
IL-1 Family Cytokines in Inflammatory Dermatoses: Pathogenetic Role and Potential Therapeutic Implications


Abstract
:1. Introduction
2. IL-1 Family Cytokines, Receptors and Co-Receptors
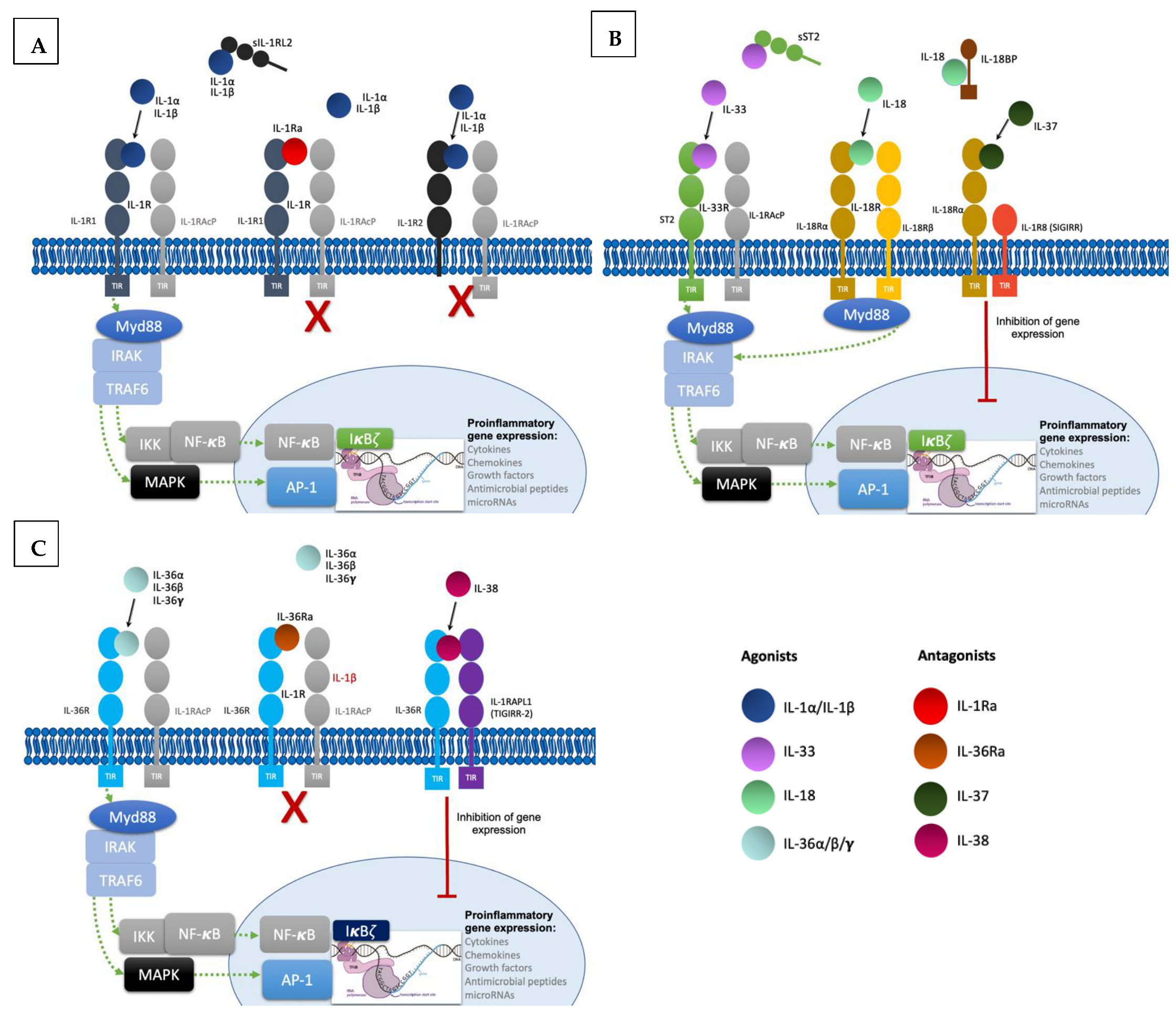
2.1. IL-1 Subfamily
2.2. IL-18 Subfamily
2.3. IL-33 Subfamily
2.4. IL-36 Subfamily
2.5. IL-37 and IL-38: Antagonist Cytokines
3. Involvement of IL-1 Family Cytokines in Inflammatory Dermatoses
3.1. Psoriasis
3.2. Atopic Dermatitis
3.3. Hidradenitis Suppurativa
3.4. Other Dermatoses
4. Therapeutic Targeting of IL-1 Family Cytokines
4.1. IL-1
4.2. IL-18
4.3. IL-33
4.4. IL-36
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACD | allergic contact dermatitis |
| AD | atopic dermatitis |
| AGEP | acute generalized exanthematous pustulosis |
| DAMPs | damage-associated molecular patterns |
| DCs | dendritic cells |
| DIRA | deficiency of interleukin-1 receptor antagonist |
| DITRA | deficiency of IL-36 receptor antagonist |
| G-CSF | granulocyte colony-stimulating factor |
| GPP | generalized pustular psoriasis |
| HS | hidradenitis supurativa |
| IL-1RAcP | interleukin 1 receptor accessory protein |
| IL-36Ra | Interleukin 1 receptor antagonist |
| IL | interleukin |
| IL1RAPL1 and IL1RAPL2 | IL-1 receptor accessory protein like 1 and 2 |
| IRAK | Interleukin 1 receptor associated kinases |
| KCs | keratinocytes |
| MAPKs | mitogen-activated protein kinases |
| Myd88 | myeloid differentiation primary response 88 |
| NF-kB | nuclear factor κB |
| PAMPs | pathogen-associated molecular patterns |
| PG | pyoderma gangrenosum |
| PP | pustulas psoriasis |
| PPP | palmoplantar pustulosis |
| PPR | pattern recognition receptors |
| SIGIRR | single immunoglobulin IL-1R-related molecule |
| SS | Sweet syndrome |
| ST2 | suppression of tumorigenicity 2 |
| TIGIRR 1 and 2 | three immunoglobulin domain-containing IL-1 receptor-related 1 and 2 |
| TIR | toll-IL1R |
| TNF | tumor necrosis factor |
References
- Boutet, M.-A.; Nerviani, A.; Pitzalis, C. IL-36, IL-37, and IL-38 Cytokines in Skin and Joint Inflammation: A Comprehensive Review of Their Therapeutic Potential. Int. J. Mol. Sci. 2019, 20, 1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez-Santana, Y.E.; Giannoudaki, E.; Leon, G.; Lucitt, M.B.; Walsh, P.T. Current Perspectives on the Interleukin-1 Family as Targets for Inflammatory Disease. Eur. J. Immunol. 2019, 49, 1306–1320. [Google Scholar] [CrossRef] [PubMed]
- Towne, J.E.; Renshaw, B.R.; Douangpanya, J.; Lipsky, B.P.; Shen, M.; Gabel, C.A.; Sims, J.E. Interleukin-36 (IL-36) Ligands Require Processing for Full Agonist (IL-36α, IL-36β, and IL-36γ) or Antagonist (IL-36Ra) Activity. J. Biol. Chem. 2011, 286, 42594–42602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mora, J.; Weigert, A. IL-1 Family Cytokines in Cancer Immunity—A Matter of Life and Death. Biol. Chem. 2016, 397, 1125–1134. [Google Scholar] [CrossRef]
- Uppala, R.; Tsoi, L.C.; Harms, P.W.; Wang, B.; Billi, A.C.; Maverakis, E.; Michelle Kahlenberg, J.; Ward, N.L.; Gudjonsson, J.E. “Autoinflammatory Psoriasis”-Genetics and Biology of Pustular Psoriasis. Cell Mol. Immunol. 2020, 18, 307–317. [Google Scholar] [CrossRef]
- Matarazzo, L.; Hernandez Santana, Y.E.; Walsh, P.T.; Fallon, P.G. The IL-1 Cytokine Family as Custodians of Barrier Immunity. Cytokine 2022, 154, 155890. [Google Scholar] [CrossRef]
- Dinarello, C.A. Overview of the IL-1 Family in Innate Inflammation and Acquired Immunity. Immunol. Rev. 2018, 281, 8–27. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, K.; Nakajima, A.; Sudo, K.; Liu, Y.; Mizoroki, A.; Ikarashi, T.; Horai, R.; Kakuta, S.; Watanabe, T.; Iwakura, Y. IL-1 Receptor Type 2 Suppresses Collagen-Induced Arthritis by Inhibiting IL-1 Signal on Macrophages. J. Immunol. 2015, 194, 3156–3168. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Humphry, M.; Maguire, J.J.; Bennett, M.R.; Clarke, M.C.H. Intracellular Interleukin-1 Receptor 2 Binding Prevents Cleavage and Activity of Interleukin-1α, Controlling Necrosis-Induced Sterile Inflammation. Immunity 2013, 38, 285–295. [Google Scholar] [CrossRef] [Green Version]
- Fields, J.K.; Günther, S.; Sundberg, E.J. Structural Basis of IL-1 Family Cytokine Signaling. Front. Immunol. 2019, 10, 1412. [Google Scholar] [CrossRef] [Green Version]
