
Sphingosine-1-Phosphate (S1P) Lyase Inhibition Aggravates Atherosclerosis and Induces Plaque Rupture in ApoE−/− Mice
 , , , ,
, , , ,  and
and
Abstract
:1. Introduction
2. Results
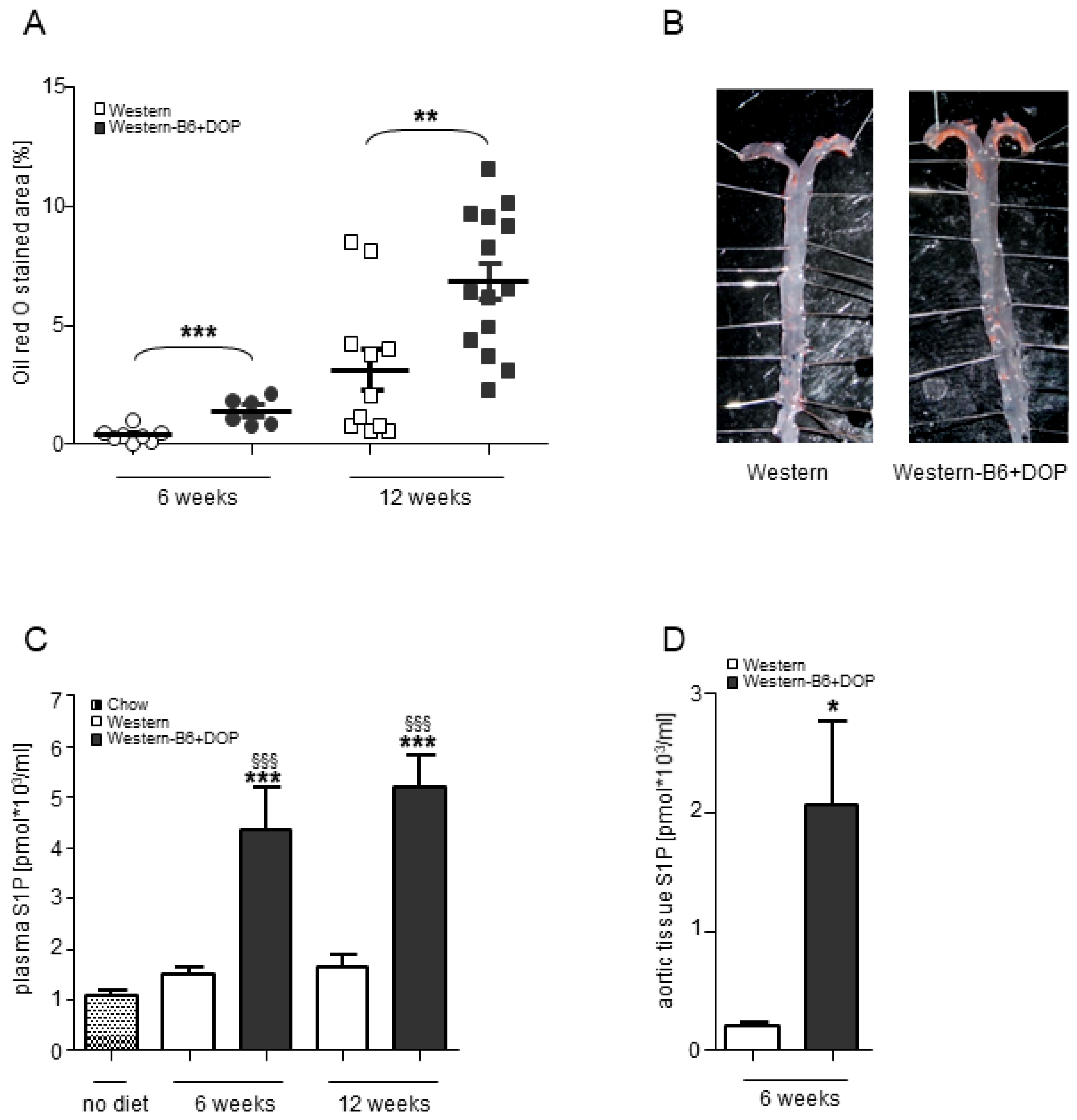
2.1. Pharmacological Inhibition of the S1P Lyase Aggravated Atherosclerosis Development in ApoE−/− Mice
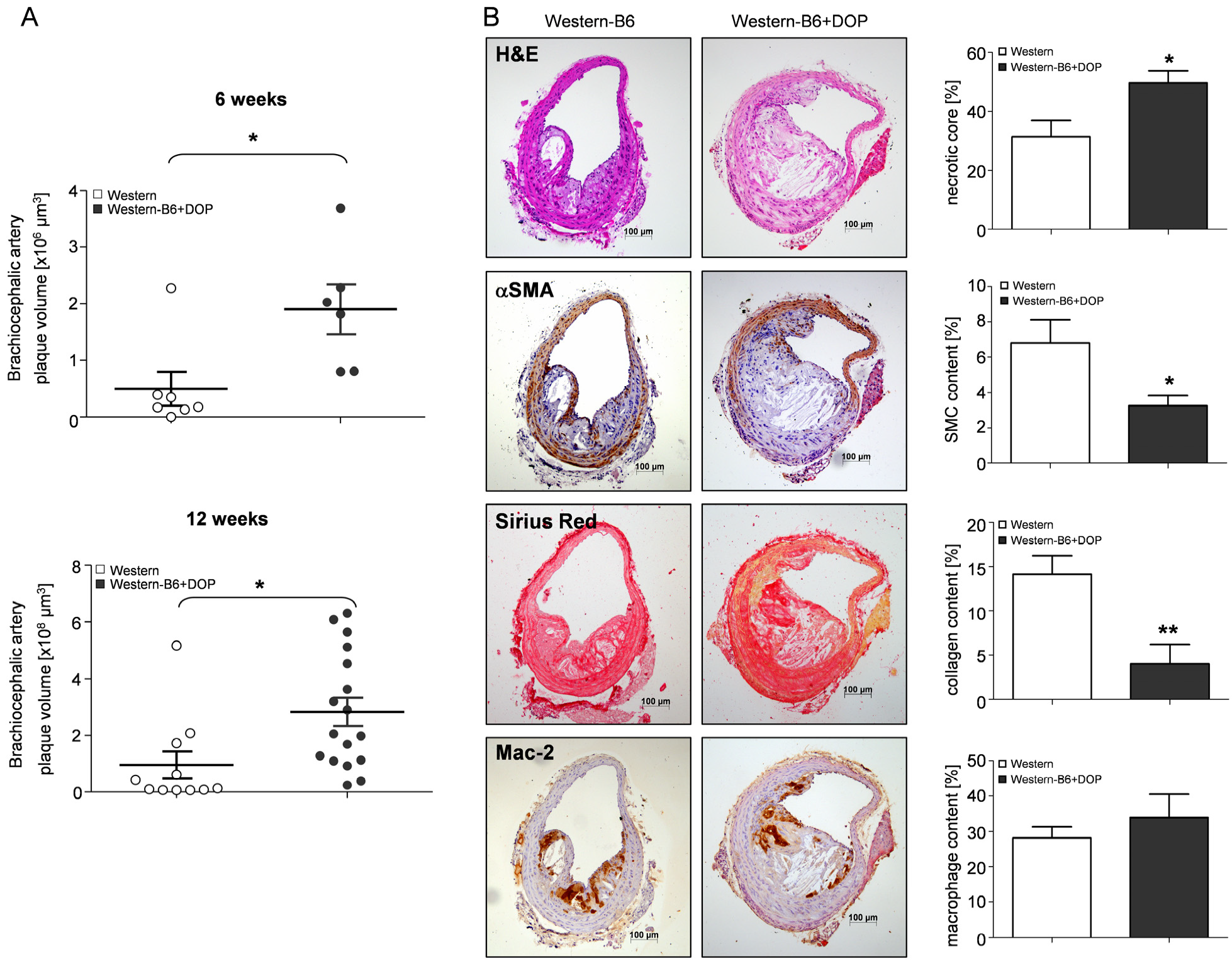
2.2. Characteristics of Atherosclerotic Lesions of S1P Lyase-Inhibited ApoE−/− Mice
2.3. Lipoprotein and Leukocyte Changes after S1P Lyase Inhibition in Cholesterol-Fed ApoE-/- Mice
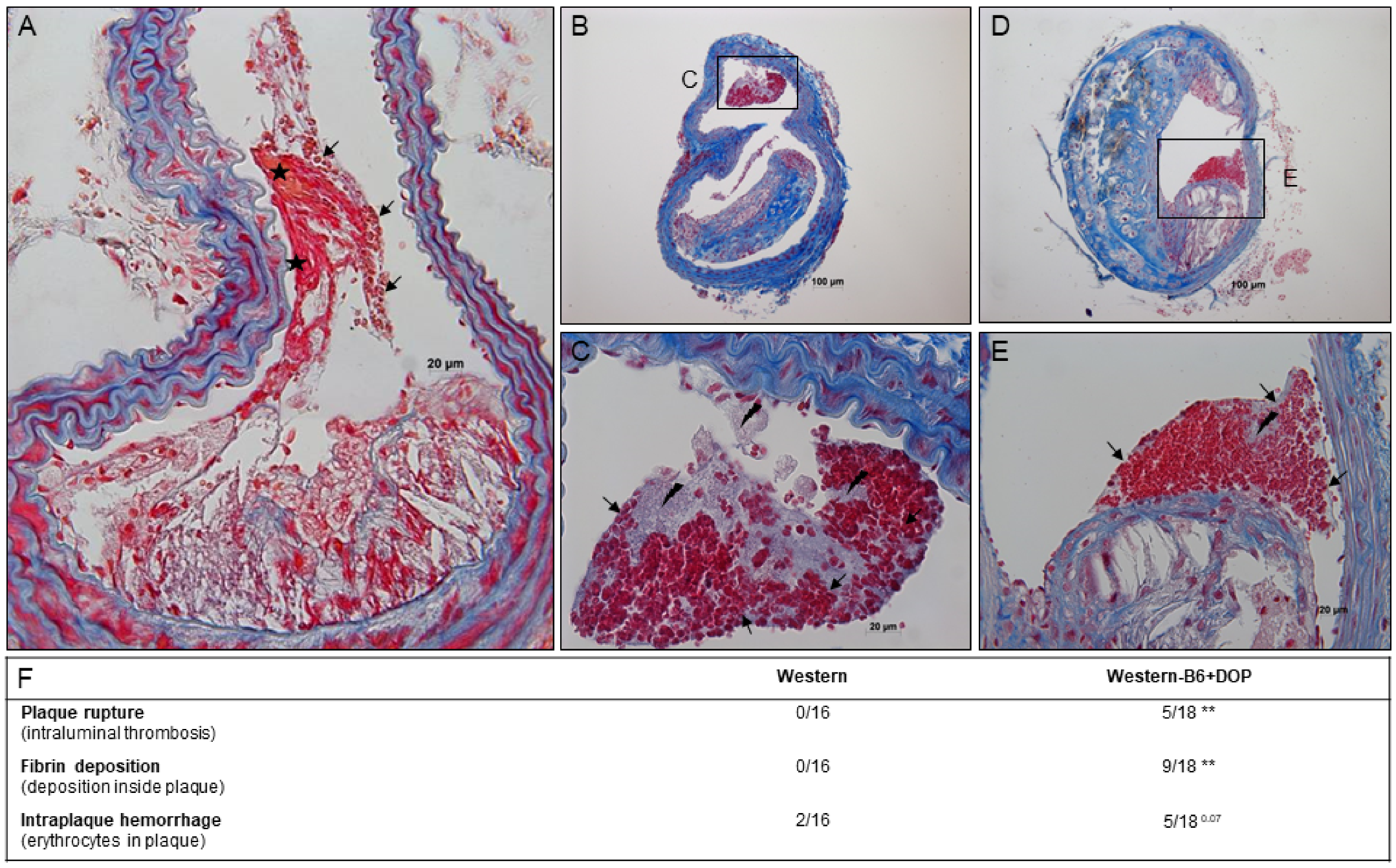
2.4. Plaque Rupture in Atherosclerotic Lesions of S1P Lyase-Inhibited ApoE−/− Mice
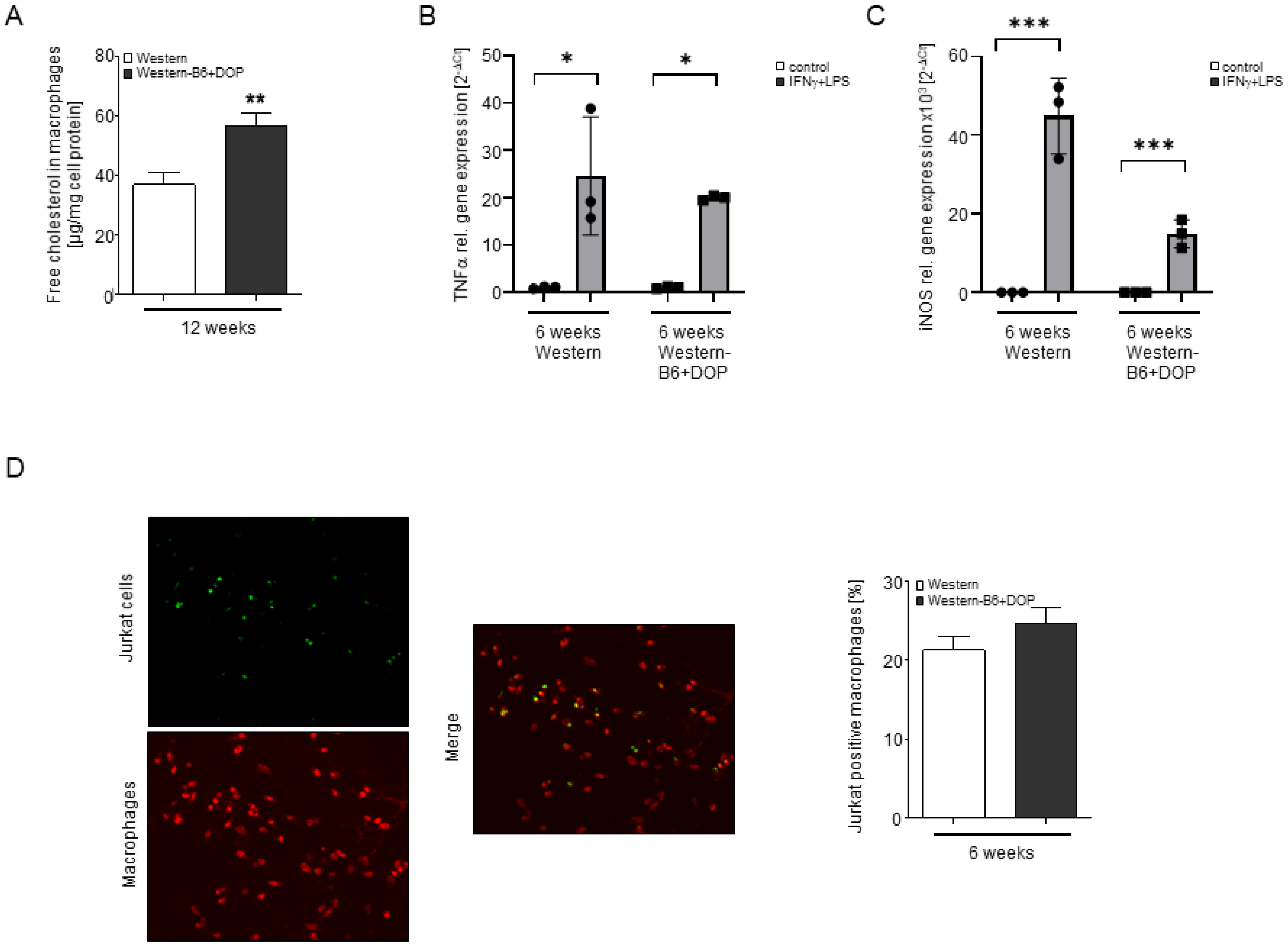
2.5. Profound Changes in Macrophage Sphingolipid and Cholesterol Content but Unaltered Macrophage Polarization, Inflammation and Efferocytosis
2.6. Genetic Deficiency and Pharmacological Inhibition of the S1P Lyase Downregulate Macrophage ABCA1 and ABCG1 Gene Expression and Impair Cholesterol Efflux to ApoA-I
2.7. Attenuated Cholesterol Efflux in S1PR3 Deficient Macrophages and Enhanced Atherosclerosis in Cholesterol-Fed ApoE−/−/S1PR3−/− Mice
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Hematology and Flow Cytometry
4.3. Plasma Lipid and Lipoprotein Analysis
4.4. Lesion Volume
4.5. Oil Red O Staining of Aortae
4.6. Immunohistochemistry
4.7. Isolation of Peritoneal Macrophages and In Vitro Gene Expression Studies
4.8. RNA Isolation and Quantitative PCR Analysis of Gene Expression
4.9. Cholesterol Efflux
4.10. Fluorimetric Cholesterol Assay
4.11. S1P Measurements
4.12. Macrophage Phagocytosis Assay
4.13. Statistics
4.14. Study Approval
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Levkau, B. HDL-S1P: Cardiovascular functions, disease-associated alterations, and therapeutic applications. Front. Pharmacol. 2015, 6, 243. [Google Scholar] [CrossRef] [PubMed]
- Waeber, C.; Walther, T. Sphingosine-1-phosphate as a potential target for the treatment of myocardial infarction. Circ. J. 2014, 78, 795–802. [Google Scholar] [CrossRef] [PubMed]
