

Apolipoprotein E in Cardiometabolic and Neurological Health and Diseases


Abstract
:1. Introduction
2. Role of ApoE in Cardiovascular Health and Disease
2.1. Atherosclerosis
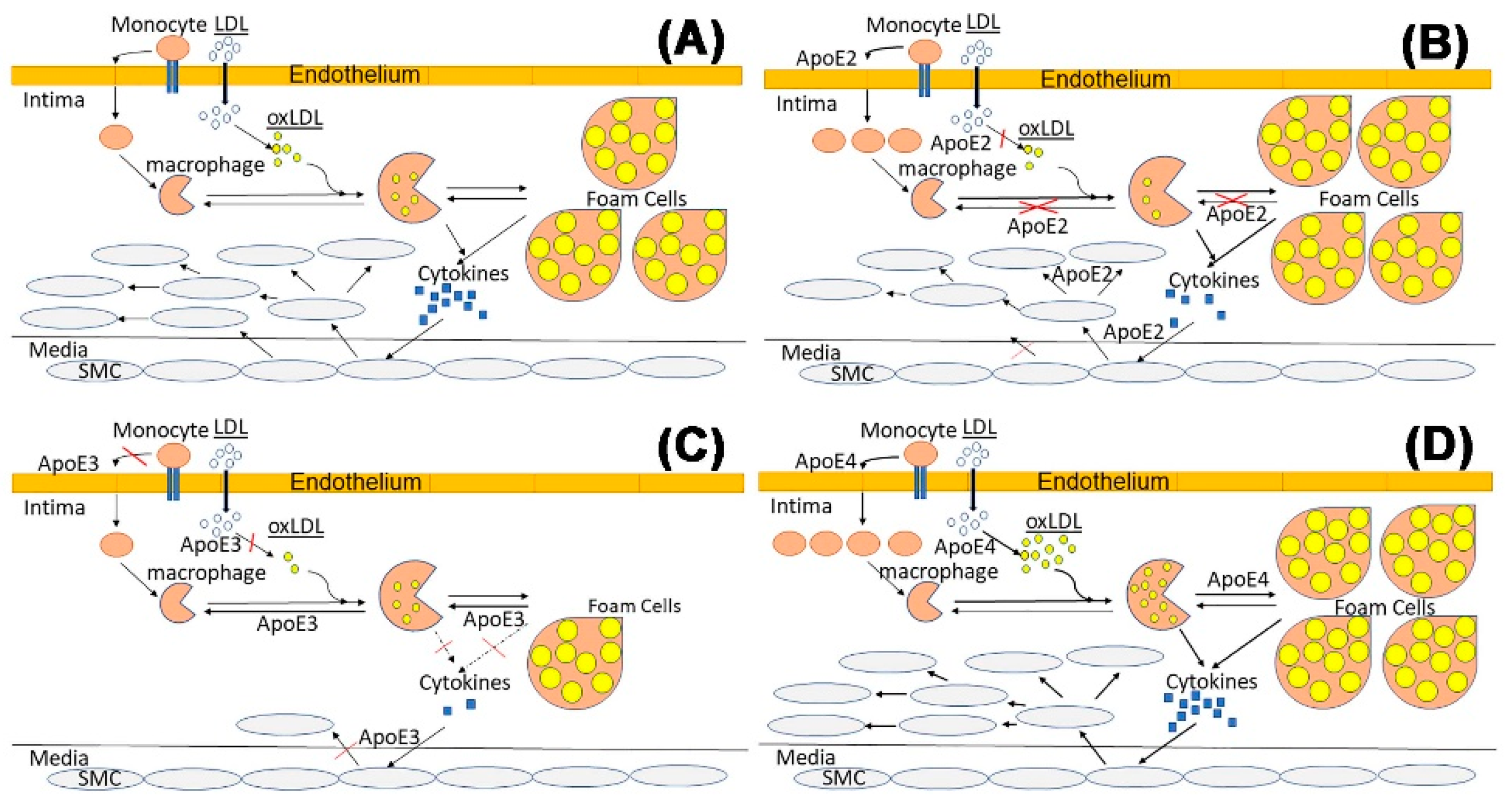
2.1.1. ApoE and Lipid Metabolism
2.1.2. ApoE and Endothelial Cells
2.1.3. ApoE and Macrophages
2.1.4. ApoE and Lymphocyte Activation
2.1.5. ApoE and Vascular Smooth Muscle Cells
2.1.6. Summary of ApoE Isoform Influence on Atherosclerosis
3. Role of ApoE in Metabolic Health and Disease
3.1. ApoE and Obesity
3.2. ApoE and Diabetes
4. Role of ApoE in Neurological Health and Disease
4.1. ApoE and Alzheimer’s Disease
4.1.1. ApoE and β-Amyloid
4.1.2. ApoE and Tau
4.1.3. ApoE and Neuroinflammation
4.1.4. ApoE and Neurotoxicity
4.2. ApoE and Parkinson’s Disease
4.3. Summary of ApoE Isoform Influence on Neurological Health and Diseases
5. Conclusions and Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ApoE | Apolipoprotein E |
| VLDL | Very low density lipoproteins |
| HDL | High density lipoproteins |
| LDL | Low density lipoproteins |
| LRP1 | LDL receptor related protein-1 |
| NO | Nitric oxide |
| eNOS | Endothelial nitric oxide synthase |
| NK | Natural Killer |
| NKT | Natural killer T cells |
| TLR | Toll-like receptor |
| ABCA1 | ATP binding cassette subfamily member 1 |
| PI3K | Phosphatidylinositol 3-kinase |
| PKCξ | Protein kinase C-ξ |
| Sp1 | Specificity protein-1 |
| IL3 | Interleukin-3 |
| GM-CSF | Granulocyte-macrophage colony stimulating factor |
| TRAF6 | TNF receptor associated factor 6 |
| IRAK1 | Interleukin 1 receptor associated kinase 1 |
| MHC-II | Major histocompatibility complex II |
| CD4 | Cluster of differentiation 4 |
| CD1b | Cluster of differentiation 1b |
| cAMP | Cyclic adenosine monophosphate |
| GLUT4 | Glucose transporter type 4 |
| Aβ | Beta-amyloid |
| TREM2 | Triggering receptor expressed on myeloid cells-2 |
| hAPPFAD | human amyloid-beta precursor protein familial Alzheimer’s disease |
| Th1 | T helper cell type 1 |
| HLA-DR | Human leukocyte antigen—DR isotype |
References
- Dolgin, E. The most popular genes in the human genome. Nature 2017, 551, 427–431. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Stampfer, M.J.; Liu, S. Meta-analysis: Apolipoprotein E genotypes and risk for coronary heart disease. Ann. Intern. Med. 2004, 141, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Couderc, R.; Mahieux, F.; Bailleul, S.; Fenelon, G.; Mary, R.; Fermanian, J. Prevalence of apolipoprotein E phenotypes in ischemic cerebrovascular disease. A case-control study. Stroke 1993, 24, 661–664. [Google Scholar] [CrossRef] [PubMed]
- Senti, M.; Nogues, X.; Pedro-Botet, J.; Rubies-Prat, J.; Vidal-Barraquer, F. Lipoprotein profile in men with peripheral vascular disease. Role of intermediate density lipoproteins and apoprotein E phenotypes. Circulation 1992, 85, 30–36. [Google Scholar] [CrossRef] [PubMed]
- de Andrade, M.; Thandi, I.; Brown, S.; Gotto, A., Jr.; Patsch, W.; Boerwinkle, E. Relationship of the apolipoprotein E polymorphism with carotid artery atherosclerosis. Am. J. Hum. Genet. 1995, 56, 1379–1390. [Google Scholar] [PubMed]
