
Regulation of Cyclooxygenase-2 Expression in Human T Cells by Glucocorticoid Receptor-Mediated Transrepression of Nuclear Factor of Activated T Cells


 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
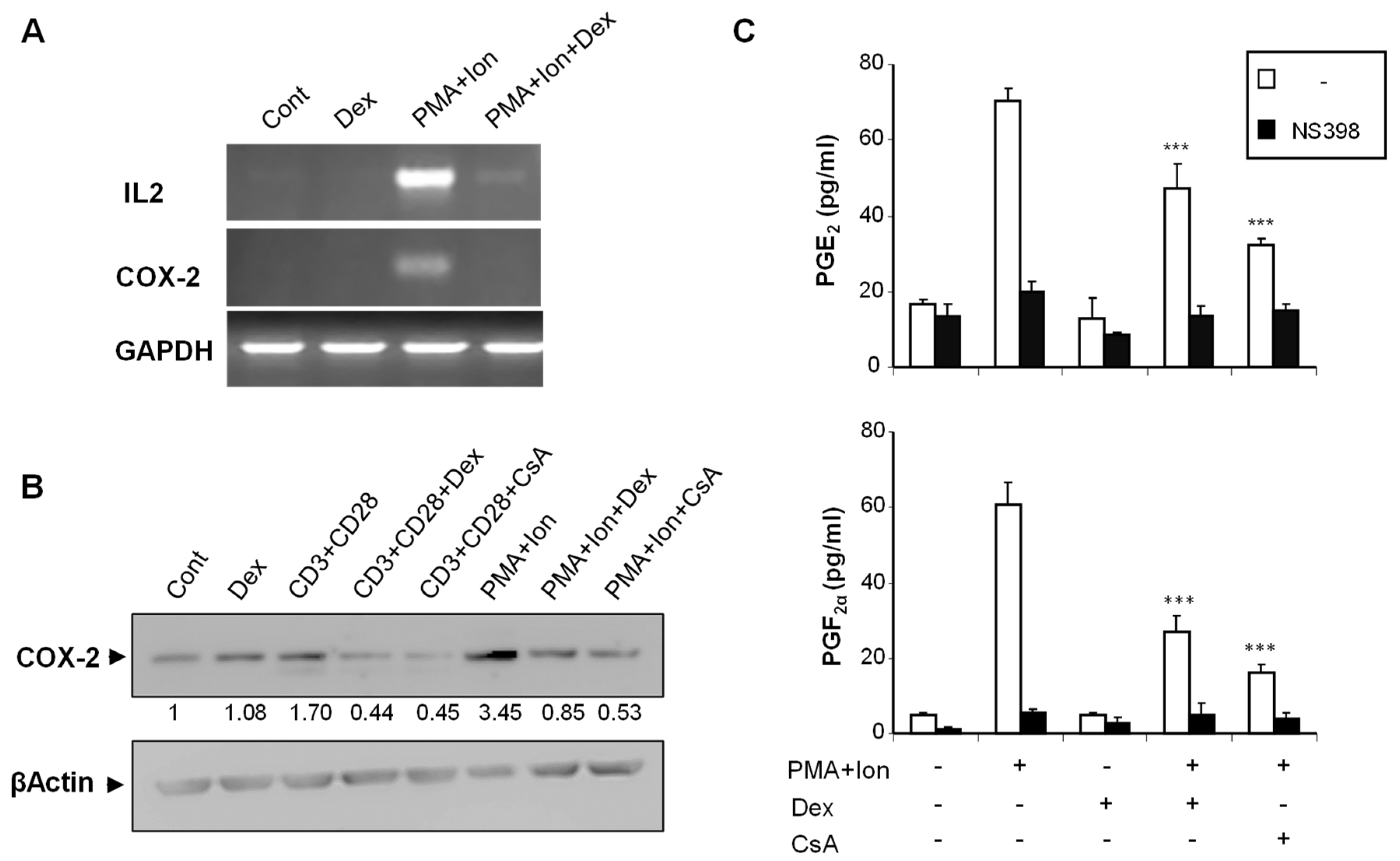
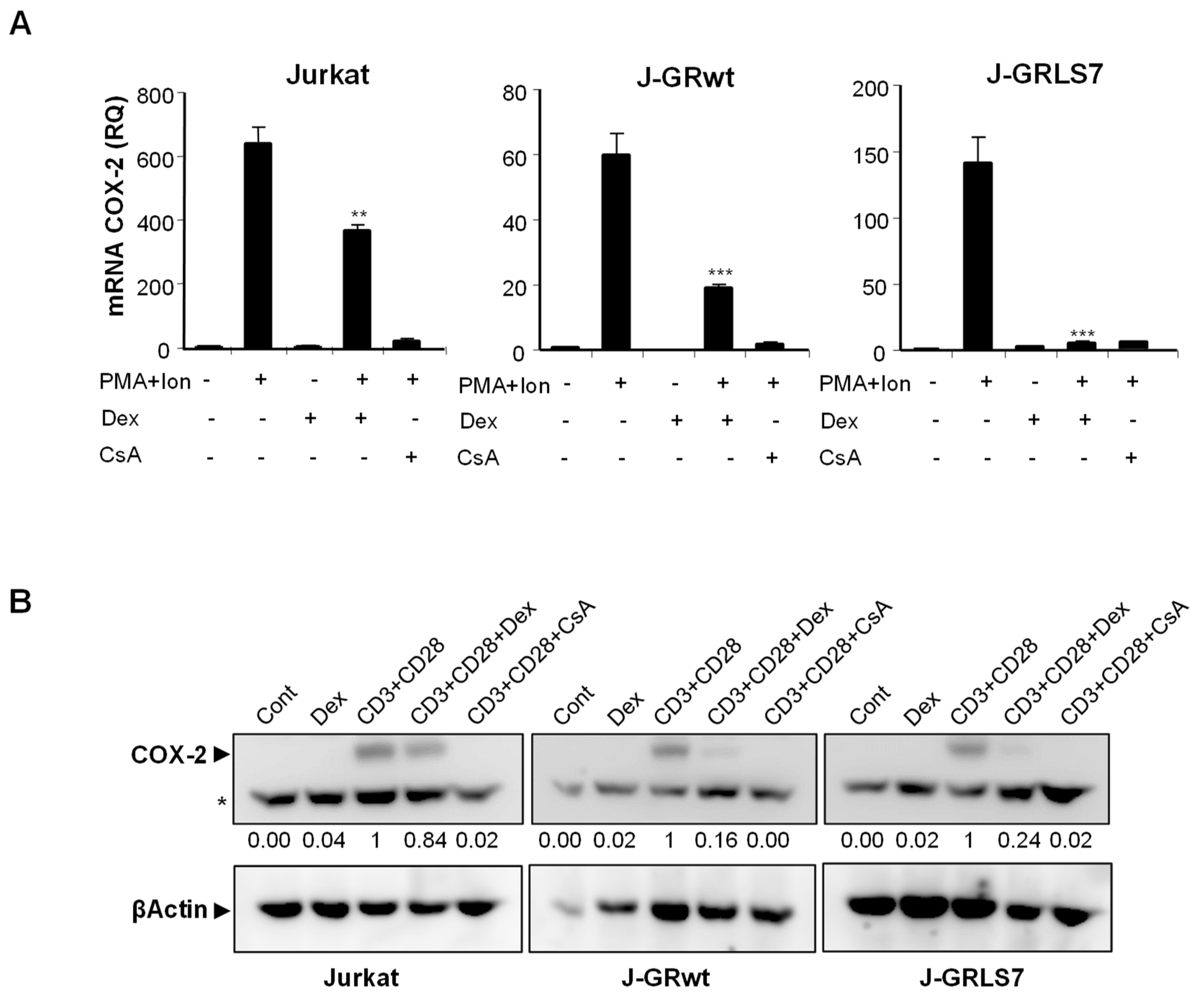
2.1. Inhibition of COX-2 Expression by Glucocorticoids
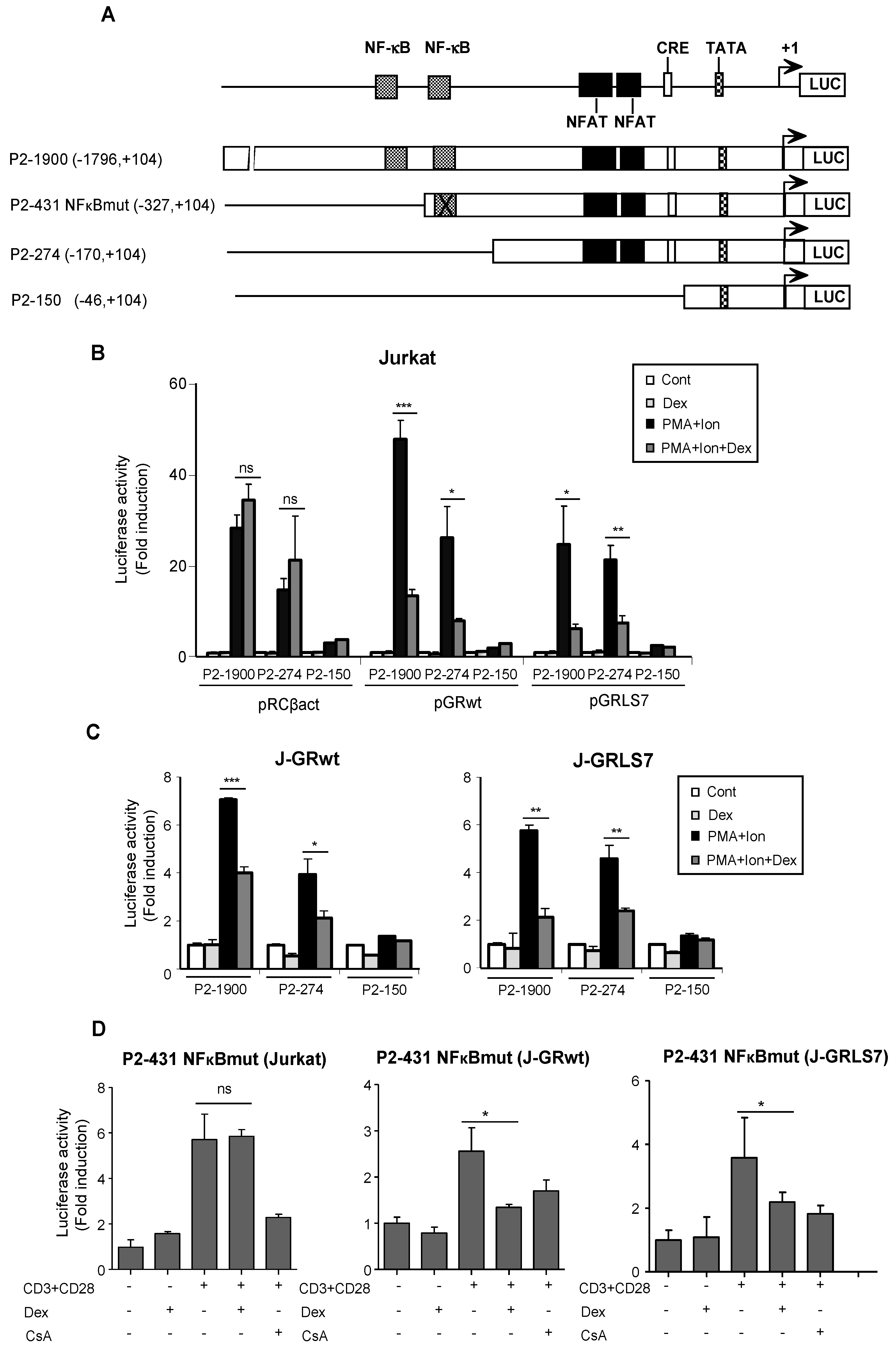
2.2. Glucocorticoids Inhibit COX-2 Promoter Activity
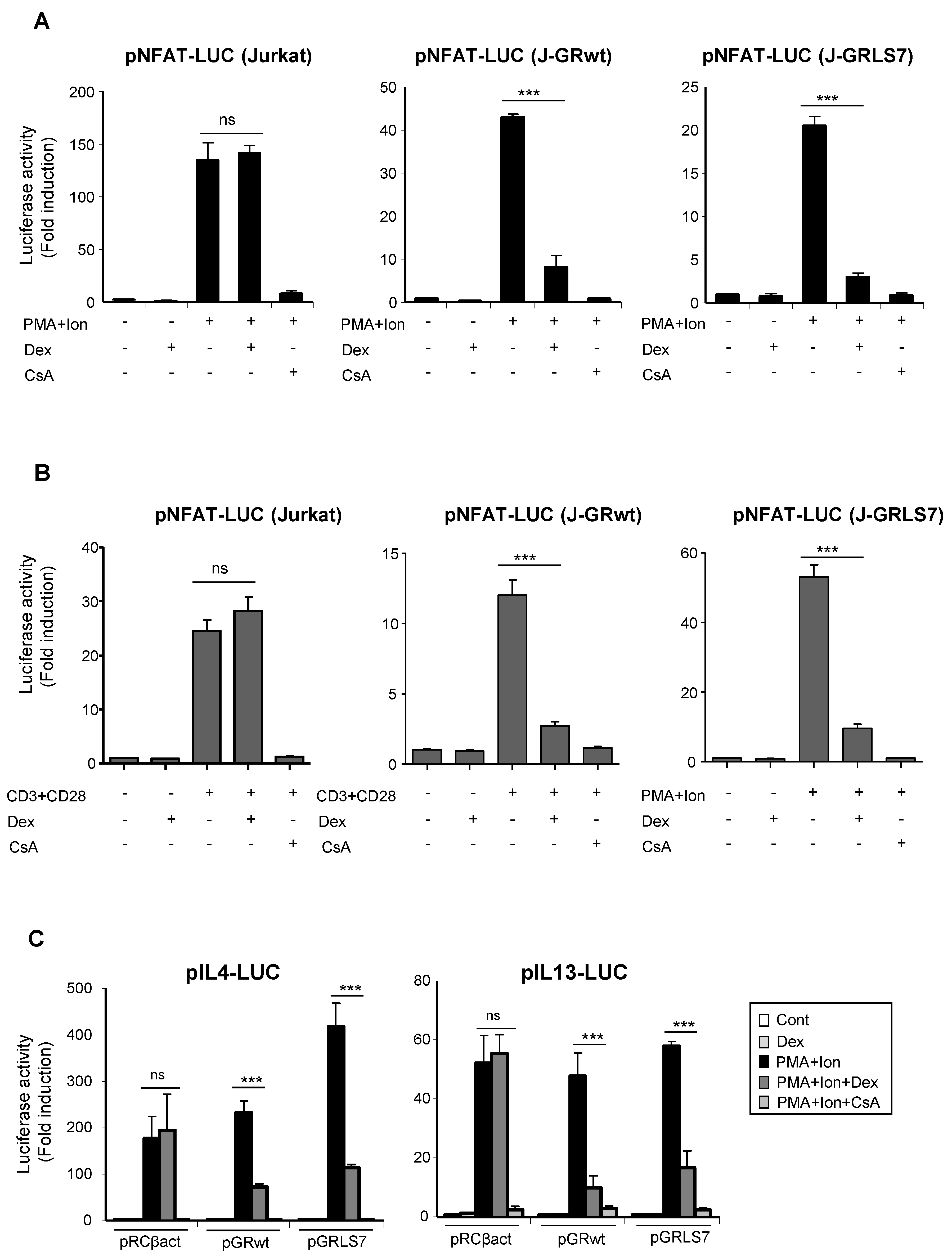
2.3. Inhibition of NFAT -Mediated Transactivation by Glucocorticoids
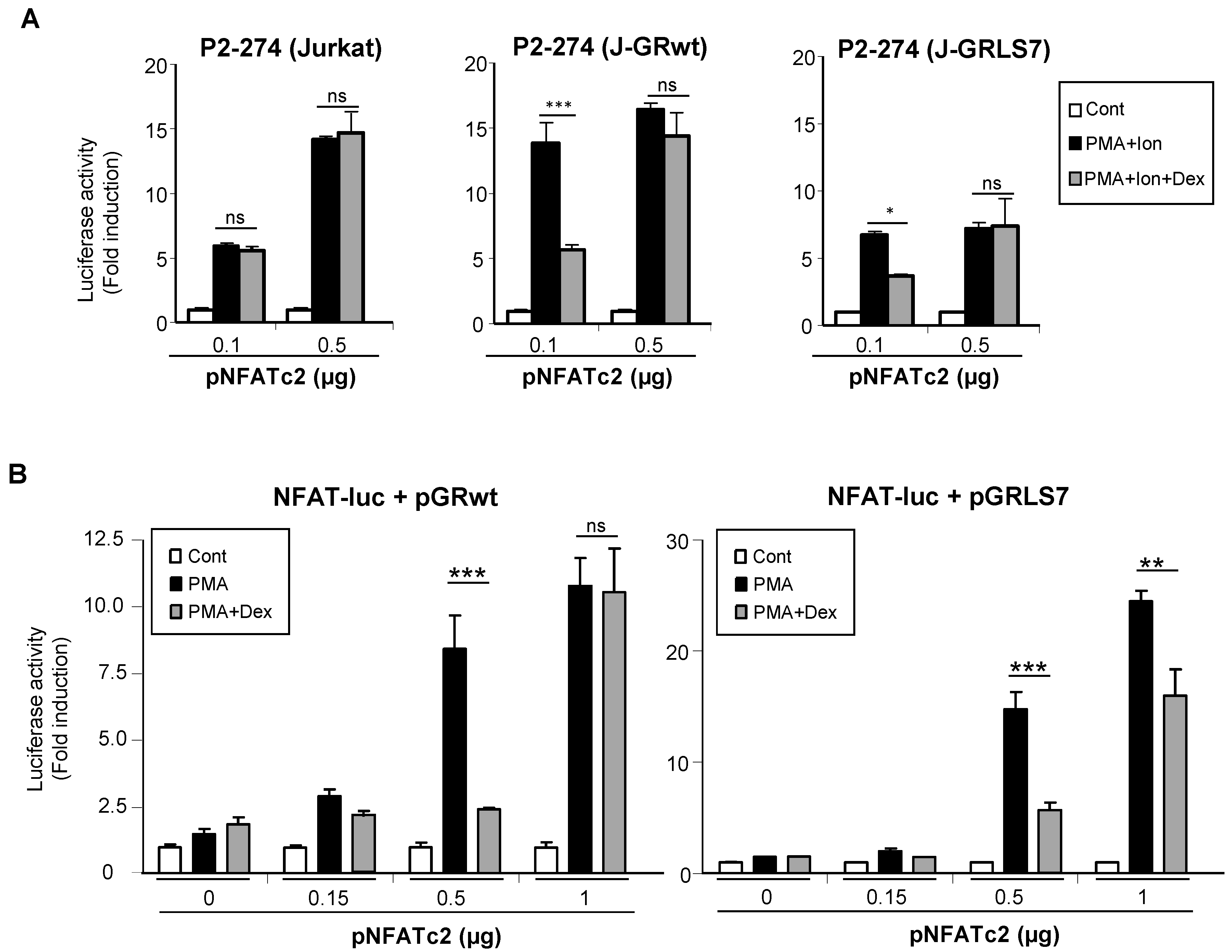
2.4. Overexpression of NFAT Restores Promoter Activity in the Presence of Dex
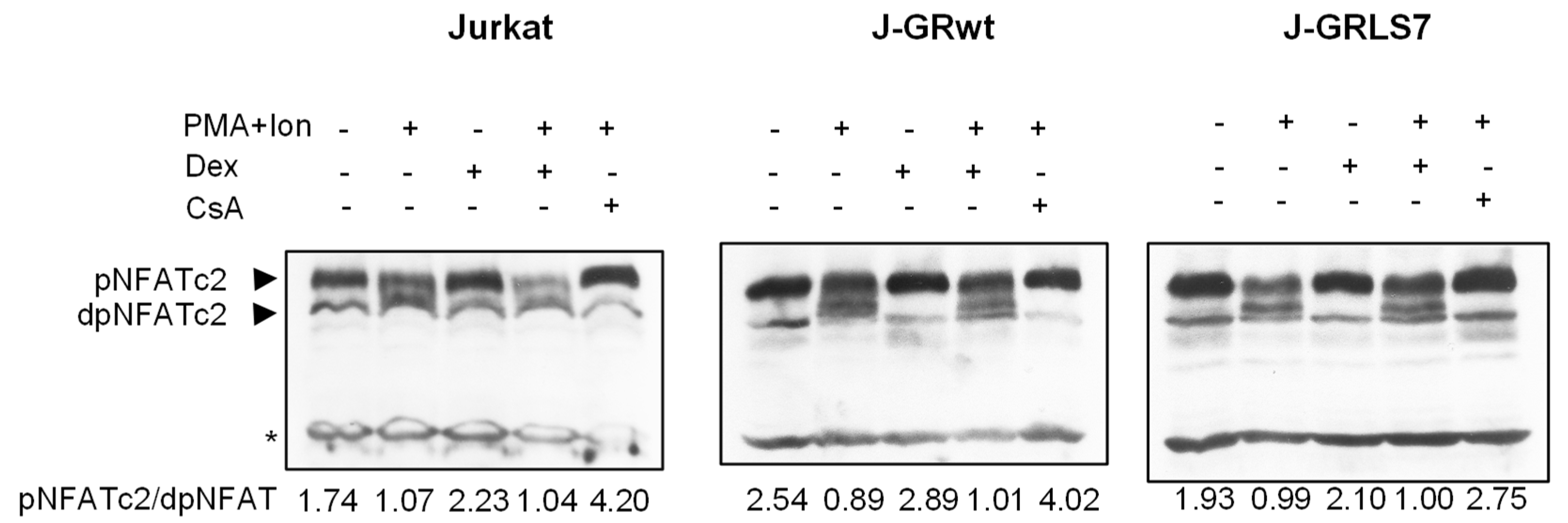
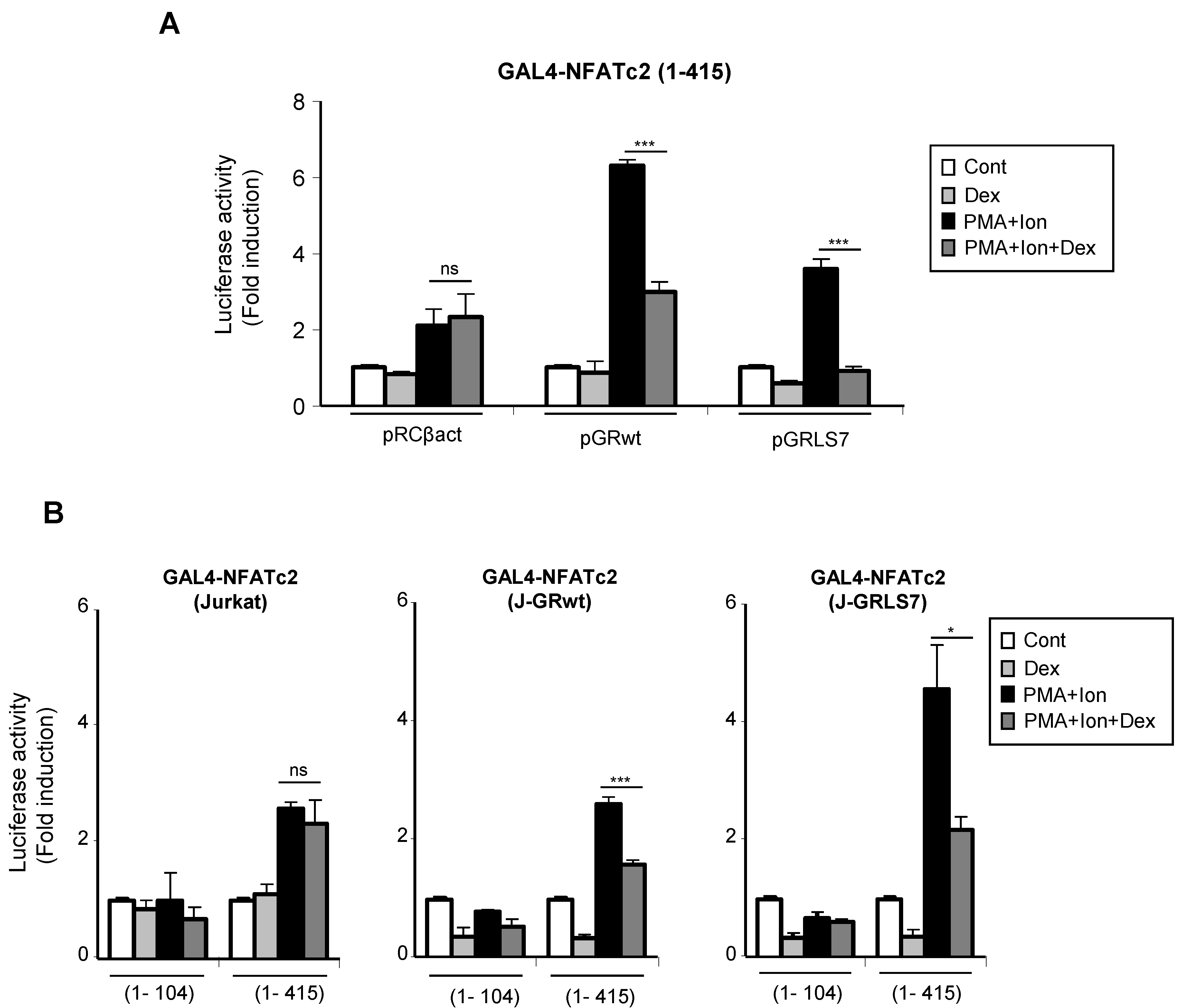
2.5. Effects of Glucocorticoids on NFAT Transactivation Activity
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Plasmid Constructs
4.3. mRNA Analysis
4.4. Immunoblot Analysis
4.5. Transfection and Luciferase Assays
4.6. Prostaglandin Measurement
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ricciotti, E.; FitzGerald, G.A. Prostaglandins and inflammation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 986–1000. [Google Scholar] [CrossRef]
- Smith, W.L.; DeWitt, D.L.; Garavito, R.M. Cyclooxygenases: Structural, cellular, and molecular biology. Annu. Rev. Biochem. 2000, 69, 145–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smyth, E.M.; Grosser, T.; Wang, M.; Yu, Y.; FitzGerald, G.A. Prostanoids in health and disease. J. Lipid Res. 2009, 50, S423–S428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeWitt, D.L.; Meade, E.A.; Smith, W.L. PGH synthase isoenzyme selectivity: The potential for safer nonsteroidal antiinflammatory drugs. Am. J. Med. 1993, 95, 40S–44S. [Google Scholar] [CrossRef]
- Mitchell, J.A.; Akarasereenont, P.; Thiemermann, C.; Flower, R.J.; Vane, J.R. Selectivity of nonsteroidal antiinflammatory drugs as inhibitors of constitutive and inducible cyclooxygenase. Proc. Natl. Acad. Sci. USA 1993, 90, 11693–11697. [Google Scholar] [CrossRef] [Green Version]
- Patrono, C.; Rocca, B. Nonsteroidal antiinflammatory drugs: Past, present and future. Pharmacol. Res. 2009, 59, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Munoz, M.D.; Osma-Garcia, I.C.; Cacheiro-Llaguno, C.; Fresno, M.; Iniguez, M.A. Coordinated up-regulation of cyclooxygenase-2 and microsomal prostaglandin E synthase 1 transcription by nuclear factor kappa B and early growth response-1 in macrophages. Cell. Signal. 2010, 22, 1427–1436. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Munoz, M.D.; Osma-Garcia, I.C.; Fresno, M.; Iniguez, M.A. Involvement of PGE2 and the cAMP signalling pathway in the up-regulation of COX-2 and mPGES-1 expression in LPS-activated macrophages. Biochem. J. 2012, 443, 451–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, H.; Nanayama, T.; Hara, S.; Yokoyama, C.; Tanabe, T. The cyclic AMP response element plays an essential role in the expression of the human prostaglandin-endoperoxide synthase 2 gene in differentiated U937 monocytic cells. FEBS Lett. 1994, 350, 51–54. [Google Scholar] [CrossRef] [Green Version]
