
Next-Generation Sequencing of Local Romanian Tomato Varieties and Bioinformatics Analysis of the Ve Locus




Abstract
:1. Introduction
2. Results
2.1. NGS Data Analysis
2.1.1. Sequencing Data Quality Control
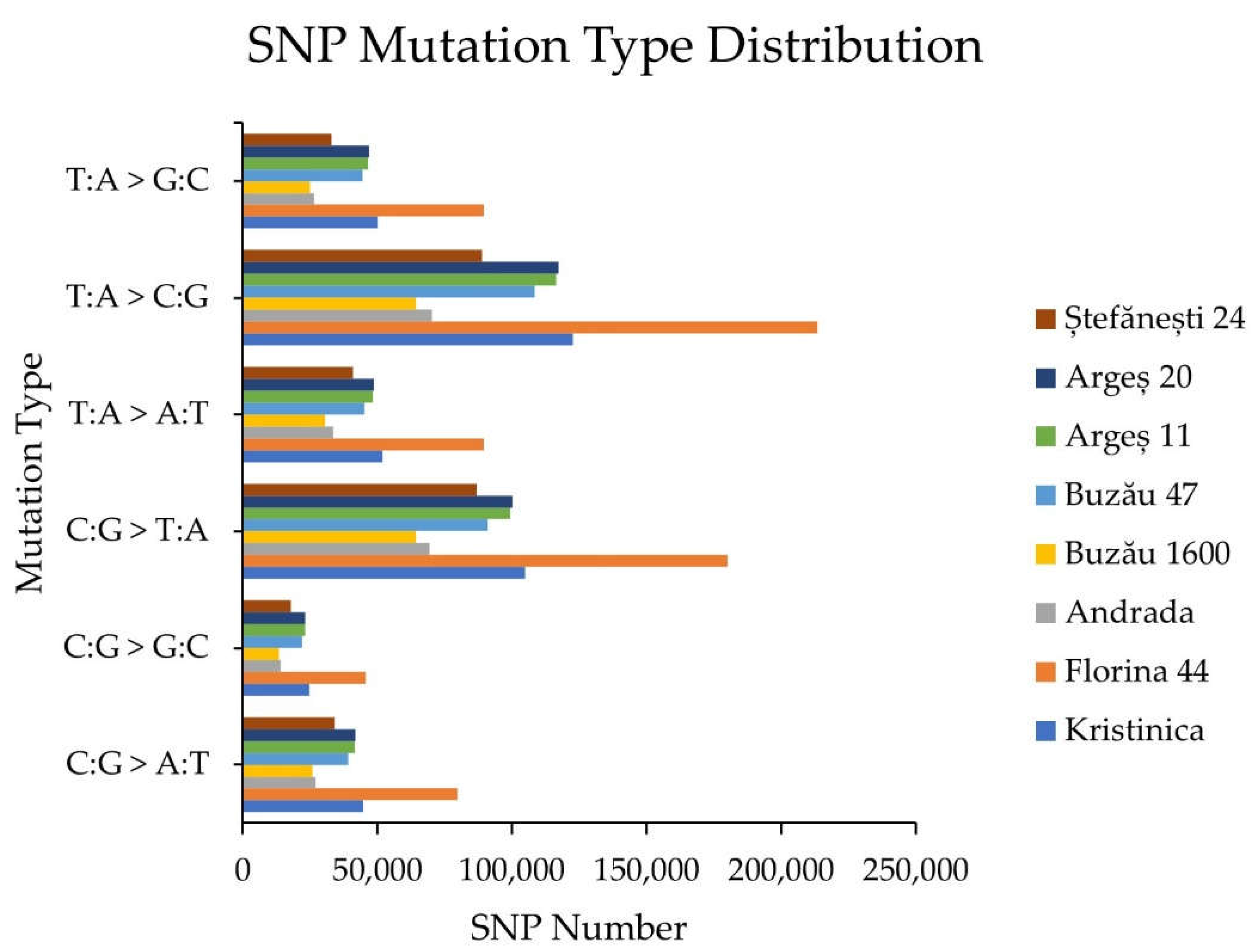
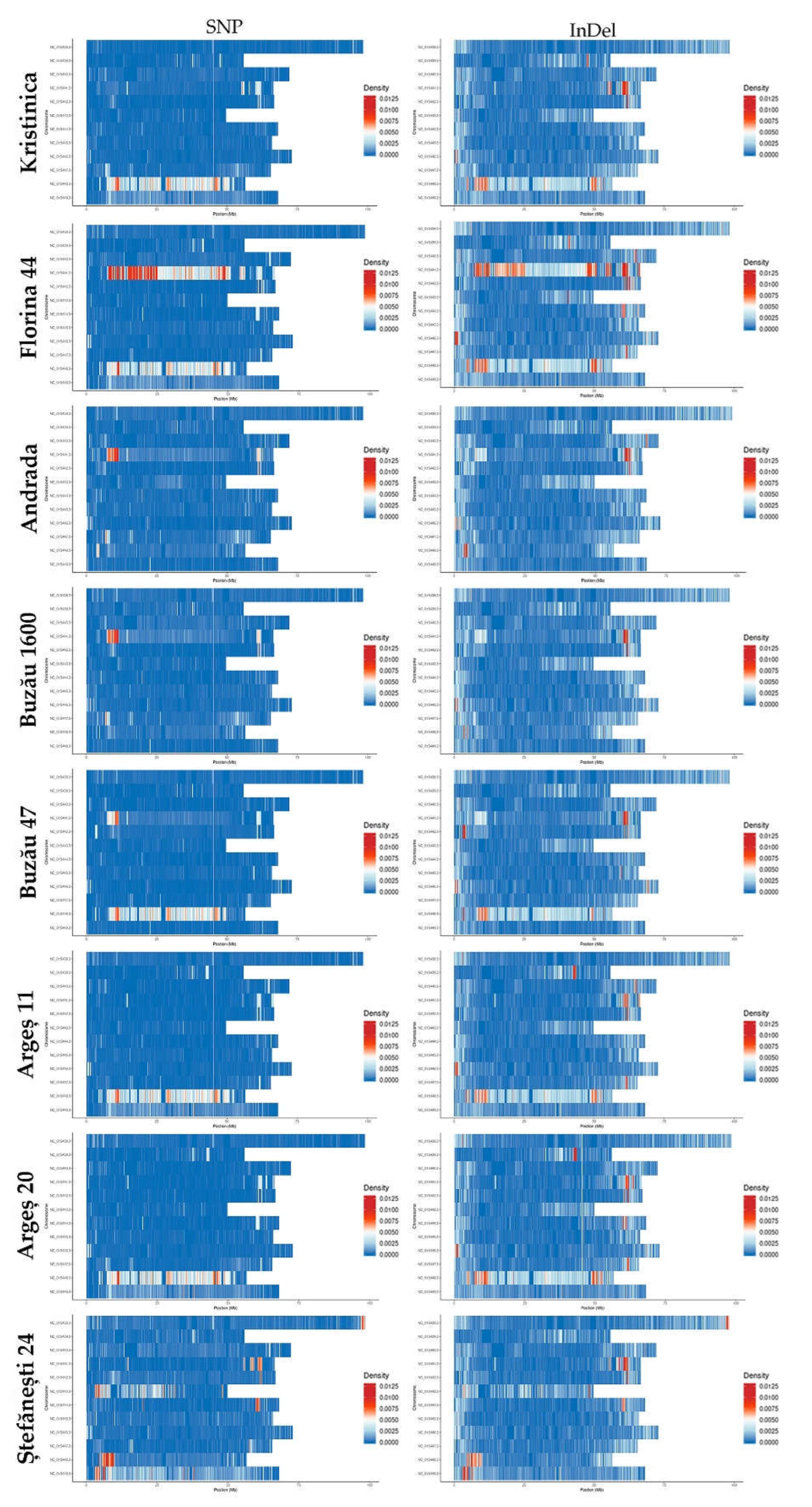
2.1.2. SNP Detection, Distribution, and Mutation Frequency
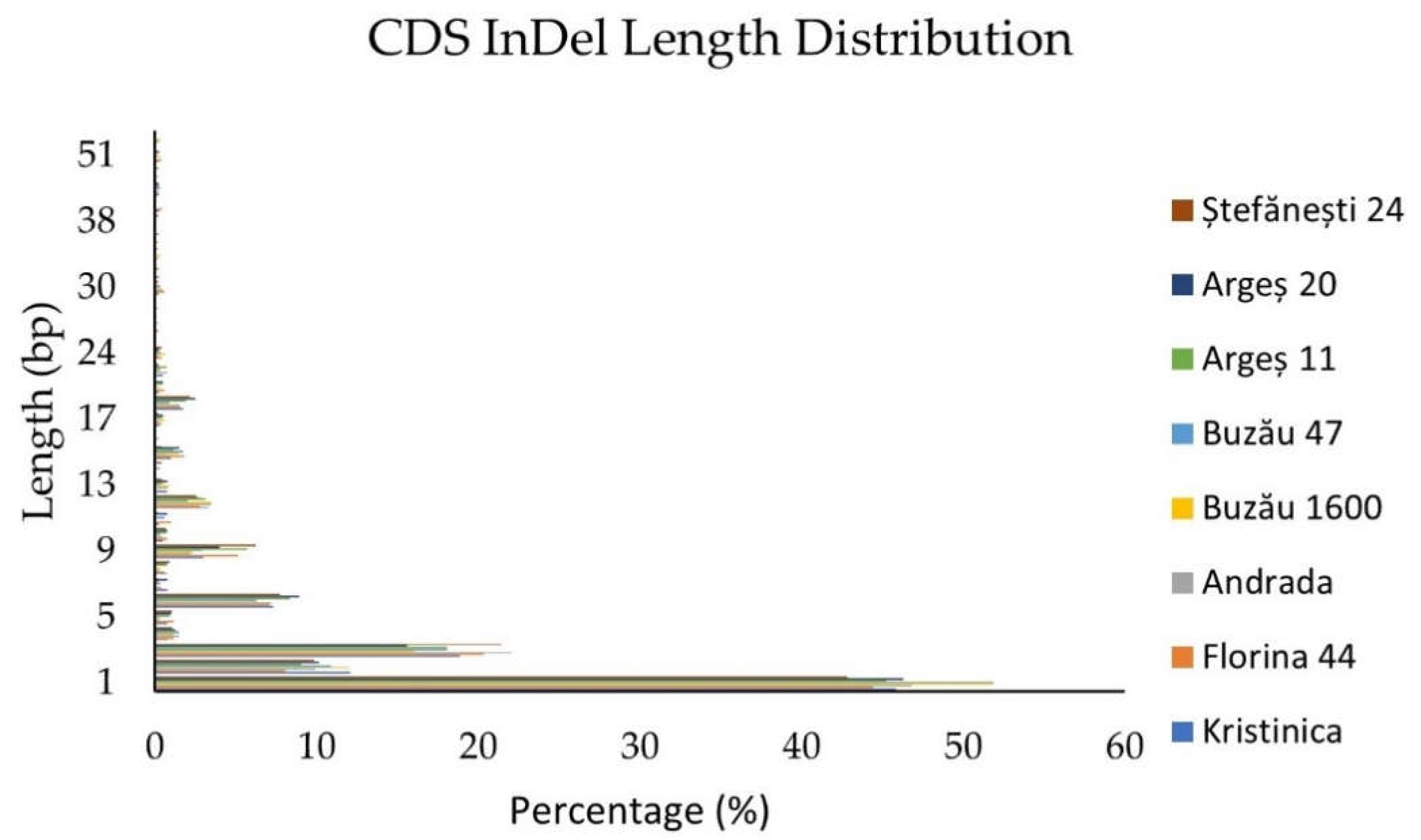
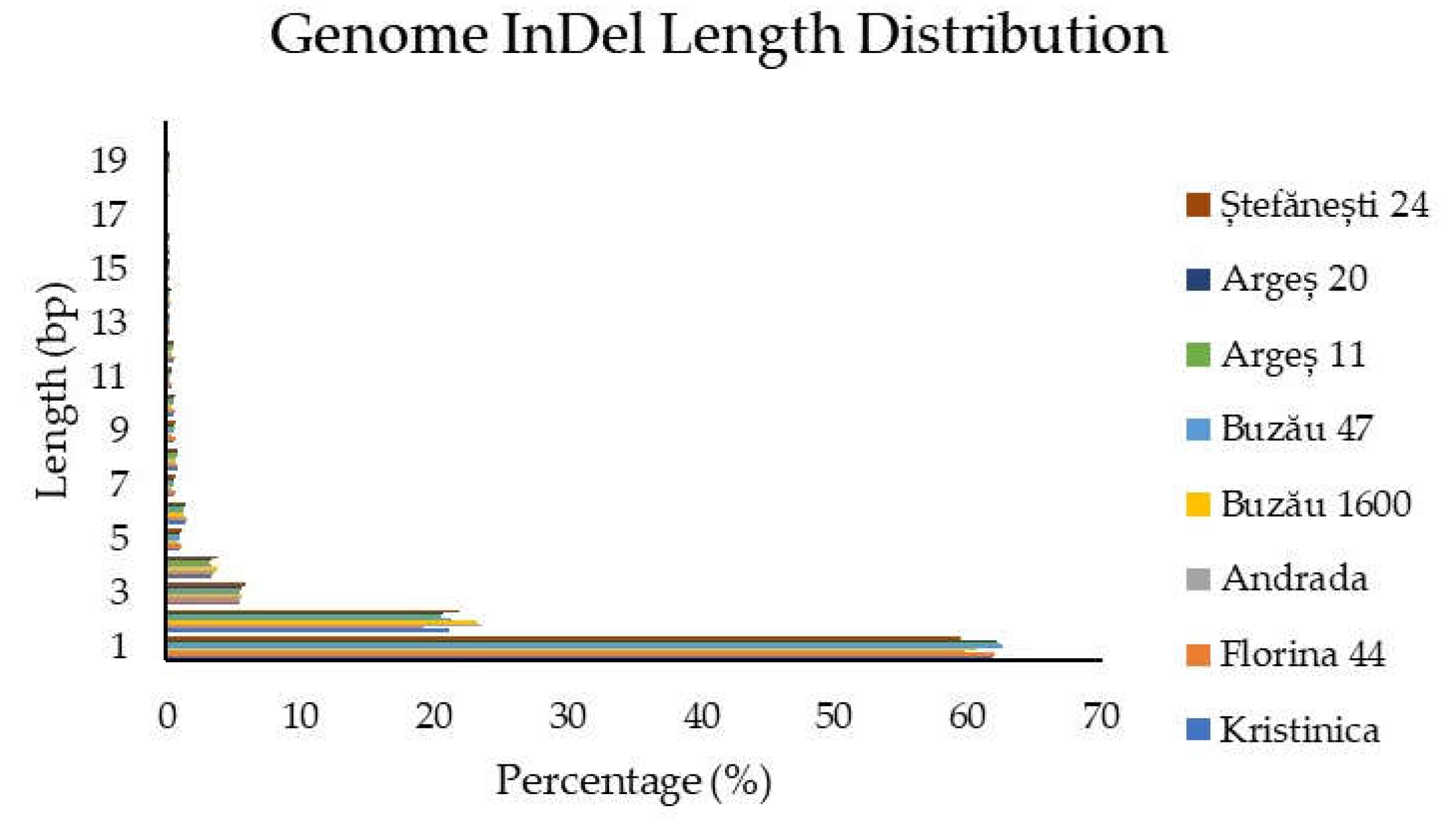
2.1.3. Insertion/Deletion Detection and Distribution
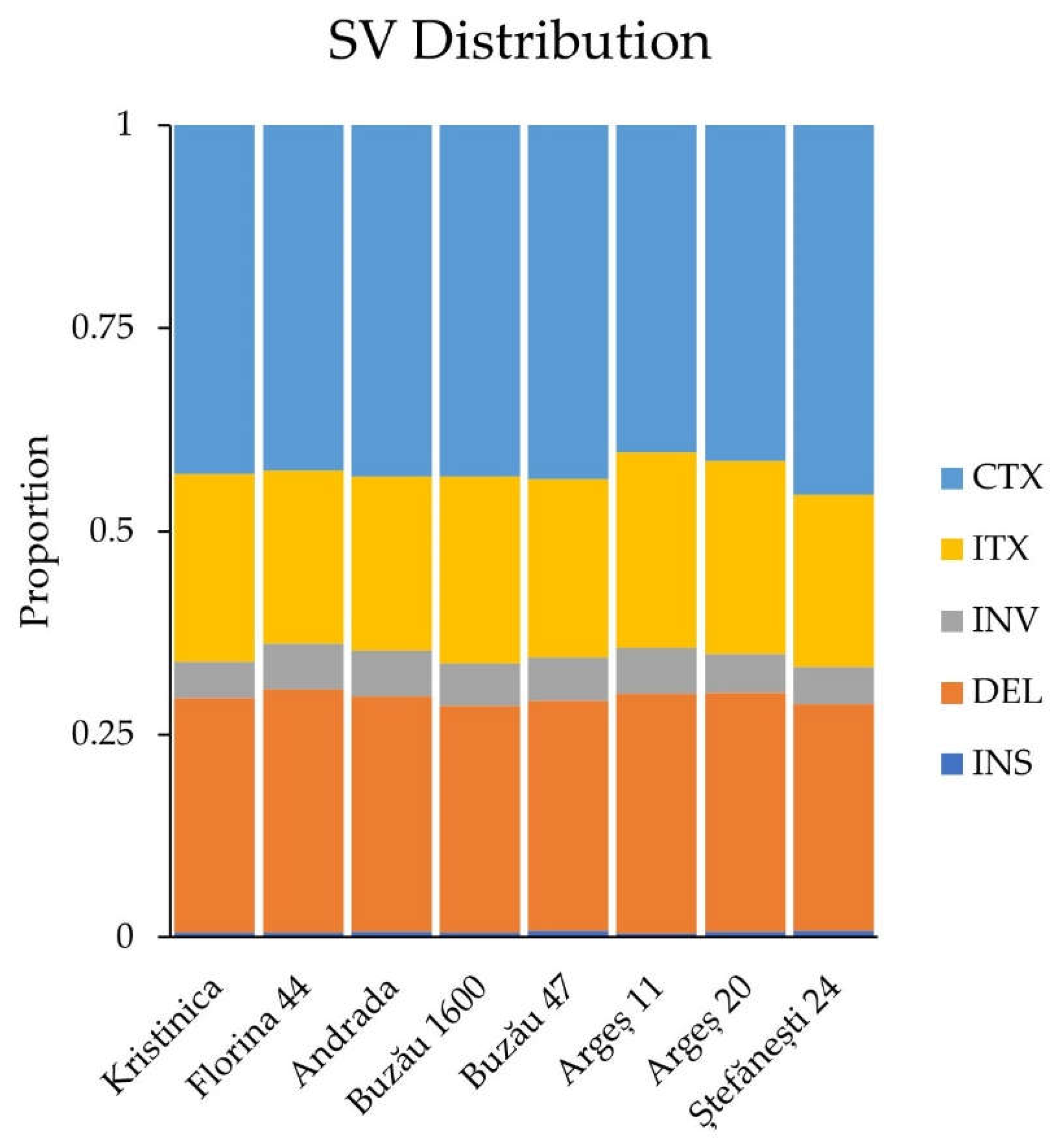
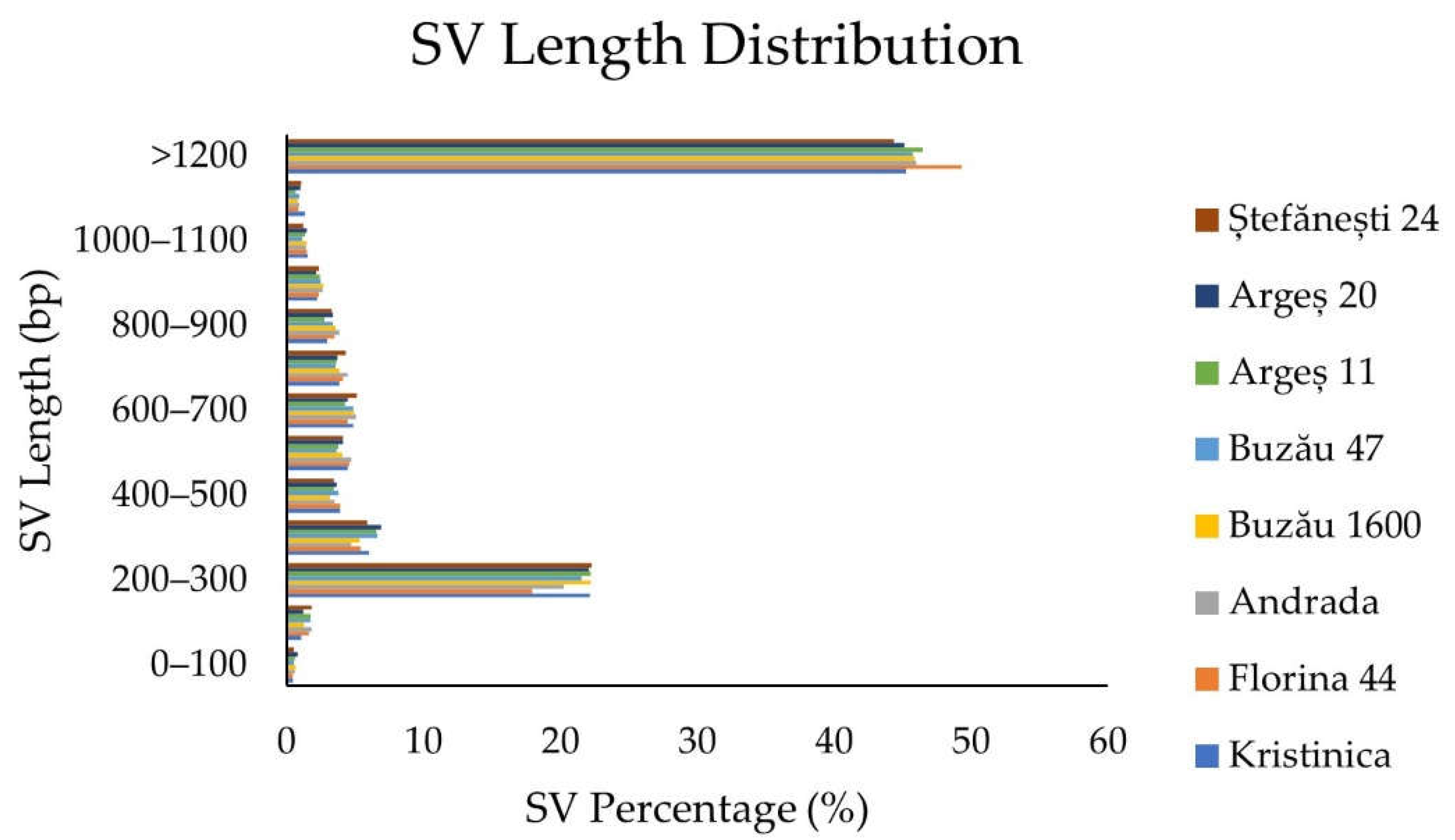
2.1.4. Structural Variant Detection and Annotation
2.1.5. Copy Number Variations Detection and Annotation
2.2. Sequence Analyses of Ve1 and Ve2 Loci in Romanian Tomato Genotypes
3. Discussion
Sequence Analyses of the Ve1 and Ve2 Loci in the Romanian Tomato Genotypes
4. Materials and Methods
4.1. Plant Material
4.2. DNA Extraction
4.3. Sequencing, Computational Data Processing, and Sequencing Analysis
4.3.1. SNP Detection and Annotation
4.3.2. Insertion/Deletion (InDel) Detection and Annotation
4.3.3. Structural Variant Detection and Annotation
4.3.4. Copy Number Variation Detection and Annotation
4.4. Sequence Analysis of the Ve Locus
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Burton-Freeman, B.; Reimers, K. Tomato Consumption and Health: Emerging Benefits. Am. J. Lifestyle Med. 2011, 5, 182–191. [Google Scholar] [CrossRef]
- Felföldi, Z.; Ranga, F.; Socaci, S.A.; Farcas, A.; Plazas, M.; Sestras, A.F.; Vodnar, D.C.; Prohens, J.; Sestras, R.E. Physico-Chemical, Nutritional, and Sensory Evaluation of Two New Commercial Tomato Hybrids and Their Parental Lines. Plants 2021, 10, 2480. [Google Scholar] [CrossRef] [PubMed]