- Davignon, J.; Gregg, R.E.; Sing, C.F. Apolipoprotein E polymorphism and atherosclerosis. Atherosclerosis 1988, 8, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Wilson, P.W.F.; Schaefer, E.J.; Larson, M.G.; Ordovas, J.M. Apolipoprotein E alleles and risk of coronary disease. A meta analysis. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 1250–1255. [Google Scholar] [CrossRef] [PubMed]
- McCarron, M.O.; Delong, D.; Alberts, M.J. APOE genotype as a risk factor for ischemic cerebrovascular disease: A meta-analysis. Neurology 1999, 53, 1308–1311. [Google Scholar] [CrossRef]
- Bennet, A.M.; Di Angelantonio, E.; Ye, Z.; Wensley, F.; Dahlin, A.; Ahlbom, A.; Keavney, B.; Collins, R.; Wiman, B.; de Faire, U.; et al. Association of apolipoprotein E genotypes with lipid levels and coronary risk. JAMA 2007, 298, 1300–1311. [Google Scholar] [CrossRef] [PubMed]
- Anthopoulos, P.G.; Hamodrakas, S.J.; Bagos, P.G. Apolipoprotein E polymorphisms and type 2 diabetes: A meta-analysis of 30 studies including 5423 cases and 8192 controls. Mol. Genet. Metab. 2010, 100, 283–291. [Google Scholar] [CrossRef]
- Sima, A.; Iordan, A.; Stancu, C. Apolipoprotein E polymorphism—A risk factor for metabolic syndrome. Clin. Chem. Lab. Med. 2007, 45, 1149–1153. [Google Scholar] [CrossRef] [PubMed]
- Vaisi-Raygani, A.; Rahimi, Z.; Nomani, H.; Tavilani, H.; Pourmotabbed, T. The presence of apolipoprotein epsilon4 and epsilon2 alleles augments the risk of coronary artery disease in type 2 diabetic patients. Clin. Biochem. 2007, 40, 1150–1156. [Google Scholar] [CrossRef]
- Ramus, S.M.; Petrovic, D. Genetic variations and subclinical markers of carotid atherosclerosis in patients with type 2 diabetes mellitus. Curr. Vasc. Pharmacol. 2019, 17, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Santos-Ferreira, C.; Baptista, R.; Oliveira-Santos, M.; Costa, R.; Moura, J.P.; Goncalves, L. Apolipoprotein E2 genotype is associated with a 2-fold increase in the incidence of type 2 diabetes mellitus: Results from a long-term observational study. J. Lipids 2019, 2019, 1698610. [Google Scholar] [CrossRef] [PubMed]
- Zeljko, H.; Skaric-Juric, T.; Narancic, N.; Tomas, Z.; Baresic, A.; Salihovic, M.; Starcevic, B.; Janicijevic, B. E2 allele of the Apolipoprotein E gene polymorphism is predictive for obesity status in Roma minority population of Croatia. Lipids Health Dis. 2011, 10, 9. [Google Scholar] [CrossRef] [PubMed]
- Strittmatter, W.J.; Roses, A.D. Apolipoprotein E and Alzheimer’s disease. Annu. Rev. Neurosci. 1996, 19, 53–77. [Google Scholar] [CrossRef] [PubMed]
- Mahley, R.W.; Weisgraber, K.H.; Huang, Y. Apolipoprotein E: Structure determines function, from atherosclerosis to Alzheimer’s disease to AIDS. J. Lipid Res. 2009, 50, S183–S188. [Google Scholar] [CrossRef] [PubMed]
