2448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratajczak, W.; Atkinson, S.D.; Kelly, C. The TWEAK/Fn14/CD163 Axis-Implications for Metabolic Disease. Rev. Endocr. Metab. Disord. 2022, 23, 449–462. [Google Scholar] [CrossRef]
- Burkly, L.C.; Dohi, T. The TWEAK/Fn14 Pathway in Tissue Remodeling: For Better or for Worse. Adv. Exp. Med. Biol. 2011, 691, 305–322. [Google Scholar] [CrossRef]
- Li, H.; Mittal, A.; Paul, P.K.; Kumar, M.; Srivastava, D.S.; Tyagi, S.C.; Kumar, A. Tumor Necrosis Factor-Related Weak Inducer of Apoptosis Augments Matrix Metalloproteinase 9 (MMP-9) Production in Skeletal Muscle through the Activation of Nuclear Factor-KappaB-Inducing Kinase and P38 Mitogen-Activated Protein Kinase: A Potential Role of. J. Biol. Chem. 2009, 284, 4439–4450. [Google Scholar] [CrossRef] [Green Version]
- Da Silva-Candal, A.; Custodia, A.; López-Dequidt, I.; Rodríguez-Yáñez, M.; Alonso-Alonso, M.L.; Ávila-Gómez, P.; Pumar, J.M.; Castillo, J.; Sobrino, T.; Campos, F.; et al. STWEAK Is a Leukoaraiosis Biomarker Associated with Neurovascular Angiopathy. Ann. Clin. Transl. Neurol. 2022, 9, 171–180. [Google Scholar] [CrossRef]
- Hervella, P.; Pérez-Mato, M.; Rodríguez-Yáñez, M.; López-Dequidt, I.; Pumar, J.M.; Sobrino, T.; Campos, F.; Castillo, J.; da Silva-Candal, A.; Iglesias-Rey, R. STWEAK as Predictor of Stroke Recurrence in Ischemic Stroke Patients Treated with Reperfusion Therapies. Front. Neurol. 2021, 12, 652867. [Google Scholar] [CrossRef]
- Da Silva-Candal, A.; Pérez-Mato, M.; Rodríguez-Yáñez, M.; López-Dequidt, I.; Pumar, J.M.; Ávila-Gómez, P.; Sobrino, T.; Campos, F.; Castillo, J.; Hervella, P.; et al. The Presence of Leukoaraiosis Enhances the Association between STWEAK and Hemorrhagic Transformation. Ann. Clin. Transl. Neurol. 2020, 7, 2103–2114. [Google Scholar] [CrossRef]
- Da Silva-Candal, A.; López-Dequidt, I.; Rodriguez-Yañez, M.; Ávila-Gómez, P.; Pumar, J.M.; Castillo, J.; Sobrino, T.; Campos, F.; Iglesias-Rey, R.; Hervella, P. STWEAK Is a Marker of Early Haematoma Growth and Leukoaraiosis in Intracerebral Haemorrhage. Stroke Vasc. Neurol. 2021, 6, 528–535. [Google Scholar] [CrossRef]
- Dimitrijevic, O.B.; Stamatovic, S.M.; Keep, R.F.; Andjelkovic, A.V. Effects of the Chemokine CCL2 on Blood-Brain Barrier Permeability during Ischemia-Reperfusion Injury. J. Cereb. Blood Flow Metab. 2006, 26, 797–810. [Google Scholar] [CrossRef] [Green Version]