- Pyne, S.; Adams, D.R.; Pyne, N.J. Sphingosine 1-phosphate and sphingosine kinases in health and disease: Recent advances. Prog. Lipid Res. 2016, 62, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Poti, F.; Simoni, M.; Nofer, J.R. Atheroprotective role of high-density lipoprotein (HDL)-associated sphingosine-1-phosphate (S1P). Cardiovasc. Res. 2014, 103, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Sattler, K.J.; Elbasan, S.; Keul, P.; Elter-Schulz, M.; Bode, C.; Graler, M.H.; Bröcker-Preuss, M.; Budde, T.; Erbel, R.; Gerd Heusch, G.; et al. Sphingosine 1-phosphate levels in plasma and HDL are altered in coronary artery disease. Basic Res. Cardiol. 2010, 105, 821–832. [Google Scholar] [CrossRef]
- Keul, P.; Polzin, A.; Kaiser, K.; Graler, M.; Dannenberg, L.; Daum, G.; Heusch, G.; Levkau, B. Potent anti-inflammatory properties of HDL in vascular smooth muscle cells mediated by HDL-S1P and their impairment in coronary artery disease due to lower HDL-S1P: A new aspect of HDL dysfunction and its therapy. FASEB J. 2019, 33, 1482–1495. [Google Scholar] [CrossRef]
- Sattler, K.; Graler, M.; Keul, P.; Weske, S.; Reimann, C.M.; Jindrova, H.; Kleinbongard, P.; Sabbadini, R.; Bröcker-Preuss, M.; Raimund Erbel, R.; et al. Defects of High-Density Lipoproteins in Coronary Artery Disease Caused by Low Sphingosine-1-Phosphate Content: Correction by Sphingosine-1-Phosphate-Loading. J. Am. Coll. Cardiol. 2015, 66, 1470–1485. [Google Scholar] [CrossRef]
- Rosenson, R.S.; Brewer, H.B., Jr.; Ansell, B.J.; Barter, P.; Chapman, M.J.; Heinecke, J.W.; Kontush, A.; Tall, A.R.; Webb, N.R. Dysfunctional HDL and atherosclerotic cardiovascular disease. Nat. Rev. Cardiol. 2016, 13, 48–60. [Google Scholar] [CrossRef]
- Vaidya, M.; Jentsch, J.A.; Peters, S.; Keul, P.; Weske, S.; Graler, M.H.; Mladenov, E.; Iliakis, G.; Heusch, G.; Levkau, B. Regulation of ABCA1-mediated cholesterol efflux by sphingosine-1-phosphate signaling in macrophages. J. Lipid Res. 2019, 60, 506–515. [Google Scholar] [CrossRef]
- Proia, R.L.; Hla, T. Emerging biology of sphingosine-1-phosphate: Its role in pathogenesis and therapy. J. Clin. Investig. 2015, 125, 1379–1387. [Google Scholar] [CrossRef] [Green Version]