- Kockx, M.; Traini, M.; Kritharides, L. Cell-specific production, secretion, and function of apolipoprotein E. J. Mol. Med. 2018, 96, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Matsunaga, A.; Fukunaga, M.; Nagahama, K.; Hara, D.; Muso, E. Apolipoprotein E-related glomerular disorders. Kidney Int. 2020, 97, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Virani, S.S.; Alonso, A.; Aparicio, H.J.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Cheng, S.; Delling, F.N.; et al. Heart disease and stroke statistics—2021 update: A report from the American Heart Association. Circulation 2021, 143, e254–e743. [Google Scholar] [CrossRef]
- Hajar, R. Risk factors for coronary artery disease: Historical perspectives. Heart Views 2017, 18, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I. 2016 Russell Ross Memorial Lecture in vascular biology: Molecular-cellular mechanisms in the progression of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 183–189. [Google Scholar] [CrossRef]
- Lahoz, C.; Schaefer, E.J.; Cupples, L.A.; Wilson, P.W.F.; Levy, D.; Osgood, D.; Parpos, S.; Pedro-Botet, J.; Daly, J.A.; Ordovas, J.M. Apolipoprotein E genotype and cardiovascular disease in the Framingham Heart Study. Atherosclerosis 2001, 154, 529–537. [Google Scholar] [CrossRef]
- Eto, M.; Watanabe, K.; Ishii, K. Reciprocal effects of apolipoprotein E alleles (epsilon 2 and epsilon 4) on plasma lipid levels in normolipidemic subjects. Clin. Genet. 1986, 29, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Koopal, C.; Geerlings, M.I.; Muller, M.; de Borst, G.J.; Algra, A.; van der Graaf, Y.; Visseren, F.L.J.; Group, S.S. The relationship between apolipoprotein E (APOE) genotype and peripheral artery disease in patients at high risk for cardiovascular disease. Atherosclerosis 2016, 246, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Blazejewska-Hyzorek, B.; Gromadzka, G.; Skowronska, M.; Czlonkowska, A. ApoE e2 allele is an independent risk factor for vulnerable carotid plaque in ischemic stroke patients. Neurol. Res. 2014, 36, 950–954. [Google Scholar] [CrossRef] [PubMed]
- Wilson, P.F.; Larson, M.G.; Ordovas, J.M.; Schaefer, E.J. Apolipoprotein E isoforms and CHD prevalence in the Framingham offspring. Circulation 1994, 272, 1666–1671. [Google Scholar]
- Duman, B.S.; Ozturk, M.; Yilmazer, S.; Hatemi, H. Apolipoprotein E polymorphism in Turkish subjects with type 2 diabetes mellitus: Allele frequency and relationship to serum lipid concentrations. Diabetes Nutr. Metab. 2004, 17, 267–274. [Google Scholar]
- Kalix, B.; Meynet, M.C.; Garin, M.C.; James, R.W. The apolipoprotein epsilon2 allele and the severity of coronary artery disease in Type 2 diabetic patients. Diabet Med. 2001, 18, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Weintraub, M.S.; Eisenberg, S.; Breslow, J.L. Dietary fat clearance in normal subjects is regulated by genetic variation in apolipoprotein E. J. Clin. Investig. 1987, 80, 1571–1577. [Google Scholar] [CrossRef] [PubMed]
- Cardia, G.; Grisorio, D.; Impedovo, G.; Lillo, A.; Regina, G. Plasma lipids as a risk factor in peripheral vascular disease. Angiology 1990, 41, 19–22. [Google Scholar] [CrossRef] [PubMed]