- Quinet, M.; Angosto, T.; Yuste-Lisbona, F.J.; Blanchard-Gros, R.; Bigot, S.; Martinez, J.-P.; Lutts, S. Tomato Fruit Development and Metabolism. Front. Plant Sci. 2019, 10, 1554. [Google Scholar] [CrossRef] [PubMed]
- Bernatzky, R.; Tanksley, S.D. Toward a saturated linkage map in tomato based on isozymes and random cDNA sequences. Genetics 1986, 112, 887–898. [Google Scholar] [CrossRef]
- Aoki, K.; Ogata, Y.; Igarashi, K.; Yano, K.; Nagasaki, H.; Kaminuma, E.; Toyoda, A. Functional genomics of tomato in a post-genome-sequencing phase. Breed. Sci. 2013, 63, 14–20. [Google Scholar] [CrossRef]
- Sato, S.; Tabata, S. Tomato Genome Sequence. In Functional Genomics and Biotechnology in Solanaceae and Cucurbitaceae Crops; Ezura, H., Ariizumi, T., Garcia-Mas, J., Rose, J., Eds.; Biotechnology in Agriculture and Forestry; Springer: Berlin, Heidelberg, 2016; pp. 1–13. ISBN 978-3-662-48535-4. [Google Scholar]
- Xu, X.; Bai, G. Whole-genome resequencing: Changing the paradigms of SNP detection, molecular mapping and gene discovery. Mol. Breed. 2015, 35, 33. [Google Scholar] [CrossRef]
- Chaudhary, J.; Khatri, P.; Singla, P.; Kumawat, S.; Kumari, A.; R, V.; Vikram, A.; Jindal, S.K.; Kardile, H.; Kumar, R.; et al. Advances in Omics Approaches for Abiotic Stress Tolerance in Tomato. Biology 2019, 8, 90. [Google Scholar] [CrossRef]
- Chaudhary, J.; Alisha, A.; Bhatt, V.; Chandanshive, S.; Kumar, N.; Mir, Z.; Kumar, A.; Yadav, S.K.; Shivaraj, S.M.; Sonah, H.; et al. Mutation Breeding in Tomato: Advances, Applicability and Challenges. Plants 2019, 8, 128. [Google Scholar] [CrossRef]