- Nie, M.; Pang, L.; Inoue, H.; Knox, A.J. Transcriptional regulation of cyclooxygenase 2 by bradykinin and interleukin-1beta in human airway smooth muscle cells: Involvement of different promoter elements, transcription factors, and histone h4 acetylation. Mol. Cell. Biol. 2003, 23, 9233–9244. [Google Scholar] [CrossRef]
- Tanabe, T.; Tohnai, N. Cyclooxygenase isozymes and their gene structures and expression. Prostaglandins Other Lipid Mediat. 2002, 68–69, 95–114. [Google Scholar] [CrossRef]
- Iniguez, M.A.; Punzon, C.; Fresno, M. Induction of cyclooxygenase-2 on activated T lymphocytes: Regulation of T cell activation by cyclooxygenase-2 inhibitors. J. Immunol. 1999, 163, 111–119. [Google Scholar] [PubMed]
- Iniguez, M.A.; Martinez-Martinez, S.; Punzon, C.; Redondo, J.M.; Fresno, M. An essential role of the nuclear factor of activated T cells in the regulation of the expression of the cyclooxygenase-2 gene in human T lymphocytes. J. Biol. Chem. 2000, 275, 23627–23635. [Google Scholar] [CrossRef] [Green Version]
- Abraham, F.; Sacerdoti, F.; De Leon, R.; Gentile, T.; Canellada, A. Angiotensin II activates the calcineurin/NFAT signaling pathway and induces cyclooxygenase-2 expression in rat endometrial stromal cells. PLoS ONE 2012, 7, e37750. [Google Scholar] [CrossRef] [Green Version]
- Corral, R.S.; Iniguez, M.A.; Duque, J.; Lopez-Perez, R.; Fresno, M. Bombesin induces cyclooxygenase-2 expression through the activation of the nuclear factor of activated T cells and enhances cell migration in Caco-2 colon carcinoma cells. Oncogene 2007, 26, 958–969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duque, J.; Fresno, M.; Iniguez, M.A. Expression and function of the nuclear factor of activated T cells in colon carcinoma cells: Involvement in the regulation of cyclooxygenase-2. J. Biol. Chem. 2005, 280, 8686–8693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flockhart, R.J.; Diffey, B.L.; Farr, P.M.; Lloyd, J.; Reynolds, N.J. NFAT regulates induction of COX-2 and apoptosis of keratinocytes in response to ultraviolet radiation exposure. FASEB J. 2008, 22, 4218–4227. [Google Scholar] [CrossRef]
- Hernandez, G.L.; Volpert, O.V.; Iniguez, M.A.; Lorenzo, E.; Martinez-Martinez, S.; Grau, R.; Fresno, M.; Redondo, J.M. Selective inhibition of vascular endothelial growth factor-mediated angiogenesis by cyclosporin A: Roles of the nuclear factor of activated T cells and cyclooxygenase 2. J. Exp. Med. 2001, 193, 607–620. [Google Scholar] [CrossRef]
