
mPR-Specific Actions Influence Maintenance of the Blood–Brain Barrier (BBB)



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. CCM2 Is the Cornerstone for the Stability and Functionality of the CSC in Vascular ECs
2.2. mPR-Specific Actions on the Performance of the CCM Signaling Complex (CSC) in nPR(−) ECs
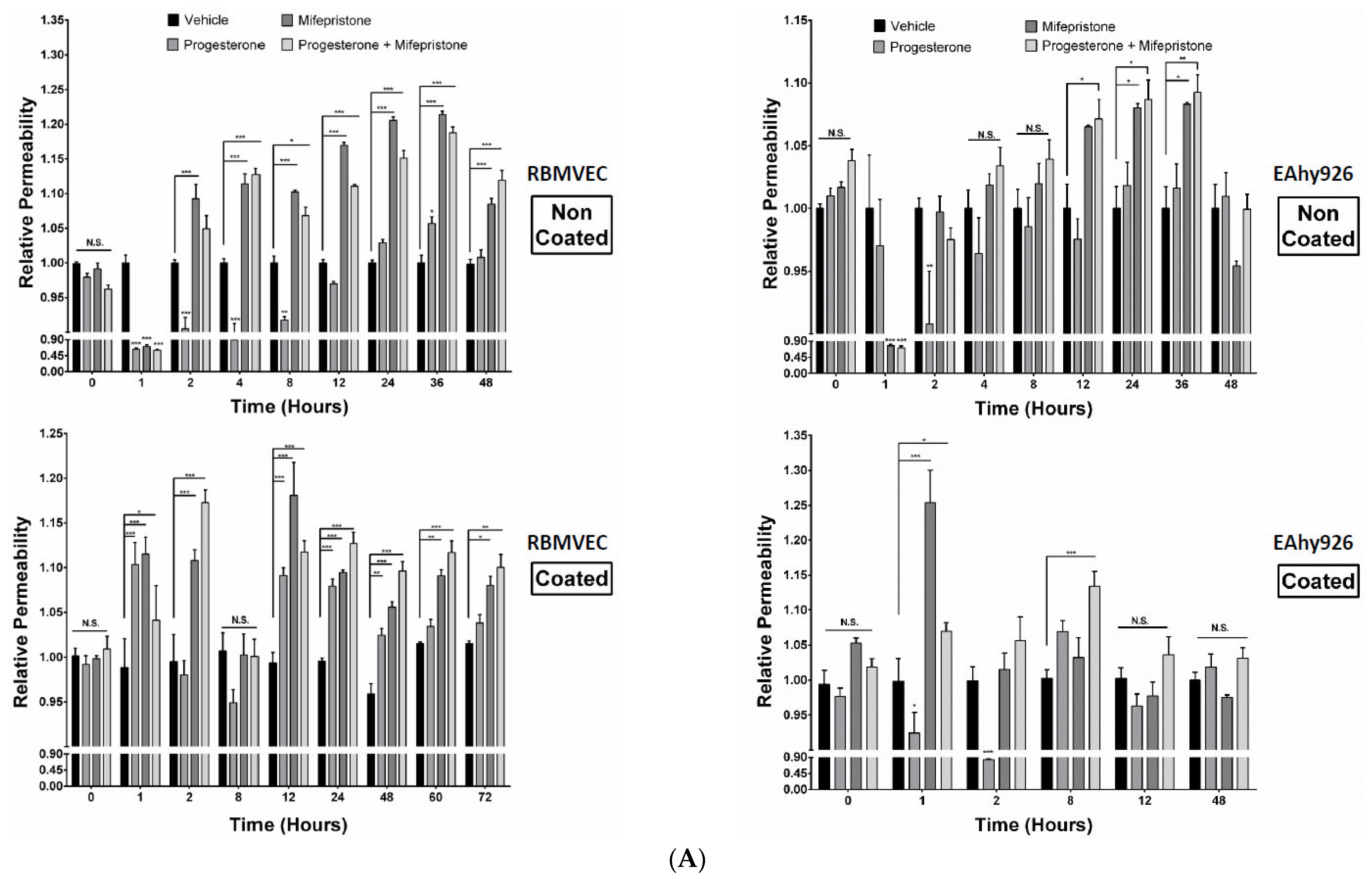
2.3. Differential Permeabilities between nPR(+/−) EC Monolayers under mPR-Specific PRG Actions In Vitro
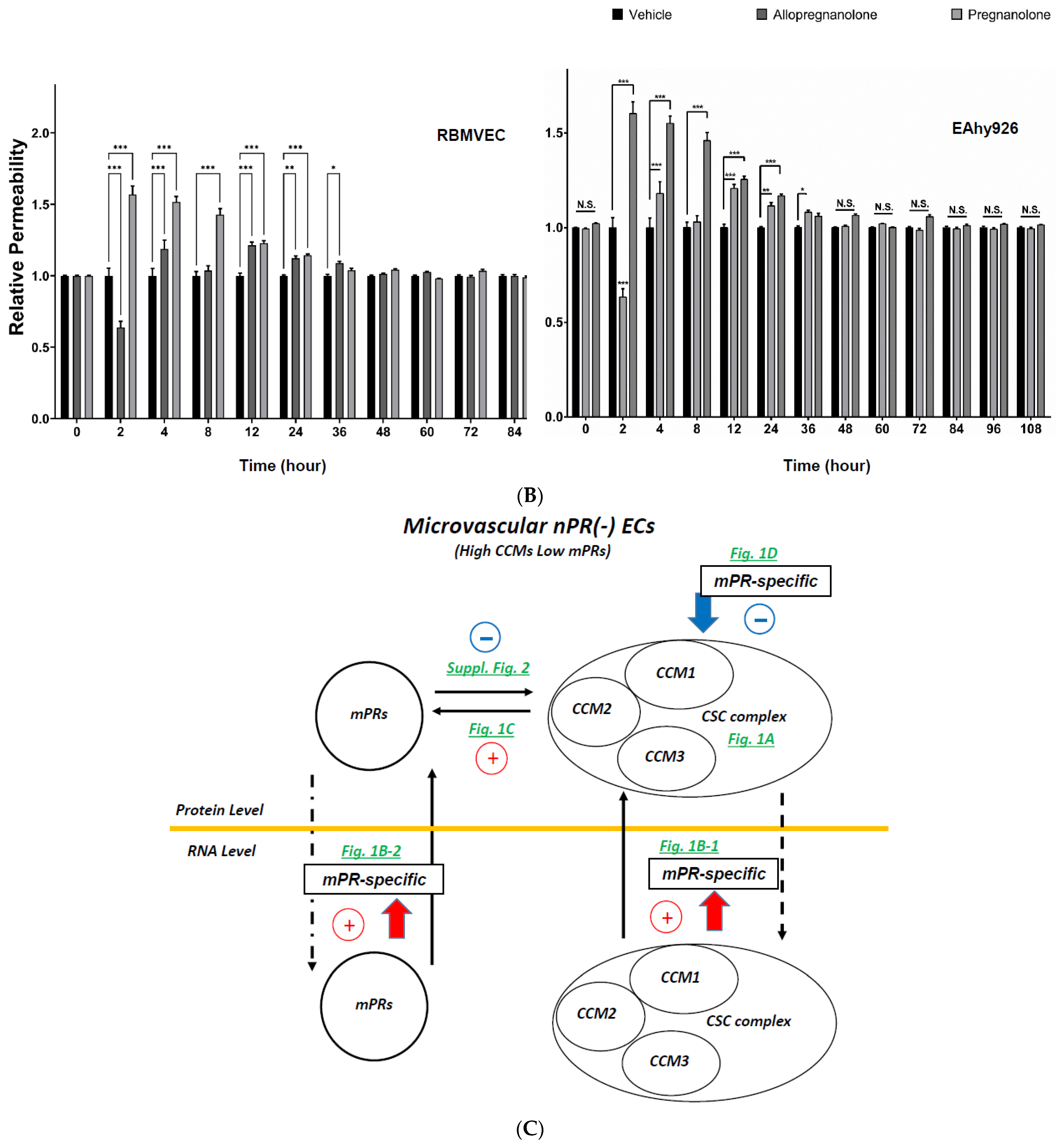
2.4. Subcellular Compartmentation of Key Factors of the CmP Network in nPR(−) RBMVECs under mPR-Specific PRG Actions
2.5. Perturbation of the CmP Signaling Network under mPR-Specific PRG Actions Is Sufficient for BBB Disruption in Ccm-Deficient Mice
2.6. Perturbation of the CmP Signaling Network under mPR-Specific PRG Actions Leads to Disrupted Angiogenesis in Ccm-Deficient Mice
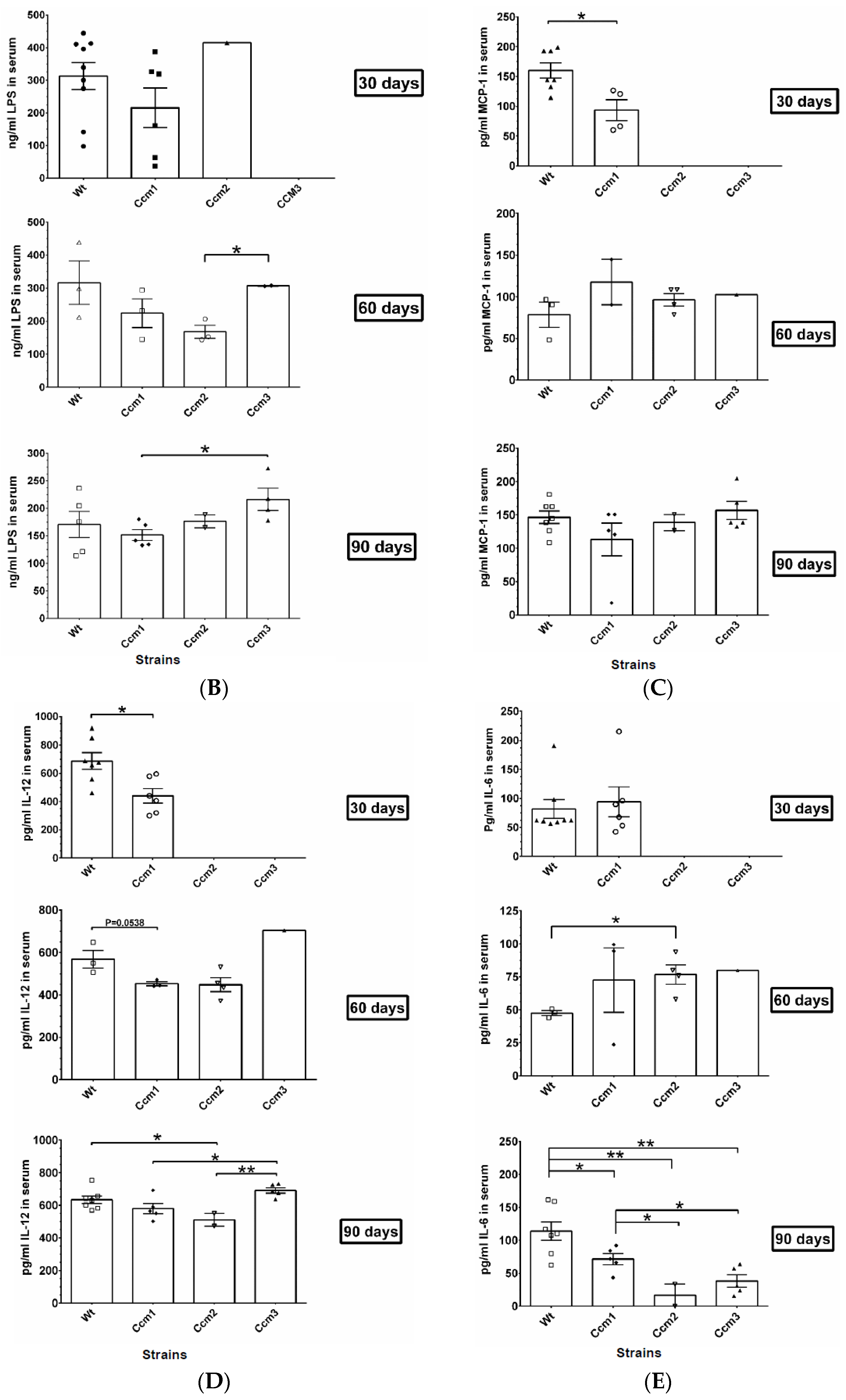
2.7. Immunosuppression Observed in Ccms Mutant Mice
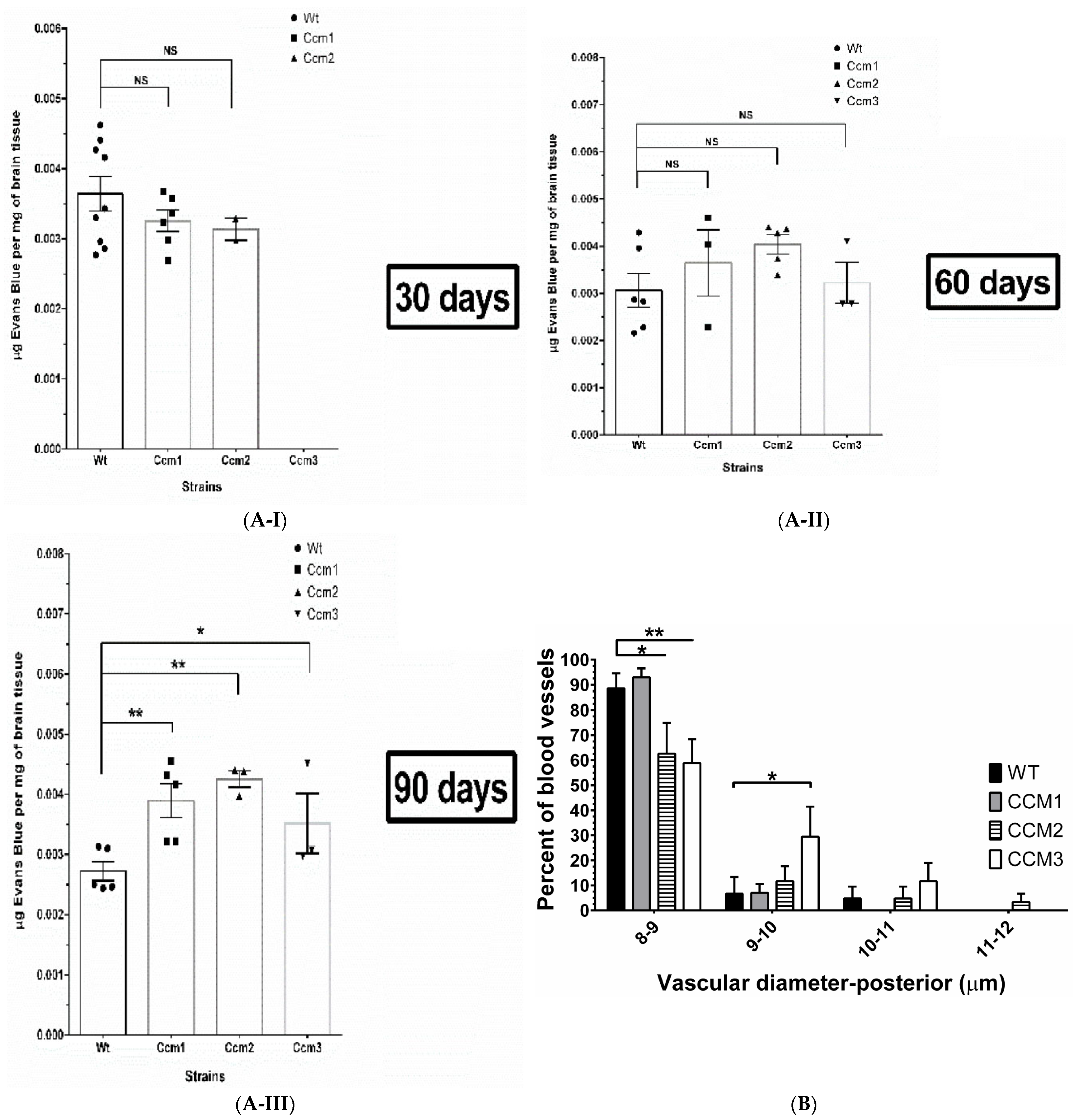
2.8. Perturbation of PRG Homeostasis Leads to a Disrupted BBB in Ccm Mutant Mice
2.9. Perturbation of Albumin Homeostasis/Biogenesis Is Observed in Ccm-Deficient Mice under mPR-Specific PRG Actions
3. Discussion
3.1. Perturbed Homeostasis of PRG Is a Casual “Trigger” for BBB Leakage
3.2. Novel Biomarkers Shed Light for the First Time to Predict the Potential Risk of BBB Disruption
4. Materials and Methods
4.1. Cell Culture and Treatment, Real-Time Quantitative PCR Analysis (RT-qPCR), and Western Blots
4.2. In Vitro EC Permeability Assays
4.3. Evaluation of Blood–Brain Barrier with Evan’s Blue Dye (EBD) from Brain Tissues
4.4. Immunofluorescent (IF) Staining Preparation for Evaluation of Subcutaneous Microvasculature in the Ears
4.5. Evaluation of Overall Angiogenic Performance of nPR(−) Aortic ECs from Ccm Mutant Mice Ex Vivo
4.6. Measurement of Cytokines and Etiological Serum Biomarkers
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CCMs | Cerebral cavernous malformations |
| CSC | CCM signaling complex |
| PRG | Progesterone |
| MIF | Mifepristone |
| nPRs | nuclear Progesterone Receptors |
| mPRs/PAQRs | membrane Progesterone Receptors or Progestin and AdipoQ Receptors |
| CmP | CSC-mPRs-PRG signaling network |
| DEGs | Differentially Expressed Genes |
| DEPs | Differentially Expressed Proteins |
| BBB | Blood–brain barrier |
References
- Padarti, A.; Zhang, J. Recent advances in cerebral cavernous malformation research. Vessel Plus 2018, 2, 21. [Google Scholar] [CrossRef]
- Abou-Fadel, J.; Vasquez, M.; Grajeda, B.; Ellis, C.; Zhang, J. Systems-wide analysis unravels the new roles of CCM signal complex (CSC). Heliyon 2019, 5, e02899. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Padarti, A.; Qu, Y.; Sheng, S.; Abou-Fadel, J.; Badr, A.; Zhang, J. Alternatively spliced isoforms reveal a novel type of PTB domain in CCM2 protein. Sci. Rep. 2019, 9, 15808. [Google Scholar] [CrossRef]
- Abou-Fadel, J.; Jiang, X.; Padarti, A.; Goswami, D.; Smith, M.; Grajeda, B.; Walker, W.; Zhang, J. CCM signaling complex (CSC) is a master regulator governing homeostasis of progestins and their mediated signaling cascades. bioRxiv 2020. [Google Scholar] [CrossRef]
- Zhang, J.; Abou-Fadel, J. Calm the raging hormone—a new therapeutic strategy involving progesterone-signaling for hemorrhagic CCMs. Vessel Plus 2021, 5, 48. [Google Scholar] [CrossRef] [PubMed]