- Chui, R.; Dorovini-Zis, K. Regulation of CCL2 and CCL3 Expression in Human Brain Endothelial Cells by Cytokines and Lipopolysaccharide. J. Neuroinflamm. 2010, 7, 1. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Hallenbeck, J.M.; Ruetzler, C.; Bol, D.; Thomas, K.; Berman, N.E.J.; Vogel, S.N. Overexpression of Monocyte Chemoattractant Protein 1 in the Brain Exacerbates Ischemic Brain Injury and Is Associated with Recruitment of Inflammatory Cells. J. Cereb. Blood Flow Metab. 2003, 23, 748–755. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Huang, J.; Li, Y.; Yang, G.-Y. Roles of Chemokine CXCL12 and Its Receptors in Ischemic Stroke. Curr. Drug Targets 2012, 13, 166–172. [Google Scholar] [CrossRef]
- Shyu, W.-C.; Lin, S.-Z.; Yen, P.-S.; Su, C.-Y.; Chen, D.-C.; Wang, H.-J.; Li, H. Stromal Cell-Derived Factor-1 Alpha Promotes Neuroprotection, Angiogenesis, and Mobilization/Homing of Bone Marrow-Derived Cells in Stroke Rats. J. Pharmacol. Exp. Ther. 2008, 324, 834–849. [Google Scholar] [CrossRef]
- Deddens, L.H.; van Tilborg, G.A.F.; van der Marel, K.; Hunt, H.; van der Toorn, A.; Viergever, M.A.; de Vries, H.E.; Dijkhuizen, R.M. In Vivo Molecular MRI of ICAM-1 Expression on Endothelium and Leukocytes from Subacute to Chronic Stages After Experimental Stroke. Transl. Stroke Res. 2017, 8, 440–448. [Google Scholar] [CrossRef] [Green Version]
- Castillo, J.; Rama, R.; Dávalos, A. Nitric Oxide–Related Brain Damage in Acute Ischemic Stroke. Stroke 2000, 31, 852–857. [Google Scholar] [CrossRef] [Green Version]
- Brea, D.; Roquer, J.; Serena, J.; Segura, T.; Castillo, J.; ARTICO STUDY. Oxidative Stress Markers Are Associated to Vascular Recurrence in Non-Cardioembolic Stroke Patients Non-Treated with Statins. BMC Neurol. 2012, 12, 65. [Google Scholar] [CrossRef] [Green Version]
- Alfieri, D.F.; Lehmann, M.F.; Flauzino, T.; de Araújo, M.C.M.; Pivoto, N.; Tirolla, R.M.; Simão, A.N.C.; Maes, M.; Reiche, E.M.V. Immune-Inflammatory, Metabolic, Oxidative, and Nitrosative Stress Biomarkers Predict Acute Ischemic Stroke and Short-Term Outcome. Neurotox. Res. 2020, 38, 330–343. [Google Scholar] [CrossRef]
- Iadecola, C.; Zhang, F.; Xu, S.; Casey, R.; Ross, M.E. Inducible Nitric Oxide Synthase Gene Expression in Brain Following Cerebral Ischemia. J. Cereb. Blood Flow Metab. 1995, 15, 378–384. [Google Scholar] [CrossRef] [Green Version]
- Leker, R.R.; Teichner, A.; Ovadia, H.; Keshet, E.; Reinherz, E.; Ben-Hur, T. Expression of Endothelial Nitric Oxide Synthase in the Ischemic Penumbra: Relationship to Expression of Neuronal Nitric Oxide Synthase and Vascular Endothelial Growth Factor. Brain Res. 2001, 909, 1–7. [Google Scholar] [CrossRef]
- Van Dyken, P.; Lacoste, B. Impact of Metabolic Syndrome on Neuroinflammation and the Blood-Brain Barrier. Front. Neurosci. 2018, 12, 930. [Google Scholar] [CrossRef] [PubMed]