- Sugimoto, T.; Haneda, M.; Sawano, H.; Isshiki, K.; Maeda, S.; Koya, D.; Inoki, K.; Yasuda, H.; Kashiwagi, A.; Kikkawa, R. Endothelin-1 induces cyclooxygenase-2 expression via nuclear factor of activated T-cell transcription factor in glomerular mesangial cells. J. Am. Soc. Nephrol. 2001, 12, 1359–1368. [Google Scholar] [CrossRef]
- Reichardt, S.D.; Amouret, A.; Muzzi, C.; Vettorazzi, S.; Tuckermann, J.P.; Luhder, F.; Reichardt, H.M. The Role of Glucocorticoids in Inflammatory Diseases. Cells 2021, 10, 2921. [Google Scholar] [CrossRef]
- Liberman, A.C.; Druker, J.; Perone, M.J.; Arzt, E. Glucocorticoids in the regulation of transcription factors that control cytokine synthesis. Cytokine Growth Factor Rev. 2007, 18, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Strickland, B.A.; Ansari, S.A.; Dantoft, W.; Uhlenhaut, N.H. How to tame your genes: Mechanisms of inflammatory gene repression by glucocorticoids. FEBS Lett. 2022, 596, 2596–2616. [Google Scholar] [CrossRef] [PubMed]
- Ratman, D.; Vanden Berghe, W.; Dejager, L.; Libert, C.; Tavernier, J.; Beck, I.M.; De Bosscher, K. How glucocorticoid receptors modulate the activity of other transcription factors: A scope beyond tethering. Mol. Cell. Endocrinol. 2013, 380, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Cain, D.W.; Cidlowski, J.A. Immune regulation by glucocorticoids. Nat. Rev. Immunol. 2017, 17, 233–247. [Google Scholar] [CrossRef]
- Yamamoto, K.R. Steroid receptor regulated transcription of specific genes and gene networks. Annu. Rev. Genet. 1985, 19, 209–252. [Google Scholar] [CrossRef]
- Van Raalte, D.H.; Ouwens, D.M.; Diamant, M. Novel insights into glucocorticoid-mediated diabetogenic effects: Towards expansion of therapeutic options? Eur. J. Clin. Investig. 2009, 39, 81–93. [Google Scholar] [CrossRef]
- Petta, I.; Dejager, L.; Ballegeer, M.; Lievens, S.; Tavernier, J.; De Bosscher, K.; Libert, C. The Interactome of the Glucocorticoid Receptor and Its Influence on the Actions of Glucocorticoids in Combatting Inflammatory and Infectious Diseases. Microbiol. Mol. Biol. Rev. 2016, 80, 495–522. [Google Scholar] [CrossRef] [Green Version]
- Adcock, I.M.; Caramori, G. Cross-talk between pro-inflammatory transcription factors and glucocorticoids. Immunol. Cell Biol. 2001, 79, 376–384. [Google Scholar] [CrossRef]
- Smoak, K.A.; Cidlowski, J.A. Mechanisms of glucocorticoid receptor signaling during inflammation. Mech. Ageing Dev. 2004, 125, 697–706. [Google Scholar] [CrossRef]
- Kujubu, D.A.; Herschman, H.R. Dexamethasone inhibits mitogen induction of the TIS10 prostaglandin synthase/cyclooxygenase gene. J. Biol. Chem. 1992, 267, 7991–7994. [Google Scholar] [CrossRef]