- Abou-Fadel, J.; Qu, Y.; Gonzalez, E.; Smith, M.; Zhang, J. Emerging roles of CCM genes during tumorigenesis with potential application as novel biomarkers across major types of cancers. Oncol. Rep. 2020, 43, 1945–1963. [Google Scholar] [CrossRef]
- Abou-Fadel, J.; Bhalli, M.; Grajeda, B.; Zhang, J. CmP Signaling Network Leads to Identification of Prognostic Biomarkers for Triple-Negative Breast Cancer in Caucasian Women. Genet. Test. Mol. Biomarkers 2022, 26, 198–219. [Google Scholar] [CrossRef]
- Abou-Fadel, J.; Grajeda, B.; Jiang, X.; La O, A.-M.D.C.-D.; Flores, E.; Padarti, A.; Bhalli, M.; Le, A.; Zhang, J. CmP signaling network unveils novel biomarkers for triple negative breast cancer in African American women. Cancer Biomarkers 2022, 34, 607–636. [Google Scholar] [CrossRef]
- Karteris, E.; Zervou, S.; Pang, Y.; Dong, J.; Hillhouse, E.W.; Randeva, H.S.; Thomas, P. Progesterone Signaling in Human Myometrium through Two Novel Membrane G Protein-Coupled Receptors: Potential Role in Functional Progesterone Withdrawal at Term. Mol. Endocrinol. 2006, 20, 1519–1534. [Google Scholar] [CrossRef]
- Tan, J.; Paria, B.C.; Dey, S.K.; Das, S.K. Differential Uterine Expression of Estrogen and Progesterone Receptors Correlates with Uterine Preparation for Implantation and Decidualization in the Mouse1. Endocrinology 1999, 140, 5310–5321. [Google Scholar] [CrossRef]
- Witiw, C.D.; Abou-Hamden, A.; Kulkarni, A.V.; Silvaggio, J.A.; Schneider, C.; Wallace, M.C. Cerebral cavernous malformations and pregnancy: Hemorrhage risk and influence on obstetrical management. Neurosurgery 2012, 71, 626–630. [Google Scholar] [CrossRef] [PubMed]
- Kalani, M.Y.; Zabramski, J.M. Risk for symptomatic hemorrhage of cerebral cavernous malformations during pregnancy. J. Neurosurg. 2013, 118, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Safavi-Abbasi, S.; Feiz-Erfan, I.; Spetzler, R.F.; Kim, L.; Dogan, S.; Porter, R.W.; Sonntag, V.K.H. Hemorrhage of cavernous malformations during pregnancy and in the peripartum period: Causal or coincidence? Case report and review of the literature. Neurosurg. Focus. 2006, 15, e12. [Google Scholar] [CrossRef] [PubMed]
- Porter, R.W.; Detwiler, P.W.; Spetzler, R.F.; Lawton, M.T.; Baskin, J.J.; Derksen, P.T.; Zabramski, J.M. Cavernous malformations of the brainstem: Experience with 100 patients. J. Neurosurg. 1999, 90, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Nakase, H.; Nakagawa, I.; Nishimura, F.; Motoyama, Y.; Park, Y.-S. Cavernous Malformations in Pregnancy. Neurol. Med. Chir. 2013, 53, 555–560. [Google Scholar] [CrossRef] [PubMed]
- Abou-Fadel, J.; Jiang, X.; Padarti, A.; Grajeda, B.; Zhang, J. CCM signaling complex (CSC) coupling both classic and non-classic progesterone receptor signaling. Cell Commun. Signal 2022, 20, 120. [Google Scholar] [CrossRef]
- Simoncini, T.; Mannella, P.; Fornari, L.; Caruso, A.; Willis, M.Y.; Garibaldi, S.; Baldacci, C.; Genazzani, A.R. Differential Signal Transduction of Progesterone and Medroxyprogesterone Acetate in Human Endothelial Cells. Endocrinology 2004, 145, 5745–5756. [Google Scholar] [CrossRef]
- Zhang, J.; Basu, S.; Clatterbuck, R.E.; Rigamonti, D.; Dietz, H.C. Pathogenesis of cerebral cavernous malformation: Depletion of Krit1 leads to perturbation of 1 integrin-mediated endothelial cell mobility and survival. Am. J. Hum. Genet. 2004, 23, 5000. [Google Scholar]