- Dinarello, C.; Novick, D.; Kim, S.; Kaplanski, G. Interleukin-18 and IL-18 Binding Protein. Front. Immunol. 2013, 4, 289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nold-Petry, C.A.; Lo, C.Y.; Rudloff, I.; Elgass, K.D.; Li, S.; Gantier, M.P.; Lotz-Havla, A.S.; Gersting, S.W.; Cho, S.X.; Lao, J.C.; et al. IL-37 Requires the Receptors IL-18Rα and IL-1R8 (SIGIRR) to Carry out Its Multifaceted Anti-Inflammatory Program upon Innate Signal Transduction. Nat. Immunol. 2015, 16, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Di Paolo, N.C.; Shayakhmetov, D.M. Interleukin 1α and the Inflammatory Process. Nat. Immunol. 2016, 17, 906–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afonina, I.S.; Tynan, G.A.; Logue, S.E.; Cullen, S.P.; Bots, M.; Lüthi, A.U.; Reeves, E.P.; McElvaney, N.G.; Medema, J.P.; Lavelle, E.C.; et al. Granzyme B-Dependent Proteolysis Acts as a Switch to Enhance the Proinflammatory Activity of IL-1α. Mol. Cell 2011, 44, 265–278. [Google Scholar] [CrossRef] [Green Version]
- Kavita, U.; Mizel, S.B. Differential Sensitivity of Interleukin-1 Alpha and -Beta Precursor Proteins to Cleavage by Calpain, a Calcium-Dependent Protease. J. Biol. Chem. 1995, 270, 27758–27765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertheloot, D.; Latz, E. HMGB1, IL-1α, IL-33 and S100 Proteins: Dual-Function Alarmins. Cell Mol. Immunol. 2017, 14, 43–64. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Dinarello, C.A.; Molgora, M.; Garlanda, C. Interleukin-1 and Related Cytokines in the Regulation of Inflammation and Immunity. Immunity 2019, 50, 778–795. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Todorovic, V. Interleukin-36: Structure, Signaling and Function. Adv. Exp. Med. Biol. 2020, 2020, 191–210. [Google Scholar] [CrossRef]
- Guma, M.; Ronacher, L.; Liu-Bryan, R.; Takai, S.; Karin, M.; Corr, M. Caspase 1-Independent Activation of Interleukin-1beta in Neutrophil-Predominant Inflammation. Arthritis Rheum. 2009, 60, 3642–3650. [Google Scholar] [CrossRef] [Green Version]
- Højen, J.F.; Kristensen, M.L.V.; McKee, A.S.; Wade, M.T.; Azam, T.; Lunding, L.P.; de Graaf, D.M.; Swartzwelter, B.J.; Wegmann, M.; Tolstrup, M.; et al. IL-1R3 Blockade Broadly Attenuates the Functions of Six Members of the IL-1 Family, Revealing Their Contribution to Models of Disease. Nat. Immunol. 2019, 20, 1138–1149. [Google Scholar] [CrossRef]
- Kaplanski, G. Interleukin-18: Biological Properties and Role in Disease Pathogenesis. Immunol. Rev. 2018, 281, 138–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasuda, K.; Nakanishi, K.; Tsutsui, H. Interleukin-18 in Health and Disease. Int. J. Mol. Sci. 2019, 20, 649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, D.; Trajkovic, V.; Hunter, D.; Leung, B.P.; Schulz, K.; Gracie, J.A.; McInnes, I.B.; Liew, F.Y. IL-18 Induces the Differentiation of Th1 or Th2 Cells Depending upon Cytokine Milieu and Genetic Background. Eur. J. Immunol. 2000, 30, 3147–3156. [Google Scholar] [CrossRef]
- Oka, N.; Markova, T.; Tsuzuki, K.; Li, W.; El-Darawish, Y.; Pencheva-Demireva, M.; Yamanishi, K.; Yamanishi, H.; Sakagami, M.; Tanaka, Y.; et al. IL-12 Regulates the Expansion, Phenotype, and Function of Murine NK Cells Activated by IL-15 and IL-18. Cancer Immunol. Immunother. 2020, 69, 1699–1712. [Google Scholar] [CrossRef]
- Quatrini, L.; Vacca, P.; Tumino, N.; Besi, F.; Pace, A.L.D.; Scordamaglia, F.; Martini, S.; Munari, E.; Mingari, M.C.; Ugolini, S.; et al. Glucocorticoids and the Cytokines IL-12, IL-15, and IL-18 Present in the Tumor Microenvironment Induce PD-1 Expression on Human Natural Killer Cells. J. Allergy Clin. Immunol. 2021, 147, 349–360. [Google Scholar] [CrossRef]
- Yoshimoto, T.; Tsutsui, H.; Tominaga, K.; Hoshino, K.; Okamura, H.; Akira, S.; Paul, W.E.; Nakanishi, K. IL-18, Although Antiallergic When Administered with IL-12, Stimulates IL-4 and Histamine Release by Basophils. Proc. Natl. Acad. Sci. USA 1999, 96, 13962–13966. [Google Scholar] [CrossRef] [Green Version]
- Niu, X.-L.; Huang, Y.; Gao, Y.-L.; Sun, Y.-Z.; Han, Y.; Chen, H.-D.; Gao, X.-H.; Qi, R.-Q. Interleukin-18 Exacerbates Skin Inflammation and Affects Microabscesses and Scale Formation in a Mouse Model of Imiquimod-Induced Psoriasis. Chin. Med. J. 2019, 132, 690–698. [Google Scholar] [CrossRef]
- Cayrol, C.; Girard, J.-P. Interleukin-33 (IL-33): A Nuclear Cytokine from the IL-1 Family. Immunol. Rev. 2018, 281, 154–168. [Google Scholar] [CrossRef]
- Hung, L.-Y.; Tanaka, Y.; Herbine, K.; Pastore, C.; Singh, B.; Ferguson, A.; Vora, N.; Douglas, B.; Zullo, K.; Behrens, E.M.; et al. Cellular Context of IL-33 Expression Dictates Impact on Anti-Helminth Immunity. Sci. Immunol. 2020, 5, eabc6259. [Google Scholar] [CrossRef]
- Cannavò, S.P.; Bertino, L.; Di Salvo, E.; Papaianni, V.; Ventura-Spagnolo, E.; Gangemi, S. Possible Roles of IL-33 in the Innate-Adaptive Immune Crosstalk of Psoriasis Pathogenesis. Mediat. Inflamm. 2019, 2019, 7158014. [Google Scholar] [CrossRef] [Green Version]