- Lasa, M.; Brook, M.; Saklatvala, J.; Clark, A.R. Dexamethasone destabilizes cyclooxygenase 2 mRNA by inhibiting mitogen-activated protein kinase p38. Mol. Cell. Biol. 2001, 21, 771–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masferrer, J.L.; Reddy, S.T.; Zweifel, B.S.; Seibert, K.; Needleman, P.; Gilbert, R.S.; Herschman, H.R. In vivo glucocorticoids regulate cyclooxygenase-2 but not cyclooxygenase-1 in peritoneal macrophages. J. Pharmacol. Exp. Ther. 1994, 270, 1340–1344. [Google Scholar] [PubMed]
- Newman, S.P.; Flower, R.J.; Croxtall, J.D. Dexamethasone suppression of IL-1 beta-induced cyclooxygenase 2 expression is not mediated by lipocortin-1 in A549 cells. Biochem. Biophys. Res. Commun. 1994, 202, 931–939. [Google Scholar] [CrossRef] [PubMed]
- O’Banion, M.K.; Winn, V.D.; Young, D.A. cDNA cloning and functional activity of a glucocorticoid-regulated inflammatory cyclooxygenase. Proc. Natl. Acad. Sci. USA 1992, 89, 4888–4892. [Google Scholar] [CrossRef] [Green Version]
- Ristimaki, A.; Narko, K.; Hla, T. Down-regulation of cytokine-induced cyclo-oxygenase-2 transcript isoforms by dexamethasone: Evidence for post-transcriptional regulation. Biochem. J. 1996, 318 Pt 1, 325–331. [Google Scholar] [CrossRef]
- Godowski, P.J.; Sakai, D.D.; Yamamoto, K.R. DNA-Protein Interactions in Transcription. In UCLA Symposia on Molecular and Cellular Biology; Gralla, J.D., Ed.; Alan R. Liss: New York, NY, USA, 1989; pp. 197–210. [Google Scholar]
- Helmberg, A.; Auphan, N.; Caelles, C.; Karin, M. Glucocorticoid-induced apoptosis of human leukemic cells is caused by the repressive function of the glucocorticoid receptor. EMBO J. 1995, 14, 452–460. [Google Scholar] [CrossRef]
- Reichardt, H.M.; Tuckermann, J.P.; Gottlicher, M.; Vujic, M.; Weih, F.; Angel, P.; Herrlich, P.; Schutz, G. Repression of inflammatory responses in the absence of DNA binding by the glucocorticoid receptor. EMBO J. 2001, 20, 7168–7173. [Google Scholar] [CrossRef] [Green Version]
- Heck, S.; Kullmann, M.; Gast, A.; Ponta, H.; Rahmsdorf, H.J.; Herrlich, P.; Cato, A.C. A distinct modulating domain in glucocorticoid receptor monomers in the repression of activity of the transcription factor AP-1. EMBO J. 1994, 13, 4087–4095. [Google Scholar] [CrossRef]
- De Bosscher, K.; Vanden Berghe, W.; Haegeman, G. The interplay between the glucocorticoid receptor and nuclear factor-kappaB or activator protein-1: Molecular mechanisms for gene repression. Endocr. Rev. 2003, 24, 488–522. [Google Scholar] [CrossRef] [Green Version]
- De Bosscher, K.; Schmitz, M.L.; Vanden Berghe, W.; Plaisance, S.; Fiers, W.; Haegeman, G. Glucocorticoid-mediated repression of nuclear factor-kappaB-dependent transcription involves direct interference with transactivation. Proc. Natl. Acad. Sci. USA 1997, 94, 13504–13509. [Google Scholar] [CrossRef]