- Castañeda-Cabral, J.L.; Colunga-Durán, A.; Ureña-Guerrero, M.E.; Beas-Zárate, C.; Nuñez-Lumbreras, M.d.L.A.; Orozco-Suárez, S.; Alonso-Vanegas, M.; Guevara-Guzmán, R.; Deli, M.A.; Valle-Dorado, M.G.; et al. Expression of VEGF- and Tight Junction-Related Proteins in the Neocortical Microvasculature of Patients with Drug-Resistant Temporal Lobe Epilepsy. Microvasc. Res. 2020, 132, 104059. [Google Scholar] [CrossRef] [PubMed]
- Asahi, M.; Asahi, K.; Jung, J.C.; del Zoppo, G.J.; Fini, M.E.; Lo, E.H. Role for Matrix Metalloproteinase 9 after Focal Cerebral Ischemia: Effects of Gene Knockout and Enzyme Inhibition with BB-94. J. Cereb. Blood Flow Metab. 2000, 20, 1681–1689. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; An, Q.; Wang, T.; Gao, S.; Zhou, G. Autophagy- and MMP-2/9-Mediated Reduction and Redistribution of ZO-1 Contribute to Hyperglycemia-Increased Blood-Brain Barrier Permeability During Early Reperfusion in Stroke. Neuroscience 2018, 377, 126–137. [Google Scholar] [CrossRef]
- Yang, Y.; Estrada, E.Y.; Thompson, J.F.; Liu, W.; Rosenberg, G.A. Matrix Metalloproteinase-Mediated Disruption of Tight Junction Proteins in Cerebral Vessels Is Reversed by Synthetic Matrix Metalloproteinase Inhibitor in Focal Ischemia in Rat. J. Cereb. Blood Flow Metab. 2007, 27, 697–709. [Google Scholar] [CrossRef]
- Douglas, A.S.; Shearer, J.A.; Okolo, A.; Pandit, A.; Gilvarry, M.; Doyle, K.M. The Relationship Between Cerebral Reperfusion and Regional Expression of Matrix Metalloproteinase-9 In Rat Brain Following Focal Cerebral Ischemia. Neuroscience 2021, 453, 256–265. [Google Scholar] [CrossRef]
- Castellanos, M.; Leira, R.; Serena, J.; Pumar, J.M.; Lizasoain, I.; Castillo, J.; Dávalos, A. Plasma Metalloproteinase-9 Concentration Predicts Hemorrhagic Transformation in Acute Ischemic Stroke. Stroke 2003, 34, 40–46. [Google Scholar] [CrossRef] [Green Version]
- Castellanos, M.; Leira, R.; Serena, J.; Blanco, M.; Pedraza, S.; Castillo, J.; Dávalos, A. Plasma Cellular-Fibronectin Concentration Predicts Hemorrhagic Transformation after Thrombolytic Therapy in Acute Ischemic Stroke. Stroke 2004, 35, 1671–1676. [Google Scholar] [CrossRef] [Green Version]
- Castellanos, M.; Sobrino, T.; Millán, M.; García, M.; Arenillas, J.; Nombela, F.; Brea, D.; Perez de la Ossa, N.; Serena, J.; Vivancos, J.; et al. Serum Cellular Fibronectin and Matrix Metalloproteinase-9 as Screening Biomarkers for the Prediction of Parenchymal Hematoma after Thrombolytic Therapy in Acute Ischemic Stroke: A Multicenter Confirmatory Study. Stroke 2007, 38, 1855–1859. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez, J.A.; Sobrino, T.; Orbe, J.; Purroy, A.; Martínez-Vila, E.; Castillo, J.; Páramo, J.A. ProMetalloproteinase-10 Is Associated with Brain Damage and Clinical Outcome in Acute Ischemic Stroke. J. Thromb. Haemost. 2013, 11, 1464–1473. [Google Scholar] [CrossRef]