- Lau, J.F.; Weinberg, M.D.; Olin, J.W. Peripheral artery disease. Part 1: Clinical evaluation and noninvasive diagnosis. Nat. Rev. Cardiol. 2011, 8, 405–418. [Google Scholar] [CrossRef] [PubMed]
- Getz, G.S.; Reardon, C.A. Apoprotein E as a lipid transport and signaling protein in the blood, liver, and artery wall. J. Lipid Res. 2009, 50, S156–S161. [Google Scholar] [CrossRef] [PubMed]
- Pendse, A.A.; Arbones-Mainar, J.M.; Johnson, L.A.; Altenburg, M.K.; Maeda, N. Apolipoprotein E knock-out and knock-in mice: Atherosclerosis, metabolic syndrome, and beyond. J. Lipid Res. 2009, 50, S178–S182. [Google Scholar] [CrossRef]
- Havel, R.J.; Kane, J.P. Primary dysbetalipoproteinemia: Predominance of a specific apoprotein species in triglyceride-rich lipoproteins. Proc. Natl. Acad. Sci. USA 1973, 70, 2015–2019. [Google Scholar] [CrossRef]
- Koopal, C.; Marais, A.D.; Visseren, F.L.J. Familial dysbetalipoproteinemia: An underdiagnosed lipid disorder. Curr. Opin. Endocrinol. Diabetes Obes. 2017, 24, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Mann, W.A.; Lohse, P.; Gregg, R.E.; Ronan, R.; Hoeg, J.M.; Zech, L.A.; Brewer, H.B. Dominant expression of type III hyperlipoproteinemia. Pathophysiological insights derived from the structural and kinetic characteristics of apoE-1 (Lys146 → Glu). J. Clin. Investig. 1995, 96, 1100–1107. [Google Scholar] [CrossRef] [PubMed]
- Innerarity, T.L.; Hui, D.Y.; Bersot, T.P.; Mahley, R.W. Type III hyperlipoproteinemia: A focus on lipoprotein receptor-apolipoprotein E2 interactions. Adv. Exp. Med. Biol. 1986, 201, 273–288. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.M.; Weisgraber, K.H. Human apolipoprotein E4 domain interaction. Arginine 61 and glutamic acid 255 interact to direct the preference for very low density lipoproteins. J. Biol. Chem. 1996, 271, 19053–19057. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.M.; Wilson, C.B.; Wardell, M.R.; Simmons, T.; Mahley, R.W.; Weisgraber, K.H.; Agard, D.A. Human apolipoprotein E. Role of arginine 61 in mediating the lipoprotein preferences of the E3 and E4 isoforms. J. Biol. Chem. 1994, 269, 22358–22365. [Google Scholar] [CrossRef]
- Dong, J.; Balestra, M.E.; Newhouse, Y.M.; Weisgraber, K.H. Human apolipoprotein E7: Lysine mutations in the carboxy-terminal domain are directly responsible for preferential binding to very low density lipoproteins. J. Lipid Res. 2000, 41, 1783–1789. [Google Scholar] [CrossRef]
- Youn, Y.C.; Lim, Y.K.; Han, S.-H.; Giau, V.V.; Lee, M.-K.; Park, K.-Y.; Kim, S.Y.; Bagyinszky, E.; An, S.S.A.; Kim, H.R. Apolipoprotein e7 allele in memory complaints: Insights through protein structure prediction. Clin. Interv. Aging 2017, 12, 1095–1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamura, T.; Dong, L.-M.; Yamamoto, A. Characterization of apolipoprotein E7 (Glu244→Lys, Glu245→Lys), a mutant apolipoprotein E associated with hyperlipidemia and atherosclerosis. J. Lipid Res. 1999, 40, 253–259. [Google Scholar] [CrossRef]