- Chen, R.; Burke, T.F.; Cumberland, J.E.; Brummet, M.; Beck, L.A.; Casolaro, V.; Georas, S.N. Glucocorticoids inhibit calcium- and calcineurin-dependent activation of the human IL-4 promoter. J. Immunol. 2000, 164, 825–832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paliogianni, F.; Raptis, A.; Ahuja, S.S.; Najjar, S.M.; Boumpas, D.T. Negative transcriptional regulation of human interleukin 2 (IL-2) gene by glucocorticoids through interference with nuclear transcription factors AP-1 and NF-AT. J. Clin. Investig. 1993, 91, 1481–1489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vacca, A.; Felli, M.P.; Farina, A.R.; Martinotti, S.; Maroder, M.; Screpanti, I.; Meco, D.; Petrangeli, E.; Frati, L.; Gulino, A. Glucocorticoid receptor-mediated suppression of the interleukin 2 gene expression through impairment of the cooperativity between nuclear factor of activated T cells and AP-1 enhancer elements. J. Exp. Med. 1992, 175, 637–646. [Google Scholar] [CrossRef] [Green Version]
- Wisniewska, M.; Stanczyk, M.; Grzelakowska-Sztabert, B.; Kaminska, B. Nuclear factor of activated T cells (NFAT) is a possible target for dexamethasone in thymocyte apoptosis. Cell Biol. Int. 1997, 21, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Macian, F.; Garcia-Rodriguez, C.; Rao, A. Gene expression elicited by NFAT in the presence or absence of cooperative recruitment of Fos and Jun. EMBO J. 2000, 19, 4783–4795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cron, R.Q.; Bort, S.J.; Wang, Y.; Brunvand, M.W.; Lewis, D.B. T cell priming enhances IL-4 gene expression by increasing nuclear factor of activated T cells. J. Immunol. 1999, 162, 860–870. [Google Scholar]
- Monticelli, S.; Solymar, D.C.; Rao, A. Role of NFAT proteins in IL13 gene transcription in mast cells. J. Biol. Chem. 2004, 279, 36210–36218. [Google Scholar] [CrossRef] [Green Version]
- Rao, A.; Luo, C.; Hogan, P.G. Transcription factors of the NFAT family: Regulation and function. Annu. Rev. Immunol. 1997, 15, 707–747. [Google Scholar] [CrossRef]
- Crabtree, G.R. Generic signals and specific outcomes: Signaling through Ca2+, calcineurin, and NF-AT. Cell 1999, 96, 611–614. [Google Scholar] [CrossRef] [Green Version]
- Hogan, P.G.; Chen, L.; Nardone, J.; Rao, A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 2003, 17, 2205–2232. [Google Scholar] [CrossRef] [Green Version]
- Luo, C.; Burgeon, E.; Rao, A. Mechanisms of transactivation by nuclear factor of activated T cells-1. J. Exp. Med. 1996, 184, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Okamura, H.; Aramburu, J.; Garcia-Rodriguez, C.; Viola, J.P.; Raghavan, A.; Tahiliani, M.; Zhang, X.; Qin, J.; Hogan, P.G.; Rao, A. Concerted dephosphorylation of the transcription factor NFAT1 induces a conformational switch that regulates transcriptional activity. Mol. Cell 2000, 6, 539–550. [Google Scholar] [CrossRef]
- Gomez-Casero, E.; San-Antonio, B.; Iniguez, M.A.; Fresno, M. Cot/Tpl2 and PKCzeta cooperate in the regulation of the transcriptional activity of NFATc2 through the phosphorylation of its amino-terminal domain. Cell. Signal. 2007, 19, 1652–1661. [Google Scholar] [CrossRef] [PubMed]