- Ouro, A.; Correa-Paz, C.; Maqueda, E.; Custodia, A.; Aramburu-Núñez, M.; Romaus-Sanjurjo, D.; Posado-Fernández, A.; Candamo-Lourido, M.; Alonso-Alonso, M.L.; Hervella, P.; et al. Involvement of Ceramide Metabolism in Cerebral Ischemia. Front. Mol. Biosci. 2022, 9, 864618. [Google Scholar] [CrossRef]
- Vutukuri, R.; Brunkhorst, R.; Kestner, R.-I.; Hansen, L.; Bouzas, N.F.; Pfeilschifter, J.; Devraj, K.; Pfeilschifter, W. Alteration of Sphingolipid Metabolism as a Putative Mechanism Underlying LPS-Induced BBB Disruption. J. Neurochem. 2018, 144, 172–185. [Google Scholar] [CrossRef] [Green Version]
- Bekhite, M.; González-Delgado, A.; Hübner, S.; Haxhikadrija, P.; Kretzschmar, T.; Müller, T.; Wu, J.M.F.; Bekfani, T.; Franz, M.; Wartenberg, M.; et al. The Role of Ceramide Accumulation in Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes on Mitochondrial Oxidative Stress and Mitophagy. Free Radic. Biol. Med. 2021, 167, 66–80. [Google Scholar] [CrossRef]
- You, Q.; Peng, Q.; Yu, Z.; Jin, H.; Zhang, J.; Sun, W.; Huang, Y. Plasma Lipidomic Analysis of Sphingolipids in Patients with Large Artery Atherosclerosis Cerebrovascular Disease and Cerebral Small Vessel Disease. Biosci. Rep. 2020, 40, BSR20193470. [Google Scholar] [CrossRef]
- Hachinski, V.C.; Potter, P.; Merskey, H. Leuko-Araiosis. Arch. Neurol. 1987, 44, 21–23. [Google Scholar] [CrossRef]
- Wardlaw, J.M.; Smith, E.E.; Biessels, G.J.; Cordonnier, C.; Fazekas, F.; Frayne, R.; Lindley, R.I.; O’Brien, J.T.; Barkhof, F.; Benavente, O.R.; et al. Neuroimaging Standards for Research into Small Vessel Disease and Its Contribution to Ageing and Neurodegeneration. Lancet Neurol. 2013, 12, 822–838. [Google Scholar] [CrossRef] [Green Version]
- Hase, Y.; Horsburgh, K.; Ihara, M.; Kalaria, R.N. White Matter Degeneration in Vascular and Other Ageing-Related Dementias. J. Neurochem. 2018, 144, 617–633. [Google Scholar] [CrossRef]
- Wardlaw, J.M.; Valdés Hernández, M.C.; Muñoz-Maniega, S. What Are White Matter Hyperintensities Made of? Relevance to Vascular Cognitive Impairment. J. Am. Heart Assoc. 2015, 4, 001140. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Li, J.; Feng, C.; Huang, X.; Wong, L.; Liu, X.; Nie, Z.; Xi, G. Blood-Brain Barrier Damage as the Starting Point of Leukoaraiosis Caused by Cerebral Chronic Hypoperfusion and Its Involved Mechanisms: Effect of Agrin and Aquaporin-4. Biomed. Res. Int. 2018, 2018, 2321797. [Google Scholar] [CrossRef] [Green Version]