- Smithgall, M.D.; Comeau, M.R.; Yoon, B.-R.P.; Kaufman, D.; Armitage, R.; Smith, D.E. IL-33 Amplifies Both Th1- and Th2-Type Responses through Its Activity on Human Basophils, Allergen-Reactive Th2 Cells, INKT and NK Cells. Int. Immunol. 2008, 20, 1019–1030. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Huard, A.; Mora, J.; da Silva, P.; Brüne, B.; Weigert, A. IL-36 Family Cytokines in Protective versus Destructive Inflammation. Cell. Signal. 2020, 75, 109773. [Google Scholar] [CrossRef] [PubMed]
- Clancy, D.M.; Henry, C.M.; Sullivan, G.P.; Martin, S.J. Neutrophil Extracellular Traps Can Serve as Platforms for Processing and Activation of IL-1 Family Cytokines. FEBS J. 2017, 284, 1712–1725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clancy, D.M.; Sullivan, G.P.; Moran, H.B.T.; Henry, C.M.; Reeves, E.P.; McElvaney, N.G.; Lavelle, E.C.; Martin, S.J. Extracellular Neutrophil Proteases Are Efficient Regulators of IL-1, IL-33, and IL-36 Cytokine Activity but Poor Effectors of Microbial Killing. Cell Rep. 2018, 22, 2937–2950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henry, C.M.; Sullivan, G.P.; Clancy, D.M.; Afonina, I.S.; Kulms, D.; Martin, S.J. Neutrophil-Derived Proteases Escalate Inflammation through Activation of IL-36 Family Cytokines. Cell Rep. 2016, 14, 708–722. [Google Scholar] [CrossRef] [Green Version]
- Johnston, A.; Xing, X.; Wolterink, L.; Barnes, D.H.; Yin, Z.; Reingold, L.; Kahlenberg, J.M.; Harms, P.W.; Gudjonsson, J.E. IL-1 and IL-36 Are Dominant Cytokines in Generalized Pustular Psoriasis. J. Allergy Clin. Immunol. 2017, 140, 109–120. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.; Wen, X.; Hao, D.; Wang, Y.; Wang, L.; He, G.; Jiang, X. The Role of IL-37 in Skin and Connective Tissue Diseases. Biomed. Pharm. 2020, 122, 109705. [Google Scholar] [CrossRef]
- Conti, P.; Pregliasco, F.E.; Bellomo, R.G.; Gallenga, C.E.; Caraffa, A.; Kritas, S.K.; Lauritano, D.; Ronconi, G. Mast Cell Cytokines IL-1, IL-33, and IL-36 Mediate Skin Inflammation in Psoriasis: A Novel Therapeutic Approach with the Anti-Inflammatory Cytokines IL-37, IL-38, and IL-1Ra. Int. J. Mol. Sci. 2021, 22, 8076. [Google Scholar] [CrossRef]
- Han, Y.; Mora, J.; Huard, A.; da Silva, P.; Wiechmann, S.; Putyrski, M.; Schuster, C.; Elwakeel, E.; Lang, G.; Scholz, A.; et al. IL-38 Ameliorates Skin Inflammation and Limits IL-17 Production from Γδ T Cells. Cell Rep. 2019, 27, 835–846.e5. [Google Scholar] [CrossRef] [Green Version]
- Navarini, A.A.; Burden, A.D.; Capon, F.; Mrowietz, U.; Puig, L.; Köks, S.; Kingo, K.; Smith, C.; Barker, J.N.; ERASPEN Network. European Consensus Statement on Phenotypes of Pustular Psoriasis. J. Eur. Acad. Dermatol. Venereol. 2017, 31, 1792–1799. [Google Scholar] [CrossRef] [Green Version]
- Gooderham, M.J.; Van Voorhees, A.S.; Lebwohl, M.G. An Update on Generalized Pustular Psoriasis. Expert Rev. Clin. Immunol. 2019, 15, 907–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boehncke, W.-H.; Schön, M.P. Psoriasis. Lancet 2015, 386, 983–994. [Google Scholar] [CrossRef]
- Sachen, K.L.; Arnold Greving, C.N.; Towne, J.E. Role of IL-36 Cytokines in Psoriasis and Other Inflammatory Skin Conditions. Cytokine 2022, 156, 155897. [Google Scholar] [CrossRef] [PubMed]
- Renne, J.; Schäfer, V.; Werfel, T.; Wittmann, M. Interleukin-1 from Epithelial Cells Fosters T Cell-Dependent Skin Inflammation. Br. J. Dermatol. 2010, 162, 1198–1205. [Google Scholar] [CrossRef]
- Cai, Y.; Xue, F.; Quan, C.; Qu, M.; Liu, N.; Zhang, Y.; Fleming, C.; Hu, X.; Zhang, H.-G.; Weichselbaum, R.; et al. A Critical Role of the IL-1β-IL-1R Signaling Pathway in Skin Inflammation and Psoriasis Pathogenesis. J. Investig. Dermatol. 2019, 139, 146–156. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Cho, D.H.; Park, H.J. IL-18 and Cutaneous Inflammatory Diseases. Int. J. Mol. Sci. 2015, 16, 29357–29369. [Google Scholar] [CrossRef] [Green Version]
- Shimoura, N.; Nagai, H.; Fujiwara, S.; Jimbo, H.; Yoshimoto, T.; Nishigori, C. Interleukin (IL)-18, Cooperatively with IL-23, Induces Prominent Inflammation and Enhances Psoriasis-like Epidermal Hyperplasia. Arch. Dermatol. Res. 2017, 309, 315–321. [Google Scholar] [CrossRef]
- Zeng, F.; Chen, H.; Chen, L.; Mao, J.; Cai, S.; Xiao, Y.; Li, J.; Shi, J.; Li, B.; Xu, Y.; et al. An Autocrine Circuit of IL-33 in Keratinocytes Is Involved in the Progression of Psoriasis. J. Investig. Dermatol. 2021, 141, 596–606.e7. [Google Scholar] [CrossRef]