- San-Antonio, B.; Iniguez, M.A.; Fresno, M. Protein kinase Czeta phosphorylates nuclear factor of activated T cells and regulates its transactivating activity. J. Biol. Chem. 2002, 277, 27073–27080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, H.; Tanabe, T. Transcriptional role of the nuclear factor kappa B site in the induction by lipopolysaccharide and suppression by dexamethasone of cyclooxygenase-2 in U937 cells. Biochem. Biophys. Res. Commun. 1998, 244, 143–148. [Google Scholar] [CrossRef]
- Brewer, J.A.; Khor, B.; Vogt, S.K.; Muglia, L.M.; Fujiwara, H.; Haegele, K.E.; Sleckman, B.P.; Muglia, L.J. T-cell glucocorticoid receptor is required to suppress COX-2-mediated lethal immune activation. Nat. Med. 2003, 9, 1318–1322. [Google Scholar] [CrossRef]
- Caldenhoven, E.; Liden, J.; Wissink, S.; Van de Stolpe, A.; Raaijmakers, J.; Koenderman, L.; Okret, S.; Gustafsson, J.A.; Van der Saag, P.T. Negative cross-talk between RelA and the glucocorticoid receptor: A possible mechanism for the antiinflammatory action of glucocorticoids. Mol. Endocrinol. 1995, 9, 401–412. [Google Scholar] [CrossRef] [Green Version]
- Reichardt, H.M.; Kaestner, K.H.; Tuckermann, J.; Kretz, O.; Wessely, O.; Bock, R.; Gass, P.; Schmid, W.; Herrlich, P.; Angel, P.; et al. DNA binding of the glucocorticoid receptor is not essential for survival. Cell 1998, 93, 531–541. [Google Scholar] [CrossRef] [Green Version]
- Liberman, A.C.; Refojo, D.; Druker, J.; Toscano, M.; Rein, T.; Holsboer, F.; Arzt, E. The activated glucocorticoid receptor inhibits the transcription factor T-bet by direct protein-protein interaction. FASEB J. 2007, 21, 1177–1188. [Google Scholar] [CrossRef] [Green Version]
- Yang-Yen, H.F.; Chambard, J.C.; Sun, Y.L.; Smeal, T.; Schmidt, T.J.; Drouin, J.; Karin, M. Transcriptional interference between c-Jun and the glucocorticoid receptor: Mutual inhibition of DNA binding due to direct protein-protein interaction. Cell 1990, 62, 1205–1215. [Google Scholar] [CrossRef]
- Langlais, D.; Couture, C.; Balsalobre, A.; Drouin, J. The Stat3/GR interaction code: Predictive value of direct/indirect DNA recruitment for transcription outcome. Mol. Cell 2012, 47, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, M.W.; Staples, K.J.; Smith, S.J.; Barnes, P.J.; Newton, R. Glucocorticoid inhibition of granulocyte macrophage-colony-stimulating factor from T cells is independent of control by nuclear factor-kappaB and conserved lymphokine element 0. Am. J. Respir. Cell Mol. Biol. 2004, 30, 555–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, P.J.; Cousins, D.J.; Jee, Y.K.; Staynov, D.Z.; Lee, T.H.; Lavender, P. Suppression of granulocyte-macrophage colony-stimulating factor expression by glucocorticoids involves inhibition of enhancer function by the glucocorticoid receptor binding to composite NF-AT/activator protein-1 elements. J. Immunol. 2001, 167, 2502–2510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macian, F.; Lopez-Rodriguez, C.; Rao, A. Partners in transcription: NFAT and AP-1. Oncogene 2001, 20, 2476–2489. [Google Scholar] [CrossRef] [Green Version]