- Zhang, J.; Basu, S.; Rigamonti, D.; Dietz, H.C.; Clatterbuck, R.E. Depletion of KRIT1 leads to perturbation of beta 1 integrin-mediated endothelial cell angiogenesis in the pathogenesis of cerebral cavernous malformation. Stroke 2005, 36, 425. [Google Scholar]
- Zhang, J.; Basu, S.; Rigamonti, D.; Dietz, H.C.; Clatterbuck, R.E. Krit1 modulates beta 1-integrin-mediated endothelial cell proliferation. Neurosurgery 2008, 63, 571–578. [Google Scholar] [CrossRef]
- Thomas, P.; Pang, Y. Anti-apoptotic Actions of Allopregnanolone and Ganaxolone Mediated Through Membrane Progesterone Receptors (PAQRs) in Neuronal Cells. Front. Endocrinol. 2020, 11, 417. [Google Scholar] [CrossRef] [PubMed]
- Patte-Mensah, C.; Meyer, L.; Taleb, O.; Mensah-Nyagan, A.G. Potential role of allopregnanolone for a safe and effective therapy of neuropathic pain. Prog. Neurobiol. 2014, 113, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Avallone, R.; Lucchi, C.; Puja, G.; Codeluppi, A.; Filaferro, M.; Vitale, G.; Rustichelli, C.; Biagini, G. BV-2 Microglial Cells Respond to Rotenone Toxic Insult by Modifying Pregnenolone, 5α-Dihydroprogesterone and Pregnanolone Levels. Cells 2020, 9, 2091. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.M.; Pinna, G.; Guidotti, A. Allopregnanolone: From molecular pathophysiology to therapeutics. A historical perspective. Neurobiol. Stress 2020, 12, 100215. [Google Scholar] [CrossRef]
- Pang, Y.; Dong, J.; Thomas, P. Characterization, neurosteroid binding and brain distribution of human membrane progesterone receptors delta and {epsilon} (mPRdelta and mPR{epsilon}) and mPRdelta involvement in neurosteroid inhibition of apoptosis. Endocrinology 2013, 154, 283–295. [Google Scholar] [CrossRef]
- Liu, H.; Rigamonti, D.; Badr, A.; Zhang, J. Ccm1 Assures Microvascular Integrity During Angiogenesis. Transl. Stroke Res. 2010, 1, 146–153. [Google Scholar] [CrossRef]
- Zhang, J.; Carr, C.; Badr, A. The Cardiovascular Triad of Dysfunctional Angiogenesis. Transl. Stroke Res. 2011, 2, 339–345. [Google Scholar] [CrossRef]
- Liu, H.; Rigamonti, D.; Badr, A.; Zhang, J. Ccm1 Regulates Microvascular Morphogenesis during Angiogenesis. J. Vasc. Res. 2010, 48, 130–140. [Google Scholar] [CrossRef]
- Boulday, G.; Blécon, A.; Petit, N.; Chareyre, F.; Garcia, L.A.; Niwa-Kawakita, M.; Giovannini, M.; Tournier-Lasserve, E. Tissue-specific conditionalCCM2knockout mice establish the essential role of endothelial CCM2 in angiogenesis: Implications for human cerebral cavernous malformations. Dis. Model. Mech. 2009, 2, 168–177. [Google Scholar] [CrossRef]
- Cohen-Gadol, A.A.; Jacob, J.T.; Edwards, D.A.; Krauss, W.E. Coexistence of intracranial and spinal cavernous malformations: A study of prevalence and natural history. J. Neurosurg. 2006, 104, 376–381. [Google Scholar] [CrossRef]
- McDonald, D.A.; Shenkar, R.; Shi, C.; Stockton, R.A.; Akers, A.L.; Kucherlapati, M.H.; Kucherlapati, R.; Brainer, J.; Ginsberg, M.H.; Awad, I.A.; et al. A novel mouse model of cerebral cavernous malformations based on the two-hit mutation hypothesis recapitulates the human disease. Hum. Mol. Genet. 2011, 20, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Detter, M.R.; Shenkar, R.; Benavides, C.R.; Neilson, C.A.; Moore, T.; Lightle, R.; Hobson, N.; Shen, L.; Cao, Y.; Girarg, R.; et al. Novel hemorrhage models of cerebral cavernous malformations. bioRxiv 2020. [Google Scholar] [CrossRef]