- Poti, F.; Bot, M.; Costa, S.; Bergonzini, V.; Maines, L.; Varga, G.; Hendrik Freise, H.; Robenek, H.; Manuela Simoni, M.; Nofer, J.R. Sphingosine kinase inhibition exerts both pro- and anti-atherogenic effects in low-density lipoprotein receptor-deficient (LDL-R(-/-)) mice. Thromb. Haemost. 2012, 107, 552–561. [Google Scholar] [CrossRef] [PubMed]
- Bot, M.; Van Veldhoven, P.P.; de Jager, S.C.; Johnson, J.; Nijstad, N.; Van Santbrink, P.J.; Westra, M.M.; Van Der Hoeven, G.; Gijbels, M.J.; Müller-Tidow, C.; et al. Hematopoietic sphingosine 1-phosphate lyase deficiency decreases atherosclerotic lesion development in LDL-receptor deficient mice. PLoS ONE. 2013, 8, e63360. [Google Scholar]
- Nofer, J.R.; Bot, M.; Brodde, M.; Taylor, P.J.; Salm, P.; Brinkmann, V.; van Berkel, T.; Assmann, G.; Biessen, E.A.L. FTY720, a synthetic sphingosine 1 phosphate analogue, inhibits development of atherosclerosis in low-density lipoprotein receptor-deficient mice. Circulation 2007, 115, 501–508. [Google Scholar] [CrossRef]
- Keul, P.; Tolle, M.; Lucke, S.; von Wnuck Lipinski, K.; Heusch, G.; Schuchardt, M.; van der Giet, M.; Levkau, B. The sphingosine-1-phosphate analogue FTY720 reduces atherosclerosis in apolipoprotein E-deficient mice. Arter. Thromb. Vasc. Biol. 2007, 27, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Klingenberg, R.; Nofer, J.R.; Rudling, M.; Bea, F.; Blessing, E.; Preusch, M.; Hermann, J.; Grone, H.J.; Katus, H.A.; Hansson, G.K.; et al. Sphingosine-1-phosphate analogue FTY720 causes lymphocyte redistribution and hypercholesterolemia in ApoE-deficient mice. Arter. Thromb. Vasc. Biol. 2007, 27, 2392–2399. [Google Scholar] [CrossRef] [PubMed]
- Poti, F.; Costa, S.; Bergonzini, V.; Galletti, M.; Pignatti, E.; Weber, C.; Simonia, M.; Nofer, J.R. Effect of sphingosine 1-phosphate (S1P) receptor agonists FTY720 and CYM5442 on atherosclerosis development in LDL receptor deficient (LDL-R(-)/(-)) mice. Vasc. Pharmacol. 2012, 57, 56–64. [Google Scholar] [CrossRef]