- Mitsui, A.; Tada, Y.; Takahashi, T.; Shibata, S.; Kamata, M.; Miyagaki, T.; Fujita, H.; Sugaya, M.; Kadono, T.; Sato, S.; et al. Serum IL-33 Levels Are Increased in Patients with Psoriasis. Clin. Exp. Dermatol. 2016, 41, 183–189. [Google Scholar] [CrossRef]
- Marrakchi, S.; Guigue, P.; Renshaw, B.R.; Puel, A.; Pei, X.-Y.; Fraitag, S.; Zribi, J.; Bal, E.; Cluzeau, C.; Chrabieh, M.; et al. Interleukin-36-Receptor Antagonist Deficiency and Generalized Pustular Psoriasis. N. Engl. J. Med. 2011, 365, 620–628. [Google Scholar] [CrossRef]
- Sehat, M.; Talaei, R.; Dadgostar, E.; Nikoueinejad, H.; Akbari, H. Evaluating Serum Levels of IL-33, IL-36, IL-37 and Gene Expression of IL-37 in Patients with Psoriasis Vulgaris. Iran. J. Allergy Asthma Immunol. 2018, 17, 179–187. [Google Scholar] [PubMed]
- Mercurio, L.; Morelli, M.; Scarponi, C.; Eisenmesser, E.Z.; Doti, N.; Pagnanelli, G.; Gubinelli, E.; Mazzanti, C.; Cavani, A.; Ruvo, M.; et al. IL-38 Has an Anti-Inflammatory Action in Psoriasis and Its Expression Correlates with Disease Severity and Therapeutic Response to Anti-IL-17A Treatment. Cell Death Dis. 2018, 9, 1104. [Google Scholar] [CrossRef] [PubMed]
- Han, M.; Yuan, X.; Shi, X.; Zhu, X.-Y.; Su, Y.; Xiong, D.-K.; Zhang, X.-M.; Zhou, H.; Wang, J.-N. The Pathological Mechanism and Potential Application of IL-38 in Autoimmune Diseases. Front. Pharm. 2021, 12, 732790. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Huang, Z.; Li, H.; Liu, X.; Zheng, S.; Su, W. IL-38: A New Player in Inflammatory Autoimmune Disorders. Biomolecules 2019, 9, 345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gugliandolo, A.; Caraffa, A.L.; Gallenga, C.E.; Kritas, S.K.; Ronconi, G.; Trubiani, O.; Conti, P.; Di Emidio, P.; Mazzon, E. Mesenchymal Stem Cells and IL-37: A Powerful Combination. J. Biol. Regul. Homeost. Agents 2019, 33, 1019–1022. [Google Scholar] [PubMed]
- Paller, A.S.; Spergel, J.M.; Mina-Osorio, P.; Irvine, A.D. The Atopic March and Atopic Multimorbidity: Many Trajectories, Many Pathways. J. Allergy Clin. Immunol. 2019, 143, 46–55. [Google Scholar] [CrossRef]
- Fujii, M. Current Understanding of Pathophysiological Mechanisms of Atopic Dermatitis: Interactions among Skin Barrier Dysfunction, Immune Abnormalities and Pruritus. Biol. Pharm. Bull. 2020, 43, 12–19. [Google Scholar] [CrossRef] [Green Version]
- Moyle, M.; Cevikbas, F.; Harden, J.L.; Guttman-Yassky, E. Understanding the Immune Landscape in Atopic Dermatitis: The Era of Biologics and Emerging Therapeutic Approaches. Exp. Dermatol. 2019, 28, 756–768. [Google Scholar] [CrossRef] [Green Version]
- Tubau, C.; Puig, L. Therapeutic Targeting of the IL-13 Pathway in Skin Inflammation. Expert Rev. Clin. Immunol. 2021, 17, 15–25. [Google Scholar] [CrossRef]
- Tokura, Y. Extrinsic and Intrinsic Types of Atopic Dermatitis. J. Dermatol. Sci. 2010, 58, 1–7. [Google Scholar] [CrossRef]
- Suárez-Fariñas, M.; Dhingra, N.; Gittler, J.; Shemer, A.; Cardinale, I.; de Guzman Strong, C.; Krueger, J.G.; Guttman-Yassky, E. Intrinsic Atopic Dermatitis Shows Similar TH2 and Higher TH17 Immune Activation Compared with Extrinsic Atopic Dermatitis. J. Allergy Clin. Immunol. 2013, 132, 361–370. [Google Scholar] [CrossRef] [Green Version]
- Tsoi, L.C.; Rodriguez, E.; Degenhardt, F.; Baurecht, H.; Wehkamp, U.; Volks, N.; Szymczak, S.; Swindell, W.R.; Sarkar, M.K.; Raja, K.; et al. Atopic Dermatitis Is an IL-13-Dominant Disease with Greater Molecular Heterogeneity Compared to Psoriasis. J. Investig. Dermatol. 2019, 139, 1480–1489. [Google Scholar] [CrossRef] [Green Version]
- Patrick, G.J.; Liu, H.; Alphonse, M.P.; Dikeman, D.A.; Youn, C.; Otterson, J.C.; Wang, Y.; Ravipati, A.; Mazhar, M.; Denny, G.; et al. Epicutaneous Staphylococcus Aureus Induces IL-36 to Enhance IgE Production and Ensuing Allergic Disease. J. Clin. Investig. 2021, 131, 143334. [Google Scholar] [CrossRef] [PubMed]
- Zea-Vera, A.F.; Estupiñan-Lopez, F.E.; Cifuentes-Burbano, J.; Vargas, M.J.; Bonelo, A. Interleukin-36 Receptor Antagonist Deficiency (DITRA) with a Novel IL36RN Homozygous Mutation c.200G > T (P.Cys67Phe) in a Young Colombian Woman. J. Clin. Immunol. 2019, 39, 261–263. [Google Scholar] [CrossRef] [PubMed]
- Tamagawa-Mineoka, R.; Okuzawa, Y.; Masuda, K.; Katoh, N. Increased Serum Levels of Interleukin 33 in Patients with Atopic Dermatitis. J. Am. Acad. Dermatol. 2014, 70, 882–888. [Google Scholar] [CrossRef] [PubMed]
- Stott, B.; Lavender, P.; Lehmann, S.; Pennino, D.; Durham, S.; Schmidt-Weber, C.B. Human IL-31 Is Induced by IL-4 and Promotes TH2-Driven Inflammation. J. Allergy Clin. Immunol. 2013, 132, 446–454.e5. [Google Scholar] [CrossRef] [PubMed]