- Meliton, A.; Meng, F.; Tian, Y.; Shah, A.A.; Birukova, A.A.; Birukov, K.G. Role of Krev Interaction Trapped-1 in Prostacyclin-Induced Protection against Lung Vascular Permeability Induced by Excessive Mechanical Forces and Thrombin Receptor Activating Peptide 6. Am. J. Respir. Cell Mol. Biol. 2015, 53, 834–843. [Google Scholar] [CrossRef] [PubMed]
- Vieceli Dalla Sega, F.; Mastrocola, R.; Aquila, G.; Fortini, F.; Fornelli, C.; Zotta, A.; Cento, A.S.; Perrelli, A.; Boda, E.; Pannuti, A.; et al. KRIT1 Deficiency Promotes Aortic Endothelial Dysfunction. Int. J. Mol. Sci. 2019, 5, 4930. [Google Scholar] [CrossRef]
- Tetel, M.J.; De Vries, G.J.; Melcangi, R.C.; Panzica, G.; O’Mahony, S.M. Steroids, stress and the gut microbiome-brain axis. J. Neuroendocr. 2017, 30, e12548. [Google Scholar] [CrossRef]
- Jang, S.-E.; Lim, S.-M.; Jeong, J.-J.; Jang, H.-M.; Lee, H.-J.; Han, M.J.; Kim, D.-H. Gastrointestinal inflammation by gut microbiota disturbance induces memory impairment in mice. Mucosal. Immunol. 2018, 11, 369–379. [Google Scholar] [CrossRef]
- Cassado Ados, A.; D’Imperio Lima, M.R.; Bortoluci, K.R. Revisiting mouse peritoneal macrophages: Heterogeneity, development, and function. Front. Immunol. 2015, 6, 225. [Google Scholar] [CrossRef]
- Okabe, Y.; Medzhitov, R. Tissue-Specific Signals Control Reversible Program of Localization and Functional Polarization of Macrophages. Cell 2014, 157, 832–844. [Google Scholar] [CrossRef]
- Szczepanik, M. Interplay between Helicobacter pylori and the immune system. Clinical implications. J. Physiol. Pharmacol. 2006, 57 (Suppl. S3), 15–27. [Google Scholar]
- Dressing, G.E.; Goldberg, J.E.; Charles, N.J.; Schwertfeger, K.L.; Lange, C.A. Membrane progesterone receptor expression in mammalian tissues: A review of regulation and physiological implications. Steroids 2011, 76, 11–17. [Google Scholar] [CrossRef]
- Chien, C.; Lai, J.; Liao, C.; Wang, O.; Lu, L.; Huang, M.; Lee, W.; Shie, M.; Chien, E. Mifepristone acts as progesterone antagonist of non-genomic responses but inhibits phytohemagglutinin-induced proliferation in human T cells. Hum. Reprod. 2009, 24, 1968–1975. [Google Scholar] [CrossRef] [PubMed]
- Henderson, T.A.; Saunders, P.; Moffett-King, A.; Groome, N.P.; Critchley, H.O.D. Steroid Receptor Expression in Uterine Natural Killer Cells. J. Clin. Endocrinol. Metab. 2003, 88, 440–449. [Google Scholar] [CrossRef] [PubMed]
- Szekeres-Bartho, J.; Philibert, D.; Chaouat, G. Progesterone Suppression of Pregnancy Lymphocytes Is not Mediated by Glucocorticoid Effect. Am. J. Reprod. Immunol. 1990, 23, 42–43. [Google Scholar] [CrossRef] [PubMed]
- Dosiou, C.; Hamilton, A.E.; Pang, Y.; Overgaard, M.T.; Tulac, S.; Dong, J.; Thomas, P.; Giudice, L.C. Expression of membrane progesterone receptors on human T lymphocytes and Jurkat cells and activation of G-proteins by progesterone. J. Endocrinol. 2008, 196, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Areia, A.; Vale-Pereira, S.; Alves, V.; Rodrigues-Santos, P.; Moura, P.; Mota-Pinto, A. Membrane progesterone receptors in human regulatory T cells: A reality in pregnancy. BJOG 2015, 122, 1544–1550. [Google Scholar] [CrossRef]
- Hammond, G.L. Potential functions of plasma steroid-binding proteins. Trends Endocrinol. Metab. 1995, 6, 298–304. [Google Scholar] [CrossRef]
- Cox, R.M.; McGlothlin, J.W.; Bonier, F. Evolutionary Endocrinology: Hormones as Mediators of Evolutionary Phenomena: An Introduction to the Symposium. Integr. Comp. Biol. 2016, 56, 121–125. [Google Scholar] [CrossRef]