- Raffai, R.L.; Dong, L.-M.; Farese, R.V.; Weisgraber, K.H. Introduction of human apolipoprotein E4 domain interaction into mouse apolipoprotein E. Proc. Natl. Acad. Sci. USA 2001, 98, 11587–11591. [Google Scholar] [CrossRef] [PubMed]
- Knouff, C.; Hinsdale, M.E.; Mezdour, H.; Altenburg, M.K.; Watanabe, M.; Quarfordt, S.H.; Sullivan, P.M.; Maeda, N. ApoE structure determines VLDL clearance and atheroslerosis risk in mice. J. Clin. Investig. 1999, 103, 1579–1586. [Google Scholar] [CrossRef]
- Malloy, S.I.; Altenburg, M.K.; Knouff, C.; Lanningham-Foster, L.; Parks, J.S.; Maeda, N. Harmful effects of increased LDLR expression in mice with human APOE*4 but not APOE*3. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 91–97. [Google Scholar] [CrossRef]
- Sullivan, P.M.; Mezdour, H.; Quarfordt, S.H.; Maeda, N. Type III hyperlipoproteinemia and spontaneous atherosclerosis in mice resulting from gene replacement of mouse apoE with human apoE*2. J. Clin. Investig. 1998, 102, 130–135. [Google Scholar] [CrossRef]
- Oppi, S.; Luscher, T.F.; Stein, S. Mouse models for atherosclerosis research—Which is my line? Front. Cardiovasc. Med. 2019, 6, 46. [Google Scholar] [CrossRef]
- Von Scheidt, M.; Zhao, Y.; Kurt, Z.; Pan, C.; Zeng, L.; Yang, X.; Schunkert, H.; Lusis, A.J. Applications and limitations of mouse models for understanding human atherosclerosis. Cell Metab. 2016, 25, 248–261. [Google Scholar] [CrossRef]
- Li, K.; Ching, D.; Luk, F.S.; Raffai, R.L. Apolipoprotein E enhances microRNA-146a in monocytes and macrophages to suppress nuclear factor-kB-driven inflammation and atherosclerosis. Circ. Res. 2015, 117, e1–e11. [Google Scholar] [CrossRef]
- Yang, R.; Powell-Braxton, L.; Ogaoawara, A.K.; Dybdal, N.; Bunting, S.; Ohneda, O.; Jin, H. Hypertension and endothelial dysfunction in apolipoprotein E knockout mice. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 2762–2768. [Google Scholar] [CrossRef]
- Fransen, P.; Van Assche, T.; Guns, P.-J.; Van Hove, C.e.; De Keulenaer, G.W.; German, A.G.; Bult, H. Endothelial function in aorta segments of apolipoprotein E-deficient mice before development of atherosclerotic lesions. Pflug. Arch. 2008, 455, 811–818. [Google Scholar] [CrossRef] [PubMed]
- Yue, L.; Bian, J.-T.; Grizelj, I.; Cavka, A.; Phillips, S.A.; Makino, A.; Mazzone, T. ApoE enhances endothelial-NO production by modulating caveolin-1 interaction with eNOS. Hypertension 2012, 60, 1040–1046. [Google Scholar] [CrossRef]
- Ulrich, V.; Konaniah, E.S.; Herz, J.; Gerard, R.D.; Jung, E.; Yuhanna, I.S.; Ahmed, M.; Hui, D.Y.; Mineo, C.; Shaul, P.W. Genetic variants of apoE and apoER2 differentially modulate endothelial function. Proc. Natl. Acad. Sci. USA 2014, 111, 13493–13498. [Google Scholar] [CrossRef] [PubMed]
- Calvier, L.; Manouchehri, N.; Sacharidou, A.; Mineo, C.; Shaul, P.W.; Hui, D.Y.; Kounnas, M.Z.; Stuve, O.; Herz, J. Apolipoprotein E receptor 2 deficiency decreases endothelial adhesion of monocytes and protects against autoimmune encephalomyelitis. Sci. Immunol. 2021, 6, eabd0931. [Google Scholar] [CrossRef]