- Tsoi, L.C.; Rodriguez, E.; Stölzl, D.; Wehkamp, U.; Sun, J.; Gerdes, S.; Sarkar, M.K.; Hübenthal, M.; Zeng, C.; Uppala, R.; et al. Progression of Acute-to-Chronic Atopic Dermatitis Is Associated with Quantitative Rather than Qualitative Changes in Cytokine Responses. J. Allergy Clin. Immunol. 2020, 145, 1406–1415. [Google Scholar] [CrossRef] [Green Version]
- Martel, B.C.; Litman, T.; Hald, A.; Norsgaard, H.; Lovato, P.; Dyring-Andersen, B.; Skov, L.; Thestrup-Pedersen, K.; Skov, S.; Skak, K.; et al. Distinct Molecular Signatures of Mild Extrinsic and Intrinsic Atopic Dermatitis. Exp. Dermatol. 2016, 25, 453–459. [Google Scholar] [CrossRef]
- Wang, W.; Yu, X.; Wu, C.; Jin, H. IL-36γ Inhibits Differentiation and Induces Inflammation of Keratinocyte via Wnt Signaling Pathway in Psoriasis. Int. J. Med. Sci. 2017, 14, 1002–1007. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.V.; Damiani, G.; Orenstein, L.A.V.; Hamzavi, I.; Jemec, G.B. Hidradenitis Suppurativa: An Update on Epidemiology, Phenotypes, Diagnosis, Pathogenesis, Comorbidities and Quality of Life. J. Eur. Acad. Dermatol. Venereol. 2021, 35, 50–61. [Google Scholar] [CrossRef]
- Sabat, R.; Jemec, G.B.E.; Matusiak, Ł.; Kimball, A.B.; Prens, E.; Wolk, K. Hidradenitis Suppurativa. Nat. Rev. Dis. Primers 2020, 6, 18. [Google Scholar] [CrossRef] [PubMed]
- Narla, S.; Azzam, M.; Townsend, S.; Vellaichamy, G.; Marzano, A.V.; Alavi, A.; Lowes, M.A.; Hamzavi, I.H. Identifying Key Components and Therapeutic Targets of the Immune System in Hidradenitis Suppurativa with an Emphasis on Neutrophils. Br. J. Dermatol. 2021, 184, 1004–1013. [Google Scholar] [CrossRef] [PubMed]
- Witte-Händel, E.; Wolk, K.; Tsaousi, A.; Irmer, M.L.; Mößner, R.; Shomroni, O.; Lingner, T.; Witte, K.; Kunkel, D.; Salinas, G.; et al. The IL-1 Pathway Is Hyperactive in Hidradenitis Suppurativa and Contributes to Skin Infiltration and Destruction. J. Investig. Dermatol. 2019, 139, 1294–1305. [Google Scholar] [CrossRef]
- Wolk, K.; Brembach, T.-C.; Šimaitė, D.; Bartnik, E.; Cucinotta, S.; Pokrywka, A.; Irmer, M.l.; Triebus, J.; Witte-Händel, E.; Salinas, G.; et al. Activity and Components of the Granulocyte Colony-Stimulating Factor Pathway in Hidradenitis Suppurativa. Br. J. Dermatol. 2021, 185, 164–176. [Google Scholar] [CrossRef] [PubMed]
- Thomi, R.; Kakeda, M.; Yawalkar, N.; Schlapbach, C.; Hunger, R.E. Increased Expression of the Interleukin-36 Cytokines in Lesions of Hidradenitis Suppurativa. J. Eur. Acad. Dermatol. Venereol. 2017, 31, 2091–2096. [Google Scholar] [CrossRef]
- Hessam, S.; Sand, M.; Gambichler, T.; Skrygan, M.; Rüddel, I.; Bechara, F.G. Interleukin-36 in Hidradenitis Suppurativa: Evidence for a Distinctive Proinflammatory Role and a Key Factor in the Development of an Inflammatory Loop. Br. J. Dermatol. 2018, 178, 761–767. [Google Scholar] [CrossRef]
- Di Caprio, R.; Balato, A.; Caiazzo, G.; Lembo, S.; Raimondo, A.; Fabbrocini, G.; Monfrecola, G. IL-36 Cytokines Are Increased in Acne and Hidradenitis Suppurativa. Arch Dermatol. Res. 2017, 309, 673–678. [Google Scholar] [CrossRef]
- Hayran, Y.; Allı, N.; Yücel, Ç.; Akdoğan, N.; Turhan, T. Serum IL-36α, IL-36β, and IL-36γ Levels in Patients with Hidradenitis Suppurativa: Association with Disease Characteristics, Smoking, Obesity, and Metabolic Syndrome. Arch Dermatol. Res. 2020, 312, 187–196. [Google Scholar] [CrossRef]
- Mattii, M.; Ayala, F.; Balato, N.; Filotico, R.; Lembo, S.; Schiattarella, M.; Patruno, C.; Marone, G.; Balato, A. The Balance between Pro- and Anti-Inflammatory Cytokines Is Crucial in Human Allergic Contact Dermatitis Pathogenesis: The Role of IL-1 Family Members. Exp. Dermatol. 2013, 22, 813–819. [Google Scholar] [CrossRef] [Green Version]
- Marzano, A.V.; Ortega-Loayza, A.G.; Heath, M.; Morse, D.; Genovese, G.; Cugno, M. Mechanisms of Inflammation in Neutrophil-Mediated Skin Diseases. Front. Immunol. 2019, 10, 1059. [Google Scholar] [CrossRef]
- Imhof, L.; Meier, B.; Frei, P.; Kamarachev, J.; Rogler, G.; Kolios, A.; Navarini, A.A.; Contassot, E.; French, L.E. Severe Sweet’s Syndrome with Elevated Cutaneous Interleukin-1β after Azathioprine Exposure: Case Report and Review of the Literature. Dermatology 2015, 230, 293–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolios, A.G.A.; Maul, J.-T.; Meier, B.; Kerl, K.; Traidl-Hoffmann, C.; Hertl, M.; Zillikens, D.; Röcken, M.; Ring, J.; Facchiano, A.; et al. Canakinumab in Adults with Steroid-Refractory Pyoderma Gangrenosum. Br. J. Dermatol. 2015, 173, 1216–1223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meier-Schiesser, B.; Feldmeyer, L.; Jankovic, D.; Mellett, M.; Satoh, T.K.; Yerly, D.; Navarini, A.; Abe, R.; Yawalkar, N.; Chung, W.-H.; et al. Culprit Drugs Induce Specific IL-36 Overexpression in Acute Generalized Exanthematous Pustulosis. J. Investig. Dermatol. 2019, 139, 848–858. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.-M.; Jin, H.-Z. Biologics in the Treatment of Pustular Psoriasis. Expert Opin. Drug Saf. 2020, 19, 969–980. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, L.; Fiocco, Z.; Satoh, T.K.; Peris, K.; French, L.E. Therapeutic Potential of Targeting Interleukin-1 Family Cytokines in Chronic Inflammatory Skin Diseases. Br. J. Dermatol. 2022, 186, 925–941. [Google Scholar] [CrossRef]