- Canavero, S. Intramedullary cavernous angiomas of the spinal cord: Clinical presentation, pathological features, and surgical management. Neurosurgery 1993, 32, 692–693. [Google Scholar] [CrossRef]
- Burkhardt, J.K.; Bozinov, O.; Nurnberg, J.; Shin, B.; Woernle, C.M.; Ulrich, N.H.; Bertalanffy, H. Neurosurgical considerations on highly eloquent brainstem cavernomas during pregnancy. Clin. Neurol. Neurosurg. 2012, 114, 1172–1176. [Google Scholar] [CrossRef]
- Gross, B.A.; Lin, N.; Du, R.; Day, A.L. The natural history of intracranial cavernous malformations. Neurosurg. Focus 2011, 30, E24. [Google Scholar] [CrossRef]
- Pastushyn, A.I.; Slin’Ko, E.I.; Mirzoyeva, G.M. Vertebral hemangiomas: Diagnosis, management, natural history and clinicopathological correlates in 86 patients. Surg. Neurol. 1998, 50, 535–547. [Google Scholar] [CrossRef]
- Babu, R.; Owens, T.R.; Karikari, I.O.; Moreno, J.; Cummings, T.J.; Gottfried, O.N.; Bagley, C.A. Spinal Cavernous and Capillary Hemangiomas in Adults. Spine 2013, 38, E423–E430. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, T.H.; Hibshoosh, H.; Riedel, C.J. Estrogen and Progesterone Receptor-negative T11 Vertebral Hemangioma Presenting as a Postpartum Compression Fracture: Case Report and Management. Neurosurgery 2000, 46, 218–221. [Google Scholar] [CrossRef]
- Morello, A.; Tumbiolo, A.; Pinto, G.; Duca, B.L. Cavernous angioma of the spinal dura. J. Neurosurg. Sci. 1991, 35, 31–35. [Google Scholar] [PubMed]
- Di Tommaso, L.; Scarpellini, F.; Salvi, F.; Ragazzini, T.; Foschini, M.P. Progesterone receptor expression in orbital cavernous hemangiomas. Virchows Arch. 2000, 436, 284–288. [Google Scholar] [CrossRef]
- Kaya, A.H.; Ulus, A.; Bayri, Y.; Topal, A.; Gun, S.; Kandemir, B.; Dagcinar, A.; Senel, A.; Iyigun, O. There are no estrogen and progesterone receptors in cerebral cavernomas: A preliminary immunohistochemical study. Surg. Neurol. 2009, 72, 263–265. [Google Scholar] [CrossRef] [PubMed]
- Perrelli, A.; Retta, S.F. Polymorphisms in genes related to oxidative stress and inflammation: Emerging links with the pathogenesis and severity of Cerebral Cavernous Malformation disease. Free Radic. Biol. Med. 2021, 172, 403–417. [Google Scholar] [CrossRef]
- Bäckström, T.; Bixo, M.; Johansson, M.; Nyberg, S.; Ossewaarde, L.; Ragagnin, G.; Savic, I.; Strömberg, J.; Timby, E.; van Broekhoven, F.; et al. Allopregnanolone and mood disorders. Prog. Neurobiol. 2014, 113, 88–94. [Google Scholar] [CrossRef]
- Epperson, C.N.; Steiner, M.; Hartlage, S.A.; Eriksson, E.; Schmidt, P.J.; Jones, I.; Yonkers, K.A. Premenstrual Dysphoric Disorder: Evidence for a New Category for DSM-5. Am. J. Psychiatry 2012, 169, 465–475. [Google Scholar] [CrossRef]
- Stoffel-Wagner, B. Neurosteroid Biosynthesis in the Human Brain and Its Clinical Implications. Ann. N. Y. Acad. Sci. 2003, 1007, 64–78. [Google Scholar] [CrossRef]
- Ottander, U.; Poromaa, I.S.; Bjurulf, E.; Skytt, Å.; Bäckström, T.; Olofsson, J.I. Allopregnanolone and pregnanolone are produced by the human corpus luteum. Mol. Cell. Endocrinol. 2005, 239, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Bäckström, T.; Andreen, L.; Birzniece, V.; Björn, I.; Johansson, I.-M.; Nordenstam-Haghjo, M.; Nyberg, S.; Sundström-Poromaa, I.; Wahlström, G.; Wang, M.; et al. The role of hormones and hormonal treatments in premenstrual syndrome. CNS Drugs 2003, 17, 325–342. [Google Scholar] [CrossRef] [PubMed]