- Poti, F.; Gualtieri, F.; Sacchi, S.; Weissen-Plenz, G.; Varga, G.; Brodde, M.; Weber, C.; Simoni, M.; Nofer, J.R. KRP-203, sphingosine 1-phosphate receptor type 1 agonist, ameliorates atherosclerosis in LDL-R-/- mice. Arter. Thromb. Vasc. Biol. 2013, 33, 1505–1512. [Google Scholar] [CrossRef]
- Galvani, S.; Sanson, M.; Blaho, V.A.; Swendeman, S.L.; Obinata, H.; Conger, H.; Dahlbäck, B.; Kono, M.; Proia, R.L.; Smith, J.D.; et al. HDL-bound sphingosine 1-phosphate acts as a biased agonist for the endothelial cell receptor S1P1 to limit vascular inflammation. Sci. Signal 2015, 8, ra79. [Google Scholar] [CrossRef]
- Skoura, A.; Michaud, J.; Im, D.S.; Thangada, S.; Xiong, Y.; Smith, J.D.; Hla, T. Sphingosine-1-phosphate receptor-2 function in myeloid cells regulates vascular inflammation and atherosclerosis. Arter. Thromb. Vasc. Biol. 2010, 31, 81–85. [Google Scholar] [CrossRef]
- Keul, P.; Lucke, S.; von Wnuck Lipinski, K.; Bode, C.; Graler, M.; Heusch, G.; Levkau, B. Sphingosine-1-phosphate receptor 3 promotes recruitment of monocyte/macrophages in inflammation and atherosclerosis. Circ. Res. 2010, 108, 314–323. [Google Scholar] [CrossRef]
- Xiong, Y.; Lee, H.J.; Mariko, B.; Lu, Y.C.; Dannenberg, A.J.; Haka, A.S.; Maxfield, F.R.; Camerer, E.; Proia, R.P.; Hla, T. Sphingosine kinases are not required for inflammatory responses in macrophages. J. Biol. Chem. 2013, 288, 32563–32573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogel, P.; Donoviel, M.S.; Read, R.; Hansen, G.M.; Hazlewood, J.; Anderson, S.J.; Weimei Sun, W.; Swaffield, J.; Oravecz, T. Incomplete inhibition of sphingosine 1-phosphate lyase modulates immune system function yet prevents early lethality and non-lymphoid lesions. PLoS ONE 2009, 4, e4112. [Google Scholar] [CrossRef] [PubMed]
- Le Stunff, H.; Milstien, S.; Spiegel, S. Generation and metabolism of bioactive sphingosine-1-phosphate. J. Cell Biochem. 2004, 92, 882–899. [Google Scholar] [CrossRef] [PubMed]
- Van Veldhoven, P.P. Sphingosine-1-phosphate lyase. Methods Enzymol. 2000, 311, 244–254. [Google Scholar]