- De Havenon, A.; Meyer, C.; McNally, J.S.; Alexander, M.; Chung, L. Subclinical Cerebrovascular Disease: Epidemiology and Treatment. Curr. Atheroscler. Rep. 2019, 21, 39. [Google Scholar] [CrossRef]
- MacGregor Sharp, M.; Saito, S.; Keable, A.; Gatherer, M.; Aldea, R.; Agarwal, N.; Simpson, J.E.; Wharton, S.B.; Weller, R.O.; Carare, R.O. Demonstrating a Reduced Capacity for Removal of Fluid from Cerebral White Matter and Hypoxia in Areas of White Matter Hyperintensity Associated with Age and Dementia. Acta Neuropathol. Commun. 2020, 8, 131. [Google Scholar] [CrossRef]
- Vagal, V.; Venema, S.U.; Behymer, T.P.; Mistry, E.A.; Sekar, P.; Sawyer, R.P.; Gilkerson, L.; Moomaw, C.J.; Haverbusch, M.; Coleman, E.R.; et al. White Matter Lesion Severity Is Associated with Intraventricular Hemorrhage in Spontaneous Intracerebral Hemorrhage. J. Stroke Cerebrovasc. Dis. 2020, 29, 104661. [Google Scholar] [CrossRef]
- Ferguson, K.J.; Cvoro, V.; MacLullich, A.M.J.; Shenkin, S.D.; Sandercock, P.A.G.; Sakka, E.; Wardlaw, J.M. Visual Rating Scales of White Matter Hyperintensities and Atrophy: Comparison of Computed Tomography and Magnetic Resonance Imaging. J. Stroke Cerebrovasc. Dis. 2018, 27, 1815–1821. [Google Scholar] [CrossRef] [Green Version]
- Kerkhofs, D.; Wong, S.M.; Zhang, E.; Staals, J.; Jansen, J.F.A.; van Oostenbrugge, R.J.; Backes, W.H. Baseline Blood-Brain Barrier Leakage and Longitudinal Microstructural Tissue Damage in the Periphery of White Matter Hyperintensities. Neurology 2021, 96, e2192–e2200. [Google Scholar] [CrossRef]
- Hinman, J.D.; Lee, M.D.; Tung, S.; Vinters, H.V.; Carmichael, S.T. Molecular Disorganization of Axons Adjacent to Human Lacunar Infarcts. Brain 2015, 138, 736–745. [Google Scholar] [CrossRef] [Green Version]
- Jha, R.M.; Kochanek, P.M.; Simard, J.M. Pathophysiology and Treatment of Cerebral Edema in Traumatic Brain Injury. Neuropharmacology 2019, 145, 230–246. [Google Scholar] [CrossRef]
- Halstead, M.R.; Geocadin, R.G. The Medical Management of Cerebral Edema: Past, Present, and Future Therapies. Neurotherapeutics 2019, 16, 1133–1148. [Google Scholar] [CrossRef]
- Stokum, J.A.; Gerzanich, V.; Simard, J.M. Molecular Pathophysiology of Cerebral Edema. J. Cereb. Blood Flow Metab. 2016, 36, 513–538. [Google Scholar] [CrossRef] [Green Version]
- Schneider, G.H.; Baethmann, A.; Kempski, O. Mechanisms of Glial Swelling Induced by Glutamate. Can. J. Physiol. Pharmacol. 1992, 70, S334–S343. [Google Scholar] [CrossRef]
- Heiss, W.-D. Malignant MCA Infarction: Pathophysiology and Imaging for Early Diagnosis and Management Decisions. Cerebrovasc. Dis. 2016, 41, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.-N.; Fang, P.-T.; Hung, C.-H.; Hsu, C.-Y.; Chou, P.-S.; Yang, Y.-H. The Association between White Matter Changes and Development of Malignant Middle Cerebral Artery Infarction. Medicine 2021, 100, e25751. [Google Scholar] [CrossRef] [PubMed]