- Girdler, S.S.; Klatzkin, R. Neurosteroids in the context of stress: Implications for depressive disorders. Pharmacol. Ther. 2007, 116, 125–139. [Google Scholar] [CrossRef] [Green Version]
- Gaffey, A.E.; Rosman, L.; Burg, M.M.; Haskell, S.G.; Brandt, C.A.; Skanderson, M.; Dziura, J.; Sico, J.J. Posttraumatic Stress Disorder, Antidepressant Use, and Hemorrhagic Stroke in Young Men and Women: A 13-Year Cohort Study. Stroke 2021, 52, 121–129. [Google Scholar] [CrossRef]
- Clatterbuck, R.E.; Eberhart, C.G.; Crain, B.J.; Rigamonti, D. Ultrastructural and immunocytochemical evidence that an incompetent blood-brain barrier is related to the pathophysiology of cavernous malformations. J. Neurol. Neurosurg. Psychiatry 2001, 71, 188–192. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Ramirez, M.A.; Pham, A.; Girard, R.; Wyseure, T.; Hale, P.; Yamashita, A.; Koskimäki, J.; Polster, S.; Saadat, L.; Romero, I.A.; et al. Cerebral cavernous malformations form an anticoagulant vascular domain in humans and mice. Blood 2019, 133, 193–204. [Google Scholar] [CrossRef]
- Amoozegar, F.; Ronksley, P.E.; Sauve, R.; Menon, B.K. Hormonal Contraceptives and Cerebral Venous Thrombosis Risk: A Systematic Review and Meta-Analysis. Front. Neurol. 2015, 6, 7. [Google Scholar] [CrossRef]
- Tang, A.T.; Choi, J.P.; Kotzin, J.J.; Yang, Y.; Hong, C.C.; Hobson, N.; Girard, R.; Zeineddine, H.A.; Lightle, R.; Moore, T.; et al. Endothelial TLR4 and the microbiome drive cerebral cavernous malformations. Nature 2017, 545, 305–310. [Google Scholar] [CrossRef]
- Argaw, A.T.; Asp, L.; Zhang, J.; Navrazhina, K.; Pham, T.; Mariani, J.N.; Mahase, S.; Dutta, D.J.; Seto, J.; Kramer, E.G.; et al. Astrocyte-derived VEGF-A drives blood-brain barrier disruption in CNS inflammatory disease. J. Clin. Investig. 2012, 122, 2454–2468. [Google Scholar] [CrossRef]
- Morita-Takemura, S.; Nakahara, K.; Tatsumi, K.; Okuda, H.; Tanaka, T.; Isonishi, A.; Wanaka, A. Changes in endothelial cell proliferation and vascular permeability after systemic lipopolysaccharide administration in the subfornical organ. J. Neuroimmunol. 2016, 298, 132–137. [Google Scholar] [CrossRef]
- Banks, W.A.; Gray, A.M.; Erickson, M.A.; Salameh, T.S.; Damodarasamy, M.; Sheibani, N.; Meabon, J.S.; Wing, E.E.; Morofuji, Y.; Cook, D.G.; et al. Lipopolysaccharide-induced blood-brain barrier disruption: Roles of cyclooxygenase, oxidative stress, neuroinflammation, and elements of the neurovascular unit. J. Neuroinflamm. 2015, 12, 223. [Google Scholar] [CrossRef] [PubMed]
- Batista, C.R.A.; Gomes, G.F.; Candelario-Jalil, E.; Fiebich, B.L.; De Oliveira, A.C.P. Lipopolysaccharide-Induced Neuroinflammation as a Bridge to Understand Neurodegeneration. Int. J. Mol. Sci. 2019, 20, 2293. [Google Scholar] [CrossRef] [PubMed]
- Moriarity, J.L.; Clatterbuck, R.E.; Rigamonti, D. The natural history of cavernous malformations. Neurosurg. Clin. N. Am. 1999, 10, 11–17. [Google Scholar] [CrossRef]
- Maraire, J.N.; Awad, I.A. Intracranial cavernous malformations: Lesion behavior and management strategies. Neurosurgery 1995, 37, 591–605. [Google Scholar] [CrossRef]
- Simpkins, A.N.; Janowski, M.; Oz, H.S.; Roberts, J.; Bix, G.; Doré, S.; Stowe, A.M. Biomarker Application for Precision Medicine in Stroke. Transl. Stroke Res. 2019, 11, 615–627. [Google Scholar] [CrossRef]
- Augello, C.J.; Noll, J.M.; Distel, T.J.; Wainright, J.D.; Stout, C.E.; Ford, B.D. Identification of novel blood biomarker panels to detect ischemic stroke in patients and their responsiveness to therapeutic intervention. Brain Res. 2018, 1698, 161–169. [Google Scholar] [CrossRef]