- Masuda, E.S.; Imamura, R.; Amasaki, Y.; Arai, K.; Arai, N. Signalling into the T-cell nucleus: NFAT regulation. Cell. Signal. 1998, 10, 599–611. [Google Scholar] [CrossRef]
- Wisniewska, M.; Pyrzynska, B.; Kaminska, B. Impaired AP-1 dimers and NFAT complex formation in immature thymocytes during in vivo glucocorticoid-induced apoptosis. Cell Biol. Int. 2004, 28, 773–780. [Google Scholar] [CrossRef]
- Garcia-Rodriguez, C.; Rao, A. Requirement for integration of phorbol 12-myristate 13-acetate and calcium pathways is preserved in the transactivation domain of NFAT1. Eur. J. Immunol. 2000, 30, 2432–2436. [Google Scholar] [CrossRef]
- Garcia-Rodriguez, C.; Rao, A. Nuclear factor of activated T cells (NFAT)-dependent transactivation regulated by the coactivators p300/CREB-binding protein (CBP). J. Exp. Med. 1998, 187, 2031–2036. [Google Scholar] [CrossRef] [Green Version]
- Rainio, E.M.; Sandholm, J.; Koskinen, P.J. Cutting edge: Transcriptional activity of NFATc1 is enhanced by the Pim-1 kinase. J. Immunol. 2002, 168, 1524–1527. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.C.; Hsu, S.C.; Shih, H.M.; Lai, M.Z. Nuclear factor of activated T cells c is a target of p38 mitogen-activated protein kinase in T cells. Mol. Cell. Biol. 2003, 23, 6442–6454. [Google Scholar] [CrossRef] [Green Version]
- Leung-Theung-Long, S.; Mondor, I.; Guiraud, M.; Lamare, C.; Nagaleekar, V.; Paulet, P.E.; Rincon, M.; Guerder, S. Impaired NFAT transcriptional activity in antigen-stimulated CD8 T cells linked to defective phosphorylation of NFAT transactivation domain. J. Immunol. 2009, 182, 6807–6814. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Perez, I.; Cano, E.; Were, F.; Villar, M.; Vazquez, J.; Redondo, J.M. c-Jun N-terminal kinase (JNK) positively regulates NFATc2 transactivation through phosphorylation within the N-terminal regulatory domain. J. Biol. Chem. 2005, 280, 20867–20878. [Google Scholar] [CrossRef] [Green Version]
- De Bosscher, K.; Haegeman, G. Minireview: Latest perspectives on antiinflammatory actions of glucocorticoids. Mol. Endocrinol. 2009, 23, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Bruna, A.; Nicolas, M.; Munoz, A.; Kyriakis, J.M.; Caelles, C. Glucocorticoid receptor-JNK interaction mediates inhibition of the JNK pathway by glucocorticoids. EMBO J. 2003, 22, 6035–6044. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Shaw, K.T.; Raghavan, A.; Aramburu, J.; Garcia-Cozar, F.; Perrino, B.A.; Hogan, P.G.; Rao, A. Interaction of calcineurin with a domain of the transcription factor NFAT1 that controls nuclear import. Proc. Natl. Acad. Sci. USA 1996, 93, 8907–8912. [Google Scholar] [CrossRef] [Green Version]