- Cro, S.; Cornelius, V.R.; Pink, A.E.; Wilson, R.; Pushpa-Rajah, A.; Patel, P.; Abdul-Wahab, A.; August, S.; Azad, J.; Becher, G.; et al. Anakinra for Palmoplantar Pustulosis: Results from a Randomized, Double-Blind, Multicentre, Two-Staged, Adaptive Placebo-Controlled Trial (APRICOT). Br. J. Dermatol. 2021, 186, 245–256. [Google Scholar] [CrossRef]
- Leslie, K.S.; Tripathi, S.V.; Nguyen, T.V.; Pauli, M.; Rosenblum, M.D. An Open-Label Study of Anakinra for the Treatment of Moderate to Severe Hidradenitis Suppurativa. J. Am. Acad. Dermatol. 2014, 70, 243–251. [Google Scholar] [CrossRef]
- Tzanetakou, V.; Kanni, T.; Giatrakou, S.; Katoulis, A.; Papadavid, E.; Netea, M.G.; Dinarello, C.A.; van der Meer, J.W.M.; Rigopoulos, D.; Giamarellos-Bourboulis, E.J. Safety and Efficacy of Anakinra in Severe Hidradenitis Suppurativa: A Randomized Clinical Trial. JAMA Dermatol. 2016, 152, 52–59. [Google Scholar] [CrossRef]
- André, R.; Marescassier, H.; Gabay, C.; Pittet, B.; Laffitte, E. Long-Term Therapy with Anakinra in Hidradenitis Suppurativa in Three Patients. Int. J. Dermatol. 2019, 58, e208–e209. [Google Scholar] [CrossRef]
- Edward Cowen, M.D. A Phase 2 Study of Anakinra in Inflammatory Pustular Dermatoses: Evaluation of Therapeutic Efficacy and Validation of Pathogenic Mechanisms. 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT01794117 (accessed on 23 June 2022).
- Gottlieb, A.; Natsis, N.E.; Kerdel, F.; Forman, S.; Gonzalez, E.; Jimenez, G.; Hernandez, L.; Kaffenberger, J.; Guido, G.; Lucas, K.; et al. A Phase II Open-Label Study of Bermekimab in Patients with Hidradenitis Suppurativa Shows Resolution of Inflammatory Lesions and Pain. J. Investig. Dermatol. 2020, 140, 1538–1545.e2. [Google Scholar] [CrossRef]
- Kanni, T.; Argyropoulou, M.; Spyridopoulos, T.; Pistiki, A.; Stecher, M.; Dinarello, C.A.; Simard, J.; Giamarellos-Bourboulis, E.J. MABp1 Targeting IL-1α for Moderate to Severe Hidradenitis Suppurativa Not Eligible for Adalimumab: A Randomized Study. J. Investig. Dermatol. 2018, 138, 795–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanni, T.; Argyropoulou, M.; Dinarello, C.A.; Simard, J.; Giamarellos-Bourboulis, E.J. MABp1 Targeting Interleukin-1α in Hidradenitis Suppurativa Ineligible for Adalimumab Treatment: Results of the Open-Label Extension Period. Clin. Exp. Dermatol. 2021, 46, 162–163. [Google Scholar] [CrossRef] [PubMed]
- Janssen Research & Development, LLC. A Phase 2a/2b, Multicenter, Randomized, Placebo and Active Comparator-Controlled, Double-Blind, Dose-Ranging Study to Evaluate the Safety and Efficacy of Bermekimab (JNJ-77474462) for the Treatment of Subjects with Moderate to Severe Hidradenitis Suppurativa. 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT04988308 (accessed on 23 June 2022).
- Carrasco, D.; Stecher, M.; Lefebvre, G.C.; Logan, A.C.; Moy, R. An Open Label, Phase 2 Study of MABp1 Monotherapy for the Treatment of Acne Vulgaris and Psychiatric Comorbidity. J. Drugs Dermatol. 2015, 14, 560–564. [Google Scholar]
- Coleman, K.M.; Gudjonsson, J.E.; Stecher, M. Open-Label Trial of MABp1, a True Human Monoclonal Antibody Targeting Interleukin 1α, for the Treatment of Psoriasis. JAMA Dermatol. 2015, 151, 555–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janssen Research & Development, LLC. A Phase 2a, Multicenter, Randomized, Placebo-Controlled, Double-Blind, Interventional Study to Assess the Efficacy, Safety, Pharmacokinetics, and Immunogenicity of Multiple IV Doses of Bermekimab for the Treatment of Adult Participants with Moderate-to-Severe Atopic Dermatitis. 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT04021862 (accessed on 23 June 2022).
- Janssen Research & Development, LLC. A Phase 2b, Multicenter, Randomized, Placebo- and Active-Comparator-Controlled, Double-Blind Study to Evaluate the Safety and Efficacy of Bermekimab (JNJ-77474462) for the Treatment of Participants with Moderate to Severe Atopic Dermatitis. 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT04791319 (accessed on 23 June 2022).