- Makris, K.; Haliassos, A.; Chondrogianni, M.; Tsivgoulis, G. Blood biomarkers in ischemic stroke: Potential role and challenges in clinical practice and research. Crit. Rev. Clin. Lab. Sci. 2018, 55, 294–328. [Google Scholar] [CrossRef]
- Girard, R.; Zeineddine, H.A.; Fam, M.D.; Mayampurath, A.; Cao, Y.; Shi, C.; Shenkar, R.; Polster, S.P.; Jesselson, M.; Duggan, R.; et al. Plasma Biomarkers of Inflammation Reflect Seizures and Hemorrhagic Activity of Cerebral Cavernous Malformations. Transl. Stroke Res. 2018, 9, 34–43. [Google Scholar] [CrossRef]
- Lambertsen, K.L.; Biber, K.; Finsen, B. Inflammatory Cytokines in Experimental and Human Stroke. J. Cereb. Blood Flow Metab. 2012, 32, 1677–1698. [Google Scholar] [CrossRef]
- Bokhari, F.A.; Shakoori, T.; Butt, A.; Ghafoor, F. TNF-alpha: A risk factor for ischemic stroke. J. Ayub Med. Coll. Abbottabad 2015, 26, 111–114. [Google Scholar]
- Georgakis, M.K.; Malik, R.; Björkbacka, H.; Pana, T.A.; Demissie, S.; Ayers, C.; Elhadad, M.A.; Fornage, M.; Beiser, A.S.; Benjamin, E.J.; et al. Circulating Monocyte Chemoattractant Protein-1 and Risk of Stroke: A Meta-Analysis of Population-Based Studies Involving 17,180 Individuals. Circ. Res. 2019, 125, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Wang, D.I.; Zhou, Y.; Xiong, H. Association between the interleukin-6-174 G/C polymorphism and risk of ischemic stroke: A meta-analysis. Genet. Mol. Res. 2015, 14, 13076–13083. [Google Scholar]
- Girard, R.; Zeineddine, H.A.; Koskimaki, J.; Fam, M.D.; Cao, Y.; Shi, C.; Moore, T.; Lightle, R.; Stadnik, A.; Chaudagar, K.; et al. Plasma Biomarkers of Inflammation and Angiogenesis Predict Cerebral Cavernous Malformation Symptomatic Hemorrhage or Lesional Growth. Circ. Res. 2018, 122, 1716–1721. [Google Scholar] [CrossRef] [PubMed]
- Weinsheimer, S.; Kim, H. Circulating plasma biomarkers associated with familial CCMs. In Proceedings of the 16th Angioma Alliance CCM Scientific Meeting, Durham, NC, USA, 12–13 November 2020; Volume 2. [Google Scholar]
- O’Connell, G.C.; Walsh, K.B.; Burrage, E.; Adeoye, O.; Chantler, P.D.; Barr, T.L. High-throughput profiling of the circulating proteome suggests sexually dimorphic corticosteroid signaling following ischemic stroke. Physiol. Genom. 2018, 50, 876–883. [Google Scholar] [CrossRef]
- He, H.; Guo, J. Serum albumin: A risk of stroke? Am. J. Emerg. Med. 2017, 35, 186–187. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Hapon, M.B.; Goyeneche, A.A.; Srinivasan, R.; Gamarra-Luques, C.D.; Callegari, E.A.; Drappeau, D.D.; Terpstra, E.J.; Pan, B.; Knapp, J.R.; et al. Mifepristone increases mRNA translation rate, triggers the unfolded protein response, increases autophagic flux, and kills ovarian cancer cells in combination with proteasome or lysosome inhibitors. Mol. Oncol. 2016, 10, 1099–1117. [Google Scholar] [CrossRef]
- Wang, C.; Deng, L.; Qiu, S.; Bian, H.; Wang, L.; Li, Y.; Liu, M.; Wu, B. Serum Albumin Is Negatively Associated with Hemorrhagic Transformation in Acute Ischemic Stroke Patients. Cerebrovasc. Dis. 2019, 47, 88–94. [Google Scholar] [CrossRef]
- Nimmagadda, A.; Park, H.P.; Prado, R.; Ginsberg, M.D. Albumin therapy improves local vascular dynamics in a rat model of primary microvascular thrombosis: A two-photon laser-scanning microscopy study. Stroke 2008, 39, 198–204. [Google Scholar] [CrossRef]