- Schwab, S.R.; Pereira, J.P.; Matloubian, M.; Xu, Y.; Huang, Y.; Cyster, J.G. Lymphocyte sequestration through S1P lyase inhibition and disruption of S1P gradients. Science 2005, 309, 1735–1739. [Google Scholar] [CrossRef]
- Gough, P.J.; Gomez, I.G.; Wille, P.T.; Raines, E.W. Macrophage expression of active MMP-9 induces acute plaque disruption in apoE-deficient mice. J. Clin. Investig. 2006, 116, 59–69. [Google Scholar] [CrossRef]
- Jagle, U.; Gasser, J.A.; Muller, M.; Kinzel, B. Conditional transgene expression mediated by the mouse beta-actin locus. Genesis 2007, 45, 659–666. [Google Scholar] [CrossRef]
- Billich, A.; Baumruker, T.; Beerli, C.; Bigaud, M.; Bruns, C.; Calzascia, T.; Isken, A.; Kinzel, B.; Loetscher, E.; Metzler, B.; et al. Partial deficiency of sphingosine-1-phosphate lyase confers protection in experimental autoimmune encephalomyelitis. PLoS ONE 2013, 8, e59630. [Google Scholar]
- Egom, E.E.; Mamas, M.A.; Chacko, S.; Stringer, S.E.; Charlton-Menys, V.; El-Omar, M.; Chirico, D.; Clarke, B.; Neyses, L.; Cruickshank, J.K.; et al. Serum sphingolipids level as a novel potential marker for early detection of human myocardial ischaemic injury. Front. Physiol. 2013, 4, 130. [Google Scholar] [CrossRef]
- Swirski, F.K.; Libby, P.; Aikawa, E.; Alcaide, P.; Luscinskas, F.W.; Weissleder, R.; Pittet, M.J. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J. Clin. Investig. 2007, 117, 195–205. [Google Scholar] [CrossRef]
- Tacke, F.; Alvarez, D.; Kaplan, T.J.; Jakubzick, C.; Spanbroek, R.; Llodra, J.; Garin, A.; Liu, J.; Mack, M.; van Rooijen, N.; et al. Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J. Clin. Investig. 2007, 117, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Williams, H.; Johnson, J.L.; Carson, K.G.; Jackson, C.L. Characteristics of intact and ruptured atherosclerotic plaques in brachiocephalic arteries of apolipoprotein E knockout mice. Arter. Thromb. Vasc. Biol. 2002, 22, 788–792. [Google Scholar] [CrossRef]
- Rosenfeld, M.E.; Carson, K.G.; Johnson, J.L.; Williams, H.; Jackson, C.L.; Schwartz, S.M. Animal models of spontaneous plaque rupture: The holy grail of experimental atherosclerosis research. Curr. Atheroscler. Rep. 2002, 4, 238–242. [Google Scholar] [CrossRef]