- Adhikari, P.; Adhikari, T.B.; Louws, F.J.; Panthee, D.R. Advances and Challenges in Bacterial Spot Resistance Breeding in Tomato (Solanum lycopersicum L.). Int. J. Mol. Sci. 2020, 21, 1734. [Google Scholar] [CrossRef]
- Shirasawa, K.; Kuwata, C.; Watanabe, M.; Fukami, M.; Hirakawa, H.; Isobe, S. Target Amplicon Sequencing for Genotyping Genome-Wide Single Nucleotide Polymorphisms Identified by Whole-Genome Resequencing in Peanut. Plant Genome 2016, 9, plantgenome2016.06.0052. [Google Scholar] [CrossRef] [Green Version]
- The 100 Tomato Genome Sequencing Consortium; Aflitos, S.; Schijlen, E.; de Jong, H.; de Ridder, D.; Smit, S.; Finkers, R.; Wang, J.; Zhang, G.; Li, N.; et al. Exploring genetic variation in the tomato (Solanum section Lycopersicon) clade by whole-genome sequencing. Plant J. 2014, 80, 136–148. [Google Scholar] [CrossRef]
- Arafa, R.A.; Rakha, M.T.; Soliman, N.E.K.; Moussa, O.M.; Kamel, S.M.; Shirasawa, K. Rapid identification of candidate genes for resistance to tomato late blight disease using next-generation sequencing technologies. PLoS ONE 2017, 12, e0189951. [Google Scholar] [CrossRef] [PubMed]
- Kawchuk, L.M.; Hachey, J.; Lynch, D.R.; Kulcsar, F.; van Rooijen, G.; Waterer, D.R.; Robertson, A.; Kokko, E.; Byers, R.; Howard, R.J.; et al. Tomato Ve disease resistance genes encode cell surface-like receptors. Proc. Natl. Acad. Sci. 2001, 98, 6511–6515. [Google Scholar] [CrossRef]
- Fradin, E.F.; Zhang, Z.; Juarez Ayala, J.C.; Castroverde, C.D.M.; Nazar, R.N.; Robb, J.; Liu, C.-M.; Thomma, B.P.H.J. Genetic Dissection of Verticillium Wilt Resistance Mediated by Tomato Ve1. Plant Physiol. 2009, 150, 320–332. [Google Scholar] [CrossRef] [PubMed]
- Illumina, I. Quality scores for next-generation sequencing. Tech. Note: Inform. 2011, 31. [Google Scholar]