- Rhoades, K.L.; Golub, S.H.; Economou, J.S. The regulation of the human tumor necrosis factor alpha promoter region in macrophage, T cell, and B cell lines. J. Biol. Chem. 1992, 267, 22102–22107. [Google Scholar] [CrossRef]
- Durand, D.B.; Shaw, J.P.; Bush, M.R.; Replogle, R.E.; Belagaje, R.; Crabtree, G.R. Characterization of antigen receptor response elements within the interleukin-2 enhancer. Mol. Cell. Biol. 1988, 8, 1715–1724. [Google Scholar] [CrossRef]
- Minden, A.; Lin, A.; Claret, F.X.; Abo, A.; Karin, M. Selective activation of the JNK signaling cascade and c-Jun transcriptional activity by the small GTPases Rac and Cdc42Hs. Cell 1995, 81, 1147–1157. [Google Scholar] [CrossRef] [Green Version]
- Szabo, S.J.; Gold, J.S.; Murphy, T.L.; Murphy, K.M. Identification of cis-acting regulatory elements controlling interleukin-4 gene expression in T cells: Roles for NF-Y and NF-ATc. Mol. Cell. Biol. 1993, 13, 4793–4805. [Google Scholar] [CrossRef]
- De Gregorio, R.; Iniguez, M.A.; Fresno, M.; Alemany, S. Cot kinase induces cyclooxygenase-2 expression in T cells through activation of the nuclear factor of activated T cells. J. Biol. Chem. 2001, 276, 27003–27009. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cacheiro-Llaguno, C.; Hernández-Subirá, E.; Díaz-Muñoz, M.D.; Fresno, M.; Serrador, J.M.; Íñiguez, M.A. Regulation of Cyclooxygenase-2 Expression in Human T Cells by Glucocorticoid Receptor-Mediated Transrepression of Nuclear Factor of Activated T Cells. Int. J. Mol. Sci. 2022, 23, 13275. https://doi.org/10.3390/ijms232113275
Cacheiro-Llaguno C, Hernández-Subirá E, Díaz-Muñoz MD, Fresno M, Serrador JM, Íñiguez MA. Regulation of Cyclooxygenase-2 Expression in Human T Cells by Glucocorticoid Receptor-Mediated Transrepression of Nuclear Factor of Activated T Cells. International Journal of Molecular Sciences. 2022; 23(21):13275. https://doi.org/10.3390/ijms232113275
Chicago/Turabian StyleCacheiro-Llaguno, Cristina, Elena Hernández-Subirá, Manuel D. Díaz-Muñoz, Manuel Fresno, Juan M. Serrador, and Miguel A. Íñiguez. 2022. "Regulation of Cyclooxygenase-2 Expression in Human T Cells by Glucocorticoid Receptor-Mediated Transrepression of Nuclear Factor of Activated T Cells" International Journal of Molecular Sciences 23, no. 21: 13275. https://doi.org/10.3390/ijms232113275
APA StyleCacheiro-Llaguno, C., Hernández-Subirá, E., Díaz-Muñoz, M. D., Fresno, M., Serrador, J. M., & Íñiguez, M. A. (2022). Regulation of Cyclooxygenase-2 Expression in Human T Cells by Glucocorticoid Receptor-Mediated Transrepression of Nuclear Factor of Activated T Cells. International Journal of Molecular Sciences, 23(21), 13275. https://doi.org/10.3390/ijms232113275