- Yvan-Charvet, L.; Pagler, T.A.; Seimon, T.A.; Thorp, E.; Welch, C.L.; Witztum, J.L.; Tabas, I.; Tall, A.R. ABCA1 and ABCG1 protect against oxidative stress-induced macrophage apoptosis during efferocytosis. Circ. Res. 2010, 106, 1861–1869. [Google Scholar] [CrossRef] [PubMed]
- Tarling, E.J.; de Aguiar Vallim, T.Q.; Edwards, P.A. Role of ABC transporters in lipid transport and human disease. Trends Endocrinol. Metab. 2013, 24, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Tall, A.R.; Yvan-Charvet, L.; Terasaka, N.; Pagler, T.; Wang, N. HDL, ABC transporters, and cholesterol efflux: Implications for the treatment of atherosclerosis. Cell Metab. 2008, 7, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Out, R.; Hoekstra, M.; Habets, K.; Meurs, I.; de Waard, V.; Hildebrand, R.B.; Wang, W.; Chimini, G.; Kuiper, J.; Van Berkel, T.J.C.; et al. Combined deletion of macrophage ABCA1 and ABCG1 leads to massive lipid accumulation in tissue macrophages and distinct atherosclerosis at relatively low plasma cholesterol levels. Arter. Thromb Vasc Biol. 2008, 28, 258–264. [Google Scholar] [CrossRef]
- Van Eck, M.; Bos, I.S.; Hildebrand, R.B.; Van Rij, B.T.; Van Berkel, T.J. Dual role for scavenger receptor class B, type I on bone marrow-derived cells in atherosclerotic lesion development. Am. J. Pathol. 2004, 165, 785–794. [Google Scholar] [CrossRef]
- Yvan-Charvet, L.; Ranalletta, M.; Wang, N.; Han, S.; Terasaka, N.; Li, R.; Welch, C.; Tall, A.R. Combined deficiency of ABCA1 and ABCG1 promotes foam cell accumulation and accelerates atherosclerosis in mice. J. Clin. Investig. 2007, 117, 3900–3908. [Google Scholar] [CrossRef]
- Aiello, R.J.; Brees, D.; Bourassa, P.A.; Royer, L.; Lindsey, S.; Coskran, T.; Haghpassand, M.; Francone, O.L. Increased atherosclerosis in hyperlipidemic mice with inactivation of ABCA1 in macrophages. Arter. Thromb. Vasc. Biol. 2002, 22, 630–637. [Google Scholar] [CrossRef]