- Sato, S.; Tabata, S.; Hirakawa, H.; Asamizu, E.; Shirasawa, K.; Isobe, S.; Kaneko, T.; Nakamura, Y.; Shibata, D.; Aoki, K.; et al. The tomato genome sequence provides insights into fleshy fruit evolution. Nature 2012, 485, 635–641. [Google Scholar] [CrossRef]
- Hamilton, J.P.; Sim, S.-C.; Stoffel, K.; Van Deynze, A.; Buell, C.R.; Francis, D.M. Single Nucleotide Polymorphism Discovery in Cultivated Tomato via Sequencing by Synthesis. Plant Genome 2012, 5, 17–29. [Google Scholar] [CrossRef]
- Sim, S.-C.; Durstewitz, G.; Plieske, J.; Wieseke, R.; Ganal, M.W.; Deynze, A.V.; Hamilton, J.P.; Buell, C.R.; Causse, M.; Wijeratne, S.; et al. Development of a Large SNP Genotyping Array and Generation of High-Density Genetic Maps in Tomato. PLoS ONE 2012, 7, e40563. [Google Scholar] [CrossRef]
- Causse, M.; Desplat, N.; Pascual, L.; Le Paslier, M.-C.; Sauvage, C.; Bauchet, G.; Bérard, A.; Bounon, R.; Tchoumakov, M.; Brunel, D.; et al. Whole genome resequencing in tomato reveals variation associated with introgression and breeding events. BMC Genom. 2013, 14, 791. [Google Scholar] [CrossRef]
- Kim, J.-E.; Oh, S.-K.; Lee, J.-H.; Lee, B.-M.; Jo, S.-H. Genome-Wide SNP Calling Using Next Generation Sequencing Data in Tomato. Mol. Cells 2014, 37, 36–42. [Google Scholar] [CrossRef]
- Gupta, P.; Reddaiah, B.; Salava, H.; Upadhyaya, P.; Tyagi, K.; Sarma, S.; Datta, S.; Malhotra, B.; Thomas, S.; Sunkum, A.; et al. Next-generation sequencing (NGS)-based identification of induced mutations in a doubly mutagenized tomato (Solanum lycopersicum) population. Plant J. 2017, 92, 495–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguilar, M.; Prieto, P. Telomeres and Subtelomeres Dynamics in the Context of Early Chromosome Interactions During Meiosis and Their Implications in Plant Breeding. Front. Plant Sci. 2021, 12, 672489. [Google Scholar] [CrossRef]