- Li, X.; Kypreos, K.; Zanni, E.E.; Zannis, V. Domains of apoE required for binding to apoE receptor 2 and to phospholipids: Implications for the functions of apoE in the brain. Biochemistry 2003, 42, 10406–10417. [Google Scholar] [CrossRef]
- Sacre, S.M.; Stannard, A.K.; Owen, J.S. Apolipoprotein E (apoE) isoforms differentially induce nitric oxide production in endothelial cells. FEBS Lett. 2003, 540, 181–187. [Google Scholar] [CrossRef]
- Feng, M.; Cui, D.; Li, Y.; Shi, J.; Xiang, L.; Bian, H.; Ma, Z.; Xia, W.; Wei, G. Carnosic acid reverses the inhibition of apoE4 on cell surface level of apoER2 and reelin signaling pathway. J. Alzheimer’s Dis 2020, 73, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Getz, G.S.; Reardon, C.A. Apoproteins E, A-I, and SAA in Macrophage Pathobiology Related to Atherogenesis. Front. Pharmacol. 2019, 10, 536. [Google Scholar] [CrossRef]
- Tabas, I.; Williams, K.J.; Boren, J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: Update and therapeutic implications. Circulation 2007, 116, 1832–1844. [Google Scholar] [CrossRef]
- Tabas, I.; Lichtman, A.H. Monocyte-macrophages and T cells in atherosclerosis. Immunity 2017, 47, 621–634. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I. Consequences and therapeutic implications of macrophage apoptosis in atherosclerosis: The importance of lesion stage and phagocytic efficiency. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2255–2264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, R. Atherosclerosis—An inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I. Macrophage apoptosis in atherosclerosis: Consequences on plaque progression and the role of endoplasmic reticulum stress. Antioxid. Redox. Signal. 2009, 11, 2333–2339. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I.; Seimon, T.; Timmins, J.; Li, G.; Lim, W. Macrophage apoptosis in advanced atherosclerosis. Ann. N. Y. Acad. Sci. 2009, 1173, E40–E45. [Google Scholar] [CrossRef]
- Zhu, Y.; Kodvawala, A.; Hui, D.Y. Apolipoprotein E inhibits Toll-like receptor (TLR)-3- and TLR-4-mediated macrophage activation through distinct mechanisms. Biochem. J. 2010, 428, 47–54. [Google Scholar] [CrossRef]
- Vedhachalam, C.; Narayanaswami, V.; Neto, N.; Forte, T.M.; Phillips, M.C.; Lund-Katz, S.; Bielicki, J.K. The C-terminal lipid-binding domain of apolipoprotein E is a highly efficient mediator of ABCA1-dependent cholesterol efflux that promotes the assembly of high-density lipoproteins. Biochemistry 2007, 46, 2583–2593. [Google Scholar] [CrossRef]
- Okoro, E.U.; Zhao, Y.; Guo, Z.M.; Zhou, L.; Lin, X.; Yang, H. Apolipoprotein E4 is deficient in inducing macrophage ABCA1 expression and stimulating the Sp1 signaling pathway. PLoS ONE 2012, 7, e44430. [Google Scholar] [CrossRef]
- Westerterp, M.; Fotakis, P.; Ouimet, M.; Bochem, A.E.; Zhang, H.; Molusky, M.M.; Wang, W.; Abramowicz, S.; la Bastide-van Gemert, S.; Wang, N.; et al. Cholesterol efflux pathways suppress inflammasome activation, NETosis, and atherogenesis. Circulation 2018, 138, 898–912. [Google Scholar] [CrossRef]