- van Eck, M.; Bos, I.S.; Kaminski, W.E.; Orso, E.; Rothe, G.; Twisk, J.; Böttcher, A.; Van Amersfoort, E.S.; Christiansen-Weber, T.A.; Fung-Leung, W.P.; et al. Leukocyte ABCA1 controls susceptibility to atherosclerosis and macrophage recruitment into tissues. Proc. Natl. Acad. Sci. USA 2002, 99, 6298–6303. [Google Scholar] [CrossRef] [PubMed]
- Bektas, M.; Allende, M.L.; Lee, B.G.; Chen, W.; Amar, M.J.; Remaley, A.T.; Saba, J.D.; Proia, R.L. Sphingosine 1-phosphate lyase deficiency disrupts lipid homeostasis in liver. J. Biol. Chem. 2010, 285, 10880–10889. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Malchinkhuu, E.; Horiuchi, Y.; Mogi, C.; Tomura, H.; Tosaka, M.; Yoshimoto, Y.; Kuwabara, A.; Okajima, F. Critical role of ABCA1 transporter in sphingosine 1-phosphate release from astrocytes. J. Neurochem. 2007, 103, 2610–2619. [Google Scholar] [CrossRef] [PubMed]
- Tall, A.R. Role of ABCA1 in cellular cholesterol efflux and reverse cholesterol transport. Arter. Thromb. Vasc. Biol. 2003, 23, 710–711. [Google Scholar] [CrossRef] [PubMed]
- Goetzl, E.J. Diverse pathways for nuclear signaling by G protein-coupled receptors and their ligands. Faseb. J. 2007, 21, 638–642. [Google Scholar] [CrossRef]
- Parham, K.A.; Zebol, J.R.; Tooley, K.L.; Sun, W.Y.; Moldenhauer, L.M.; Cockshell, M.P.; Gliddon, B.L.; Moretti, P.A.; Tigyi, G.; Pitson, S.M. Sphingosine 1-phosphate is a ligand for peroxisome proliferator-activated receptor-gamma that regulates neoangiogenesis. Faseb. J. 2015, 29, 3638–3653. [Google Scholar] [CrossRef]
- Carstairs, K.C. The identification of platelets and platelet antigens in histological sections. J. Pathol Bacteriol. 1965, 90, 225–231. [Google Scholar] [CrossRef]
- Robinet, P.; Wang, Z.; Hazen, S.L.; Smith, J.D. A simple and sensitive enzymatic method for cholesterol quantification in macrophages and foam cells. J. Lipid Res. 2010, 51, 3364–3369. [Google Scholar] [CrossRef]
- Hanel, P.; Andreani, P.; Graler, M.H. Erythrocytes store and release sphingosine 1-phosphate in blood. Faseb. J. 2007, 21, 1202–1209. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Macrophages [pmol/Cell × 106] | Plasma [pmol/mL] | |||||
|---|---|---|---|---|---|---|