- Edwards, D.; Forster, J.W.; Chagné, D.; Batley, J. What Are SNPs? In Association Mapping in Plants; Oraguzie, N.C., Rikkerink, E.H.A., Gardiner, S.E., De Silva, H.N., Eds.; Springer: New York, NY, USA, 2007; pp. 41–52. ISBN 978-0-387-36011-9. [Google Scholar]
- Yuan, Y.; Bayer, P.E.; Batley, J.; Edwards, D. Current status of structural variation studies in plants. Plant Biotechnol. J. 2021, 19, 2153–2163. [Google Scholar] [CrossRef] [PubMed]
- Francia, E.; Pecchioni, N.; Policriti, A.; Scalabrin, S. CNV and Structural Variation in Plants: Prospects of NGS Approaches. In Advances in the Understanding of Biological Sciences Using Next Generation Sequencing (NGS) Approaches; Sablok, G., Kumar, S., Ueno, S., Kuo, J., Varotto, C., Eds.; Springer International Publishing: Cham, Switzerland, 2015; pp. 211–232. ISBN 978-3-319-17157-9. [Google Scholar]
- Robb, E.J.; Nazar, R.N. Tomato Ve-resistance locus: Resilience in the face of adversity? Planta 2021, 254, 126. [Google Scholar] [CrossRef] [PubMed]
- Nazar, R.N.; Xu, X.; Kurosky, A.; Robb, J. Antagonistic function of the Ve R-genes in tomato. Plant Mol. Biol. 2018, 98, 67–79. [Google Scholar] [CrossRef]
- Cock, P.J.A.; Fields, C.J.; Goto, N.; Heuer, M.L.; Rice, P.M. The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants. Nucleic Acids Res. 2010, 38, 1767–1771. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Chen, K.; Wallis, J.W.; McLellan, M.D.; Larson, D.E.; Kalicki, J.M.; Pohl, C.S.; McGrath, S.D.; Wendl, M.C.; Zhang, Q.; Locke, D.P.; et al. BreakDancer: An algorithm for high-resolution mapping of genomic structural variation. Nat. Methods 2009, 6, 677–681. [Google Scholar] [CrossRef]