- Sun, N.Z.; Ro, T.; Jolly, P.; Sayed, C.J. Non-Response to Interleukin-1 Antagonist Canakinumab in Two Patients with Refractory Pyoderma Gangrenosum and Hidradenitis Suppurativa. J. Clin. Aesthet. Dermatol. 2017, 10, 36–38. [Google Scholar]
- Houriet, C.; Seyed Jafari, S.M.; Thomi, R.; Schlapbach, C.; Borradori, L.; Yawalkar, N.; Hunger, R.E. Canakinumab for Severe Hidradenitis Suppurativa: Preliminary Experience in 2 Cases. JAMA Dermatol. 2017, 153, 1195–1197. [Google Scholar] [CrossRef]
- Skendros, P.; Papagoras, C.; Lefaki, I.; Giatromanolaki, A.; Kotsianidis, I.; Speletas, M.; Bocly, V.; Theodorou, I.; Dalla, V.; Ritis, K. Successful Response in a Case of Severe Pustular Psoriasis after Interleukin-1β Inhibition. Br. J. Dermatol. 2017, 176, 212–215. [Google Scholar] [CrossRef]
- Mansouri, B.; Kivelevitch, D.; Campa, M.; Menter, A. Palmoplantar Pustular Psoriasis Unresponsive to the Interleukin-1β Antagonist Canakinumab. Clin. Exp. Dermatol. 2016, 41, 324–326. [Google Scholar] [CrossRef]
- Gabay, C.; Fautrel, B.; Rech, J.; Spertini, F.; Feist, E.; Kötter, I.; Hachulla, E.; Morel, J.; Schaeverbeke, T.; Hamidou, M.A.; et al. Open-Label, Multicentre, Dose-Escalating Phase II Clinical Trial on the Safety and Efficacy of Tadekinig Alfa (IL-18BP) in Adult-Onset Still’s Disease. Ann. Rheum. Dis. 2018, 77, 840–847. [Google Scholar] [CrossRef]
- Chen, Y.-L.; Gutowska-Owsiak, D.; Hardman, C.S.; Westmoreland, M.; MacKenzie, T.; Cifuentes, L.; Waithe, D.; Lloyd-Lavery, A.; Marquette, A.; Londei, M.; et al. Proof-of-Concept Clinical Trial of Etokimab Shows a Key Role for IL-33 in Atopic Dermatitis Pathogenesis. Sci. Transl. Med. 2019, 11, eaax2945. [Google Scholar] [CrossRef]
- AnaptysBio, Inc. A Phase 2, Double-Blind, Placebo-Controlled, Parallel Group, Multiple Dose Study to Investigate Etokimab (ANB020) in Adult Subjects with Chronic Rhinosinusitis with Nasal Polyposis. 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT03614923 (accessed on 23 June 2022).
- Regeneron Pharmaceuticals. A Randomized, Double-Blind, Placebo-Controlled, Phase 2a Study to Assess the Efficacy and Safety of REGN3500 Monotherapy and Combination of REGN3500 Plus Dupilumab in Adult Patients with Moderate-to-Severe Atopic Dermatitis. 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT03736967 (accessed on 23 June 2022).
- Regeneron Pharmaceuticals. A Phase 2b, Randomized, Double-Blind, Placebo-Controlled, Parallel-Group, Dose-Ranging Study Investigating the Efficacy, Safety, and Pharmacokinetic Profiles of REGN3500 Administered to Adult Patients with Moderate-to- Severe Atopic Dermatitis. 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT03738423 (accessed on 23 June 2022).
- Pfizer. A Phase 1, Randomized, Double-Blind, Third-Party Open, Placebo-Controlled, Dose Escalating Study to Evaluate the Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of Single and/or Multiple Intravenous and/or Subcutaneous Doses of Pf-06817024 in Healthy Subjects Who May Be Mildly Atopic, Subjects with Chronic Rhinosinusitis with Nasal Polyps, and Subjects with Moderate-Severe Atopic Dermatitis. 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT01989143 (accessed on 23 June 2022).
- Nnane, I.; Frederick, B.; Yao, Z.; Raible, D.; Shu, C.; Badorrek, P.; van den Boer, M.; Branigan, P.; Duffy, K.; Baribaud, F.; et al. The First-in-Human Study of CNTO 7160, an Anti-Interleukin-33 Receptor Monoclonal Antibody, in Healthy Subjects and Patients with Asthma or Atopic Dermatitis. Br. J. Clin. Pharm. 2020, 86, 2507–2518. [Google Scholar] [CrossRef] [PubMed]
- Bachelez, H.; Choon, S.-E.; Marrakchi, S.; Burden, A.D.; Tsai, T.-F.; Morita, A.; Turki, H.; Hall, D.B.; Shear, M.; Baum, P.; et al. Inhibition of the Interleukin-36 Pathway for the Treatment of Generalized Pustular Psoriasis. N. Engl. J. Med. 2019, 380, 981–983. [Google Scholar] [CrossRef] [PubMed]
- Bachelez, H.; Choon, S.-E.; Marrakchi, S.; Burden, A.D.; Tsai, T.-F.; Morita, A.; Navarini, A.A.; Zheng, M.; Xu, J.; Turki, H.; et al. Trial of Spesolimab for Generalized Pustular Psoriasis. N. Engl. J. Med. 2021, 385, 2431–2440. [Google Scholar] [CrossRef] [PubMed]
- Mrowietz, U.; Burden, A.D.; Pinter, A.; Reich, K.; Schäkel, K.; Baum, P.; Datsenko, Y.; Deng, H.; Padula, S.J.; Thoma, C.; et al. Spesolimab, an Anti-Interleukin-36 Receptor Antibody, in Patients with Palmoplantar Pustulosis: Results of a Phase Iia, Multicenter, Double-Blind, Randomized, Placebo-Controlled Pilot Study. Dermatol. Ther. 2021, 11, 571–585. [Google Scholar] [CrossRef]
- Boehringer Ingelheim. Randomized, Double-Blind, Placebo-Controlled, Study of Spesolimab in Patients with Moderate to Severe Hidradenitis Suppurativa. 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT04762277 (accessed on 4 July 2022).
- Boehringer Ingelheim. An Open-Label, Long-Term Extension Trial of Spesolimab Treatment in Adult Patients with Hidradenitis Suppurativa (HS). 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT04876391 (accessed on 4 July 2022).
- Boehringer Ingelheim. Multi-Centre, Open-Label, Expanded Access Trial of Spesolimab i.v. in Patients with Generalized Pustular Psoriasis (GPP) Presenting with a Flare. 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT05200247 (accessed on 4 July 2022).
- Boehringer Ingelheim. Multi-Center, Double-Blind, Randomized, Placebo-Controlled, Phase IIa Trial to Evaluate Spesolimab (BI 655130) Efficacy in Patients with Fibrostenotic Crohn’s Disease. 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT05013385 (accessed on 4 July 2022).
- Boehringer Ingelheim. An Open Label, Long Term Safety Trial of Spesolimab Treatment in Patients with Fistulising Crohn’s Disease Who Have Completed Previous Spesolimab Trials. 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT04362254 (accessed on 4 July 2022).
- Boehringer Ingelheim. A Phase II/III Randomized, Double-Blind, Placebo-Controlled, Multicenter Study to Evaluate the Safety and Efficacy of BI655130 (SPESOLIMAB) Induction Therapy in Patients with Moderate-to-Severely Active Ulcerative Colitis Who Have Failed Previous Biologics Therapy. 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT03482635 (accessed on 4 July 2022).
- Boehringer Ingelheim. An Open Label, Long Term Safety Trial of BI 65513 (SPESOLIMAB) Treatment in Patients with Moderate to Severely Active Ulcerative Colitis Who Have Completed Previous BI 655130 Trials. 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT03648541 (accessed on 4 July 2022).
- Boehringer Ingelheim. EffisayilTM 2: Multi-Center, Randomized, Parallel Group, Double Blind, Placebo Controlled, Phase IIb Dose-Finding Study to Evaluate Efficacy and Safety of BI 655130 (Spesolimab) Compared to Placebo in Preventing Generalized Pustular Psoriasis (GPP) Flares in Patients with History of GPP. 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT04399837 (accessed on 4 July 2022).