- Hoehn-Berlage, M.; Norris, D.G.; Kohno, K.; Mies, G.; Leibfritz, D.; Hossmann, K.A. Evolution of Regional Changes in Apparent Diffusion Coefficient during Focal Ischemia of Rat Brain: The Relationship of Quantitative Diffusion NMR Imaging to Reduction in Cerebral Blood Flow and Metabolic Disturbances. J. Cereb. Blood Flow Metab. 1995, 15, 1002–1011. [Google Scholar] [CrossRef] [PubMed]
- Fischer, S.; Wobben, M.; Marti, H.H.; Renz, D.; Schaper, W. Hypoxia-Induced Hyperpermeability in Brain Microvessel Endothelial Cells Involves VEGF-Mediated Changes in the Expression of Zonula Occludens-1. Microvasc. Res. 2002, 63, 70–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, T.H.; Ståhl, N.; Schalén, W.; Reinstrup, P.; Toft, P.; Nordström, C.H. Recirculation Usually Precedes Malignant Edema in Middle Cerebral Artery Infarcts. Acta Neurol. Scand. 2012, 126, 404–410. [Google Scholar] [CrossRef]
- Von Kummer, R.; Meyding-Lamadé, U.; Forsting, M.; Rosin, L.; Rieke, K.; Hacke, W.; Sartor, K. Sensitivity and Prognostic Value of Early CT in Occlusion of the Middle Cerebral Artery Trunk. AJNR. Am. J. Neuroradiol. 1994, 15, 9–15; discussion 16–18. [Google Scholar]
- Pluta, R.; Lossinsky, A.S.; Wis´niewski, H.M.; Mossakowski, M.J. Early Blood-Brain Barrier Changes in the Rat Following Transient Complete Cerebral Ischemia Induced by Cardiac Arrest. Brain Res. 1994, 633, 41–52. [Google Scholar] [CrossRef]
- Pluta, R.; Lossinsky, A.S.; Walski, M.; Wisniewski, H.M.; Mossakowski, M.J. Platelet Occlusion Phenomenon after Short- and Long-Term Survival Following Complete Cerebral Ischemia in Rats Produced by Cardiac Arrest. J. Hirnforsch. 1994, 35, 463–471. [Google Scholar]
- Van Vliet, E.A.; Ndode-Ekane, X.E.; Lehto, L.J.; Gorter, J.A.; Andrade, P.; Aronica, E.; Gröhn, O.; Pitkänen, A. Long-Lasting Blood-Brain Barrier Dysfunction and Neuroinflammation after Traumatic Brain Injury. Neurobiol. Dis. 2020, 145, 105080. [Google Scholar] [CrossRef]
- Song, H.W.; Foreman, K.L.; Gastfriend, B.D.; Kuo, J.S.; Palecek, S.P.; Shusta, E.V. Transcriptomic Comparison of Human and Mouse Brain Microvessels. Sci. Rep. 2020, 10, 12358. [Google Scholar] [CrossRef]
- Sobrino, T.; Hurtado, O.; Moro, M.A.; Rodríguez-Yáñez, M.; Castellanos, M.; Brea, D.; Moldes, O.; Blanco, M.; Arenillas, J.F.; Leira, R.; et al. The Increase of Circulating Endothelial Progenitor Cells after Acute Ischemic Stroke Is Associated with Good Outcome. Stroke 2007, 38, 2759–2764. [Google Scholar] [CrossRef] [Green Version]