- Abyzov, A.; Urban, A.E.; Snyder, M.; Gerstein, M. CNVnator: An approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 2011, 21, 974–984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stothard, P. The Sequence Manipulation Suite: JavaScript Programs for Analyzing and Formatting Protein and DNA Sequences. BioTechniques 2000, 28, 1102–1104. [Google Scholar] [CrossRef]
- Corpet, F. Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 1988, 16, 10881–10890. [Google Scholar] [CrossRef] [PubMed]
- Luria, N.; Smith, E.; Reingold, V.; Bekelman, I.; Lapidot, M.; Levin, I.; Elad, N.; Tam, Y.; Sela, N.; Abu-Ras, A.; et al. A New Israeli Tobamovirus Isolate Infects Tomato Plants Harboring Tm-22 Resistance Genes. PLoS ONE 2017, 12, e0170429. [Google Scholar] [CrossRef]
- Kumar, R.; Khurana, A. Functional genomics of tomato: Opportunities and challenges in post-genome NGS era. J. Biosci. 2014, 39, 917–929. [Google Scholar] [CrossRef] [PubMed]
- Zamfir, B.; Hoza, D.; Vînătoru, C.; Lagunovschi, V.; Bratu, C. Research on conservation, evaluation and genetic heritage exploitation of tomato. Sci. Papers Ser. B Horticulture 2017, LXI, 307–312. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SNP Position | DNA Sequence | Amino Acid Sequence | Genotypes | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Heinz 1706 | Craigella GCR218 | Motelle | VFN-8 | Ailsa Craig | Craigella GCR26 | Moneymaker | Kristinica, Florina 44, Buzău 1600, Argeș 11, Argeș 20 | Andrada, Buzău 47 | Ștefănești 24 | |||