| Western 1 wk | Western-B6+DOP 1 wk | Western 6 wk | Western-B6+DOP 6 wk | Western 6 wk | Western-B6+DOP 6 wk | |
| S1P | 0.48 ± 0.10 | 462.62 ± 18.36 ** | 0.17 ± 0.17 | 142.13 ± 31.46 § | 1411 ± 221 | 3854 ± 702 * |
| Sph | 2.9 ± 0.9 | 268.3 ± 26.5 ** | 3.5 ± 0.6 | 51.5 ± 9.5 § | 138 ± 22 | 255 ± 48 |
| Cer-16 | 42.7 ± 16.2 | 658.1 ± 94.7 * | 77.8 ± 7.6 | 245.0 ± 29.7 §§ | 3533 ± 1436 | 8031 ± 1504 * |
| SM | 1139 ± 287 | 2589 ± 405 * | 800 ± 125 | 737 ± 93 | 413,845 ± 168,542 | 414,410 ± 70,540 |
| PC | 282 ± 87 | 719 ± 96 * | 510 ± 86 | 433 ± 60 | 225,378 ± 30,820 | 315,700 ± 14,504 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Keul, P.; Peters, S.; von Wnuck Lipinski, K.; Schröder, N.H.; Nowak, M.K.; Duse, D.A.; Polzin, A.; Weske, S.; Gräler, M.H.; Levkau, B. Sphingosine-1-Phosphate (S1P) Lyase Inhibition Aggravates Atherosclerosis and Induces Plaque Rupture in ApoE−/− Mice. Int. J. Mol. Sci. 2022, 23, 9606. https://doi.org/10.3390/ijms23179606
Keul P, Peters S, von Wnuck Lipinski K, Schröder NH, Nowak MK, Duse DA, Polzin A, Weske S, Gräler MH, Levkau B. Sphingosine-1-Phosphate (S1P) Lyase Inhibition Aggravates Atherosclerosis and Induces Plaque Rupture in ApoE−/− Mice. International Journal of Molecular Sciences. 2022; 23(17):9606. https://doi.org/10.3390/ijms23179606
Chicago/Turabian StyleKeul, Petra, Susann Peters, Karin von Wnuck Lipinski, Nathalie H. Schröder, Melissa K. Nowak, Dragos A. Duse, Amin Polzin, Sarah Weske, Markus H. Gräler, and Bodo Levkau. 2022. "Sphingosine-1-Phosphate (S1P) Lyase Inhibition Aggravates Atherosclerosis and Induces Plaque Rupture in ApoE−/− Mice" International Journal of Molecular Sciences 23, no. 17: 9606. https://doi.org/10.3390/ijms23179606
APA StyleKeul, P., Peters, S., von Wnuck Lipinski, K., Schröder, N. H., Nowak, M. K., Duse, D. A., Polzin, A., Weske, S., Gräler, M. H., & Levkau, B. (2022). Sphingosine-1-Phosphate (S1P) Lyase Inhibition Aggravates Atherosclerosis and Induces Plaque Rupture in ApoE−/− Mice. International Journal of Molecular Sciences, 23(17), 9606. https://doi.org/10.3390/ijms23179606