- Campos, F.; Qin, T.; Castillo, J.; Seo, J.H.; Arai, K.; Lo, E.H.; Waeber, C. Fingolimod Reduces Hemorrhagic Transformation Associated with Delayed Tissue Plasminogen Activator Treatment in a Mouse Thromboembolic Model. Stroke 2013, 44, 505–511. [Google Scholar] [CrossRef] [Green Version]
- Jordán, J.; Segura, T.; Brea, D.; Galindo, M.F.; Castillo, J. Inflammation as Therapeutic Objective in Stroke. Curr. Pharm. Des. 2008, 14, 3549–3564. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.-D.; Zhao, Y.; Liu, Y. Role of the TWEAK/Fn14 Pathway in Autoimmune Diseases. Immunol. Res. 2016, 64, 44–50. [Google Scholar] [CrossRef]
- Sieow, B.F.-L.; Wun, K.S.; Yong, W.P.; Hwang, I.Y.; Chang, M.W. Tweak to Treat: Reprograming Bacteria for Cancer Treatment. Trends Cancer 2021, 7, 447–464. [Google Scholar] [CrossRef]
- Méndez-Barbero, N.; Gutiérrez-Muñoz, C.; Blázquez-Serra, R.; Martín-Ventura, J.; Blanco-Colio, L. Tumor Necrosis Factor-Like Weak Inducer of Apoptosis (TWEAK)/Fibroblast Growth Factor-Inducible 14 (Fn14) Axis in Cardiovascular Diseases: Progress and Challenges. Cells 2020, 9, 405. [Google Scholar] [CrossRef] [Green Version]
- Da Silva-Candal, A.; Argibay, B.; Iglesias-Rey, R.; Vargas, Z.; Vieites-Prado, A.; López-Arias, E.; Rodríguez-Castro, E.; López-Dequidt, I.; Rodríguez-Yáñez, M.; Piñeiro, Y.; et al. Vectorized Nanodelivery Systems for Ischemic Stroke: A Concept and a Need. J. Nanobiotechnol. 2017, 15, 30. [Google Scholar] [CrossRef]
- Bony, B.A.; Tarudji, A.W.; Miller, H.A.; Gowrikumar, S.; Roy, S.; Curtis, E.T.; Gee, C.C.; Vecchio, A.; Dhawan, P.; Kievit, F.M. Claudin-1-Targeted Nanoparticles for Delivery to Aging-Induced Alterations in the Blood-Brain Barrier. ACS Nano. 2021, 15, 18520–18531. [Google Scholar] [CrossRef]
- Yang, H.; Luo, Y.; Hu, H.; Yang, S.; Li, Y.; Jin, H.; Chen, S.; He, Q.; Hong, C.; Wu, J.; et al. PH-Sensitive, Cerebral Vasculature-Targeting Hydroxyethyl Starch Functionalized Nanoparticles for Improved Angiogenesis and Neurological Function Recovery in Ischemic Stroke. Adv. Healthc. Mater. 2021, 10, 2100028. [Google Scholar] [CrossRef]
- Correa-Paz, C.; Navarro Poupard, M.F.; Polo, E.; Rodríguez-Pérez, M.; Migliavacca, M.; Iglesias-Rey, R.; Ouro, A.; Maqueda, E.; Hervella, P.; Sobrino, T.; et al. Sonosensitive Capsules for Brain Thrombolysis Increase Ischemic Damage in a Stroke Model. J. Nanobiotechnol. 2022, 20, 46. [Google Scholar] [CrossRef]
- Correa-Paz, C.; da Silva-Candal, A.; Polo, E.; Parcq, J.; Vivien, D.; Maysinger, D.; Pelaz, B.; Campos, F. New Approaches in Nanomedicine for Ischemic Stroke. Pharmaceutics 2021, 13, 757. [Google Scholar] [CrossRef]