| 29/35 | CCTATGGTT | PMV | - | - | - | + | - | - | - | - | - | |
| CTATGGCTT | LWL | + | + | + | + | - | + | + | + | + | + | |
| 246 | GTG | Silent | + | + | + | + | - | - | - | + | - | + |
| GTC | - | - | - | - | + | + | + | - | + | - | ||
| 380 | GAC | D | - | - | + | - | + | + | - | - | + | - |
| GCC | A | + | + | - | + | - | - | + | + | - | + | |
| 610 | ACT | T | + | + | + | + | - | - | - | + | + | + |
| TCT | S | - | - | - | - | + | + | + | - | - | - | |
| 706 | ACT | T | - | - | - | - | + | + | + | - | + | + |
| TCT | S | + | + | + | + | - | - | - | + | - | - | |
| 1220 | TCAGAG | SE | + | + | + | + | - | - | - | + | - | - |
| TAGAG | STOP | - | - | - | - | + | + | + | - | + | + | |
| 1548 | AAC | N | + | + | + | + | - | - | - | + | - | + |
| AAG | K * | - | - | - | - | + | + | + | - | + | - | |
| 1888 | GAC | D | + | + | + | + | - | - | - | + | - | - |
| AAC | N * | - | - | - | - | + | + | + | - | + | + | |
| SNP Position | DNA Sequence | Amino Acid Sequence | Genotypes | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Heinz 1706 | Craigella GCR218 | Motelle | VFN-8 | Ailsa Craig | Craigella GCR26 | Moneymaker | Kristinica, Florina 44, Buzău 1600, Argeș 11, Argeș 20 | Andrada, Buzău 47 | Ștefănești 24 | |||
| 1385 | ACA | T | - | - | - | - | - | - | - | - | + | - |
| AGA | R | + | + | + | + | + | + | + | + | - | + | |
| 1811 | GTA | V | - | - | - | - | + | + | + | - | + | |
| GCA | A | + | + | + | + | - | - | - | + | - | + | |
| 2761 | GAC | D | + | + | + | + | + | + | - | + | + | + |
| AAC | N | - | - | - | - | - | - | + | - | - | - | |
| 2771 | AGA | R | - | - | - | - | - | + | + | - | + | - |
| ACA | T | + | + | + | + | + | - | - | + | - | + | |