- Boehringer Ingelheim. An Open-Label, Long Term Safety Trial of Spesolimab Treatment in Patients with Palmoplantar Pustulosis (PPP) Who Have Completed Previous BI Spesolimab Trials. 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT04493424 (accessed on 4 July 2022).
- AnaptysBio, Inc. AnaptysBio Reports Imsidolimab POPLAR Phase 2 Clinical Trial in Moderate-to-Severe Palmoplantar Pustulosis (PPP) Did Not Meet Primary Endpoint. Available online: https://ir.anaptysbio.com/news-releases/news-release-details/anaptysbio-reports-imsidolimab-poplar-phase-2-clinical-trial/ (accessed on 17 January 2022).
- AnaptysBio, Inc. A Phase 2, Randomized, Placebo-Controlled, Double-Blind, Multiple Dose Study to Evaluate the Efficacy and Safety of ANB019 in Subjects with Palmoplantar Pustulosis. 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT03633396 (accessed on 4 July 2022).
- AnaptysBio, Inc. A Single Arm Multiple Dose Study to Assess the Efficacy and Safety of ANB019 in Subjects with Generalized Pustular Psoriasis. 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT03619902 (accessed on 4 July 2022).
- AnaptysBio, Inc. A Phase 3, Long-Term Extension Study to Evaluate the Safety and Efficacy of Imsidolimab (ANB019) in the Treatment of Adult Subjects with Generalized Pustular Psoriasis. 2022. Available online: https://www.clinicaltrials.gov/ct2/show/NCT05352893 (accessed on 4 July 2022).
- AnaptysBio, Inc. A Phase 3, Randomized, Double-Blind, Placebo-Controlled Study to Evaluate the Efficacy and Safety of Imsidolimab (ANB019) in the Treatment of Adult Subjects with Generalized Pustular Psoriasis. 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT05366855 (accessed on 4 July 2022).
- AnaptysBio, Inc. A Phase 2, Randomized, Double-Blind, Placebo Controlled Study to Evaluate the Efficacy and Safety of Imsidolimab (ANB019) in the Treatment of Subjects with Hidradenitis Suppurativa. 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT04856930 (accessed on 4 July 2022).
- Klementiev, B.; Li, S.; Korshunova, I.; Dmytriyeva, O.; Pankratova, S.; Walmod, P.S.; Kjær, L.K.; Dahllöf, M.S.; Lundh, M.; Christensen, D.P.; et al. Anti-Inflammatory Properties of a Novel Peptide Interleukin 1 Receptor Antagonist. J. Neuroinflamm. 2014, 11, 27. [Google Scholar] [CrossRef] [Green Version]
- Todorović, V.; Su, Z.; Putman, C.B.; Kakavas, S.J.; Salte, K.M.; McDonald, H.A.; Wetter, J.B.; Paulsboe, S.E.; Sun, Q.; Gerstein, C.E.; et al. Small Molecule IL-36γ Antagonist as a Novel Therapeutic Approach for Plaque Psoriasis. Sci. Rep. 2019, 9, 9089. [Google Scholar] [CrossRef]
- Cro, S.; Smith, C.H. Response to: “Anakinra for Palmoplantar Pustulosis: Results from a Randomized, Double-Blind, Multicentre, Two-Staged, Adaptive Placebo-Controlled Trial (APRICOT)”: Reply from the Authors. Br. J. Dermatol. 2022, 186, 909–910. [Google Scholar] [CrossRef]


{kind=link}
| Cytokines | Receptors (Other Names) | Co-Receptors (Other Names) | Function | |
|---|---|---|---|---|
| IL-1 | IL-1α | IL-1R1 | IL-1RAcP (IL1-R3) | Pro-inflammatory |
| IL-1β | IL-1R2 | |||
| IL-1Ra | IL-1R1 | N/A | Antagonist | |
| IL-18 | IL-18Rα (IL1-R5) | IL-18Rβ (IL-18RAcP or IL1R7) | Pro-inflammatory | |
| IL-33 | ST2 (IL-33R or IL1-R4) | IL-1RAcP | Pro-inflammatory Th2 responses | |
| IL-36 | IL-36α | IL-36R (IL-1Rrp2 or IL-R6) | IL-1RAcP (IL1-R3) | Pro-inflammatory |
| IL-36β | ||||
| IL-36γ | ||||
| IL-36Ra | IL-36R (IL-1Rrp2 or IL-R6) | N/A | Antagonist | |
| IL-37 | IL-18Rα (IL1-R5) | IL-1R8 (SIGIRR or TIR8) | Antagonist | |
| IL-38 | IL-36R (IL-1Rrp2 or IL1-R6) IL-R9 | IL1RAPL1 (TIGIRR-2) IL1RAPL2 (TIGIRR-1) | Antagonist/anti-inflammatory | |
| Drug (Mechanism of Action) | Disease | Clinical Trial Number |
|---|---|---|
| Spesolimab (Effiyasil™) (BI655130) Humanized monoclonal antibody targeting IL-36R | GPP | NCT02978690 |
| PPP | NCT03100903 | |
| PPP | NCT03135548 | |
| Ulcerative colitis | NCT03482635 | |
| GPP | NCT03782792 | |
| PPP | NCT04015518 | |
| Crohn’s disease | NCT04362254 | |
| GPP | NCT04399837 | |
| PPP | NCT04493424 | |
| HS | NCT04762277 | |
| HS | NCT04876391 | |
| Crohn’s disease | NCT05013385 | |
| GPP | NCT05200247 | |
| GPP | NCT05239039 | |
| Imsidolimab (ANB019) Humanized monoclonal antibody targeting IL-36R | PPP | NCT03633396 |
| Acne vulgaris | NCT04856917 | |
| HS | NCT04856930 | |
| GPP | NCT05352893 | |
| GPP | NCT05366855 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iznardo, H.; Puig, L. IL-1 Family Cytokines in Inflammatory Dermatoses: Pathogenetic Role and Potential Therapeutic Implications. Int. J. Mol. Sci. 2022, 23, 9479. https://doi.org/10.3390/ijms23169479
Iznardo H, Puig L. IL-1 Family Cytokines in Inflammatory Dermatoses: Pathogenetic Role and Potential Therapeutic Implications. International Journal of Molecular Sciences. 2022; 23(16):9479. https://doi.org/10.3390/ijms23169479
Chicago/Turabian StyleIznardo, Helena, and Luís Puig. 2022. "IL-1 Family Cytokines in Inflammatory Dermatoses: Pathogenetic Role and Potential Therapeutic Implications" International Journal of Molecular Sciences 23, no. 16: 9479. https://doi.org/10.3390/ijms23169479
APA StyleIznardo, H., & Puig, L. (2022). IL-1 Family Cytokines in Inflammatory Dermatoses: Pathogenetic Role and Potential Therapeutic Implications. International Journal of Molecular Sciences, 23(16), 9479. https://doi.org/10.3390/ijms23169479