- Vargas-Osorio, Z.; da Silva-Candal, A.; Piñeiro, Y.; Iglesias-Rey, R.; Sobrino, T.; Campos, F.; Castillo, J.; Rivas, J. Multifunctional Superparamagnetic Stiff Nanoreservoirs for Blood Brain Barrier Applications. Nanomaterials 2019, 9, 449. [Google Scholar] [CrossRef] [Green Version]
- Da Silva-Candal, A.; Brown, T.; Krishnan, V.; Lopez-Loureiro, I.; Ávila-Gómez, P.; Pusuluri, A.; Pérez-Díaz, A.; Correa-Paz, C.; Hervella, P.; Castillo, J.; et al. Shape Effect in Active Targeting of Nanoparticles to Inflamed Cerebral Endothelium under Static and Flow Conditions. J. Control. Release 2019, 309, 94–105. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
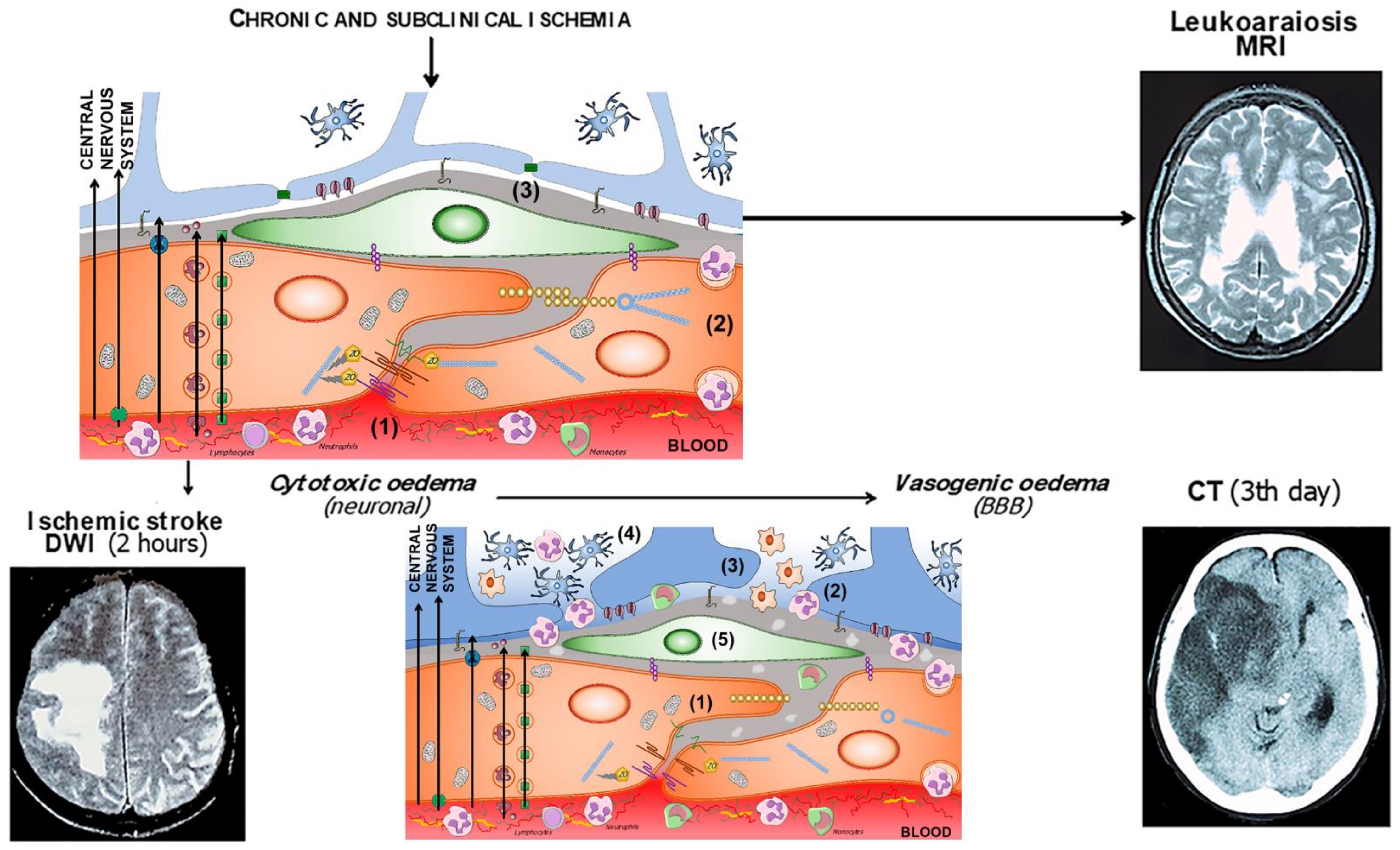{kind=link}
| Blood-Brain Barrier Components | Change/Event | References |
|---|---|---|
| Glycocalyx | Degradation | [94,95,96,97] |
| Endothelial cells | Interaction with leukocytes Increase in caveolae | [98,99,100,101,102] |
| Tight junctions | Decrease the expression/ change location of proteins | [103,104,105,106] |
| Pericytes | Increase in caveolae Astrocytic endfeet oedema | [102,107,108] |
| Basement membrane | ||
| Astrocytic endfeet |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alonso-Alonso, M.L.; Sampedro-Viana, A.; Fernández-Rodicio, S.; Bazarra-Barreiros, M.; Ouro, A.; Sobrino, T.; Campos, F.; Castillo, J.; Hervella, P.; Iglesias-Rey, R. Need for a Paradigm Shift in the Treatment of Ischemic Stroke: The Blood-Brain Barrier. Int. J. Mol. Sci. 2022, 23, 9486. https://doi.org/10.3390/ijms23169486
Alonso-Alonso ML, Sampedro-Viana A, Fernández-Rodicio S, Bazarra-Barreiros M, Ouro A, Sobrino T, Campos F, Castillo J, Hervella P, Iglesias-Rey R. Need for a Paradigm Shift in the Treatment of Ischemic Stroke: The Blood-Brain Barrier. International Journal of Molecular Sciences. 2022; 23(16):9486. https://doi.org/10.3390/ijms23169486
Chicago/Turabian StyleAlonso-Alonso, Maria Luz, Ana Sampedro-Viana, Sabela Fernández-Rodicio, Marcos Bazarra-Barreiros, Alberto Ouro, Tomás Sobrino, Francisco Campos, José Castillo, Pablo Hervella, and Ramón Iglesias-Rey. 2022. "Need for a Paradigm Shift in the Treatment of Ischemic Stroke: The Blood-Brain Barrier" International Journal of Molecular Sciences 23, no. 16: 9486. https://doi.org/10.3390/ijms23169486
APA StyleAlonso-Alonso, M. L., Sampedro-Viana, A., Fernández-Rodicio, S., Bazarra-Barreiros, M., Ouro, A., Sobrino, T., Campos, F., Castillo, J., Hervella, P., & Iglesias-Rey, R. (2022). Need for a Paradigm Shift in the Treatment of Ischemic Stroke: The Blood-Brain Barrier. International Journal of Molecular Sciences, 23(16), 9486. https://doi.org/10.3390/ijms23169486