| 2893 | CCA | P | - | + | + | + | - | - | - | - | + | - |
| TCA | S | + | - | - | - | + | + | + | + | - | + | |
| 2934 | CTC | Silent | - | - | - | - | + | + | + | - | - | - |
| CTT | + | + | + | + | - | - | - | + | + | + | ||
| 3243 | GGT | Silent | - | - | - | - | + | + | + | - | + | - |
| GGG | + | + | + | + | - | - | - | + | - | + | ||
| 3380 | TTT | T | - | - | - | - | + | - | - | - | - | - |
| TCT | S | + | + | + | + | - | + | + | + | + | + | |
| 3383 | TTT | T | - | - | - | - | + | - | - | - | - | - |
| TCT | S | + | + | + | + | - | + | + | + | + | + | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Udriște, A.-A.; Iordachescu, M.; Ciceoi, R.; Bădulescu, L. Next-Generation Sequencing of Local Romanian Tomato Varieties and Bioinformatics Analysis of the Ve Locus. Int. J. Mol. Sci. 2022, 23, 9750. https://doi.org/10.3390/ijms23179750
Udriște A-A, Iordachescu M, Ciceoi R, Bădulescu L. Next-Generation Sequencing of Local Romanian Tomato Varieties and Bioinformatics Analysis of the Ve Locus. International Journal of Molecular Sciences. 2022; 23(17):9750. https://doi.org/10.3390/ijms23179750
Chicago/Turabian StyleUdriște, Anca-Amalia, Mihaela Iordachescu, Roxana Ciceoi, and Liliana Bădulescu. 2022. "Next-Generation Sequencing of Local Romanian Tomato Varieties and Bioinformatics Analysis of the Ve Locus" International Journal of Molecular Sciences 23, no. 17: 9750. https://doi.org/10.3390/ijms23179750
APA StyleUdriște, A. -A., Iordachescu, M., Ciceoi, R., & Bădulescu, L. (2022). Next-Generation Sequencing of Local Romanian Tomato Varieties and Bioinformatics Analysis of the Ve Locus. International Journal of Molecular Sciences, 23(17), 9750. https://doi.org/10.3390/ijms23179750