- Hill, M.D.; Moy, C.S.; Palesch, Y.Y.; Martin, R.; Dillon, C.R.; Waldman, B.D.; Patterson, L.; Mendez, I.M.; Ryckborst, K.J.; Tamariz, D.; et al. The Albumin in Acute Stroke Trial (ALIAS); Design and Methodology. Int. J. Stroke 2007, 2, 214–219. [Google Scholar] [CrossRef]
- Yousuf, S.; Atif, F.; Sayeed, I.; Wang, J.; Stein, D.G. Neuroprotection by progesterone after transient cerebral ischemia in stroke-prone spontaneously hypertensive rats. Horm. Behav. 2016, 84, 29–40. [Google Scholar] [CrossRef]
- Singh, M.; Su, C. Progesterone and neuroprotection. Horm Behav. 2013, 63, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, M.; Denier, C.; Oudinet, J.-P.; Adams, D.; Guennoun, R. Progesterone neuroprotection: The background of clinical trial failure. J. Steroid Biochem. Mol. Biol. 2016, 160, 53–66. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Clatterbuck, R.E.; Rigamonti, D.; Chang, D.D.; Dietz, H.C. Novel insights regarding the pathogenesis of cerebral cavernous malformation (CCM). Am. J. Hum. Genet. 2001, 69, 178. [Google Scholar]
- Zhang, J.; Rigamonti, D.; Dietz, H.C.; Clatterbuck, R.E. Interaction between krit1 and malcavernin: Implications for the pathogenesis of cerebral cavernous malformations. Neurosurgery 2007, 60, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Mikelis, C.M.; Simaan, M.; Ando, K.; Fukuhara, S.; Sakurai, A.; Amornphimoltham, P.; Masedunskas, A.; Weigert, R.; Chavakis, T.; Adams, R.H.; et al. RhoA and ROCK mediate histamine-induced vascular leakage and anaphylactic shock. Nat. Commun. 2015, 6, 6725. [Google Scholar] [CrossRef]
- Yamazaki, T.; Li, W.; Mukouyama, Y.-S. Whole-mount Confocal Microscopy for Adult Ear Skin: A Model System to Study Neuro-vascular Branching Morphogenesis and Immune Cell Distribution. J. Vis. Exp. 2018, 133, e57406. [Google Scholar] [CrossRef]
- Nicosia, R.F. The aortic ring model of angiogenesis: A quarter century of search and discovery. J. Cell. Mol. Med. 2009, 13, 4113–4136. [Google Scholar] [CrossRef]
- Walker, W.E. Methods to Study the Innate Immune Response to Sepsis. Methods Mol. Biol. 2018, 1717, 189–206. [Google Scholar] [CrossRef]












Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abou-Fadel, J.; Jiang, X.; Padarti, A.; Goswami, D.G.; Smith, M.; Grajeda, B.; Bhalli, M.; Le, A.; Walker, W.E.; Zhang, J. mPR-Specific Actions Influence Maintenance of the Blood–Brain Barrier (BBB). Int. J. Mol. Sci. 2022, 23, 9684. https://doi.org/10.3390/ijms23179684
Abou-Fadel J, Jiang X, Padarti A, Goswami DG, Smith M, Grajeda B, Bhalli M, Le A, Walker WE, Zhang J. mPR-Specific Actions Influence Maintenance of the Blood–Brain Barrier (BBB). International Journal of Molecular Sciences. 2022; 23(17):9684. https://doi.org/10.3390/ijms23179684
Chicago/Turabian StyleAbou-Fadel, Johnathan, Xiaoting Jiang, Akhil Padarti, Dinesh G. Goswami, Mark Smith, Brian Grajeda, Muaz Bhalli, Alexander Le, Wendy E. Walker, and Jun Zhang. 2022. "mPR-Specific Actions Influence Maintenance of the Blood–Brain Barrier (BBB)" International Journal of Molecular Sciences 23, no. 17: 9684. https://doi.org/10.3390/ijms23179684
APA StyleAbou-Fadel, J., Jiang, X., Padarti, A., Goswami, D. G., Smith, M., Grajeda, B., Bhalli, M., Le, A., Walker, W. E., & Zhang, J. (2022). mPR-Specific Actions Influence Maintenance of the Blood–Brain Barrier (BBB). International Journal of Molecular Sciences, 23(17), 9684. https://doi.org/10.3390/ijms23179684