- Krimbou, L.; Denis, M.; Haidar, B.; Carrier, M.; Marcil, M.; Genest, J., Jr. Molecular interactions between apoE and ABCA1: Impact on apoE lipidation. J. Lipid Res. 2004, 45, 839–848. [Google Scholar] [CrossRef]
- Lin, C.-Y.; Duan, H.; Mazzone, T. Apolipoprotein E-dependent cholesterol efflux from macrophages: Kinetic study and divergent mechanisms for endogenous versus exogenous apolipoprotein E. J. Lipid Res. 1999, 40, 1618–1626. [Google Scholar] [CrossRef]
- Dove, D.E.; Linton, M.F.; Fazio, S. ApoE-mediated cholesterol efflux from macrophages: Separation of autocrine and paracrine effects. Am. J. Physiol. Cell Physiol. 2005, 288, C586–C592. [Google Scholar] [CrossRef] [PubMed]
- Zanotti, I.; Pedrelli, M.; Poti, F.; Stomeo, G.; Gomaraschi, M.; Calabresi, L.; Bernini, F. Macrophage, but not systemic, apolipoprotein E is necessary for macrophage reverse cholesterol transport in vivo. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Bellosta, S.; Langer, C.; Bernini, F.; Pitas, R.E.; Mahley, R.W.; Assmann, G.; von Eckardstein, A. Low dose expression of a human apolipoprotein E transgene in macrophages restores cholesterol efflux capacity of apolipoprotein E-deficient mouse plasma. Proc. Natl. Acad. Sci. USA 1998, 95, 7585–7590. [Google Scholar] [CrossRef]
- Fan, D.; Qiu, S.; Overton, C.D.; Yancey, P.G.; Swift, L.L.; Jerome, W.G.; Linton, M.F.; Fazio, S. Impaired secretion of apolipoprotein E2 from macrophages. J. Biol. Chem. 2007, 282, 13746–13753. [Google Scholar] [CrossRef]
- Igel, E.; Haller, A.; Wolfkiel, P.R.; Orr-Asman, M.; Jaeschke, A.; Hui, D.Y. Distinct pro-inflammatory properties of myeloid cell-derived apolipoprotein E2 and E4 in atherosclerosis promotion. J. Biol. Chem. 2021, 297, 101106. [Google Scholar] [CrossRef]
- Murphy, A.J.; Akhtari, M.; Tolani, S.; Pagler, T.A.; Bijl, N.; Kuo, C.-L.; Wang, M.; Sanson, M.; Abramowicz, S.; Welch, C.; et al. ApoE regulates hematopoietic stem cell proliferation, monocytosis, and monocyte accumulation in atherosclerotic lesions in mice. J. Clin. Investig. 2011, 121, 4138–4149. [Google Scholar] [CrossRef]
- Miyata, M.; Smith, J.D. Apolipoprotein E allele-specific antioxidant activity and effects on cytotoxicity by oxidative insults and beta-amyloid peptides. Nat. Genet. 1996, 14, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Mabile, L.; Lefebvre, C.; Lavigne, J.; Boulet, L.; Davignon, J.; Lussier-Cacan, S.; Bernier, L. Secreted apolipoprotein E reduces macrophages-mediated LDL oxidation in an isoform-dependent way. J. Cell Biochem. 2003, 90, 766–776. [Google Scholar] [CrossRef]
- Jofre-Monseny, L.; de Pascual-Teresa, S.; Plonka, E.; Huebbe, P.; Boesch-Saadatmandi, C.; Minihane, A.M.; Rimbach, G. Differential effects of apolipoprotein E3 and E4 on markers of oxidative status in macrophages. Br. J. Nutr. 2007, 97, 864–871. [Google Scholar] [CrossRef] [PubMed]
- Cash, J.G.; Kuhel, D.G.; Basford, J.E.; Jaeschke, A.; Chatterjee, T.K.; Weintraub, N.L.; Hui, D.Y. Apolipoprotein E4 impairs macrophage efferocytosis and potentiates apoptosis by accelerating endoplasmic reticulum stress. J. Biol. Chem. 2012, 287, 27876–27884. [Google Scholar] [CrossRef] [PubMed]
- Hui, D.Y.; Harmony, J.A.K.; Innerarity, T.L.; Mahley, R.W. Immunoregulatory plasma lipoproteins: Role of apoprotein E and B. J. Biol. Chem. 1980, 255, 11775–11781. [Google Scholar] [CrossRef]
- Mistry, M.J.; Clay, M.A.; Kelly, M.E.; Steiner, M.A.; Harmony, J.A.K. Apolipoprotein E restricts interleukin-dependent T lymphocyte proliferation at the G1A/G1B boundary. Cell Immunol. 1995, 160, 14–23. [Google Scholar] [CrossRef]
- de Bont, N.; Netea, M.G.; Demacker, P.N.; Verschueren, I.; Kullberg, B.J.; Van Dijk, K.W.; Van der Meer, J.W.M.; Stalenhoef, A.F. Apolipoprotein E knockout mice are highly susceptible to endotoxemia and Klebsiella pneumoniae infection. J. Lipid Res. 1999, 40, 680–685. [Google Scholar] [CrossRef]
- Vonk, A.G.; De Bont, N.; Netea, M.G.; Demacker, P.N.M.; Van der Meer, J.W.M.; Stalenhoef, A.F.H.; Kullberg, B.J. Apolipoprotein E deficient mice exhibit an increased susceptibility to disseminated candidiasis. Med. Mycol. 2004, 42, 341–348. [Google Scholar] [CrossRef]
- Miller, R.M.; Federoff, H.J. Isoform-specific effects of apoE on HSV immediate early gene expression and establishment of latency. Neurobiol. Aging 2008, 29, 71–77. [Google Scholar] [CrossRef]
- Burt, T.D.; Agan, B.K.; Marconi, V.C.; He, W.; Kulkarni, H.; Mold, J.E.; Cavrois, M.; Huang, Y.; Mahley, R.W.; Dolan, M.J.; et al. Apolipoprotein (apo) E4 enhances HIV-1 cell entry in vitro, and the APOE epsilon4/epsilon4 genotype accelerates HIV disease progression. Proc. Natl. Acad. Sci. USA 2008, 105, 8718–8723. [Google Scholar] [CrossRef] [PubMed]
- Bonacina, F.; Coe, D.; Wang, G.; Longhi, M.P.; Baragetti, A.; Moregola, A.; Garlaschelli, K.; Uboldi, P.; Pellegatta, F.; Grigore, L.; et al. Myeloid apolipoprotein E controls dendritic cell antigen presentation and T cell activation. Nat. Commun. 2018, 9, 3083. [Google Scholar] [CrossRef] [PubMed]
- Frostegard, J.; Ulfgren, A.-K.; Nyberg, P.; Hedin, U.; Swedenborg, J.; Andersson, U.; Hansson, G.K. Cytokine expression in advanced human atherosclerotic plaques: Dominance of pro-inflammatory (Th1) and macrophage-stimulating cytokines. Atherosclerosis 1999, 145, 33–43. [Google Scholar] [CrossRef]
- Allan, L.L.; Hoefl, K.; Zheng, D.-J.; Chung, B.K.; Kozak, F.K.; Tan, R.; van den Elzen, P. Apolipoprotein-mediated lipid antigen presentation in B cells provides a pathway for innate help by NKT cells. Blood 2009, 114, 2411–2416. [Google Scholar] [CrossRef] [PubMed]
- Major, A.S.; Joyce, S.; Van Kaer, L. Lipid metabolism, atherogenesis and CD1-restricted antigen presentation. Trends Mol. Med. 2006, 12, 270–278. [Google Scholar] [CrossRef] [PubMed]
- van den Elzen, P.; Garg, S.; Leon, L.; Brigl, M.; Leadbetter, E.A.; Gumperz, J.E.; Dascher, C.C.; Cheng, T.-Y.; Sacks, F.M.; Illarionov, P.A.; et al. Apolipoprotein-mediated pathways of lipid antigen presentation. Nature 2005, 437, 906–910. [Google Scholar] [CrossRef] [PubMed]
- Getz, G.S.; VanderLaan, P.A.; Reardon, C.A. Natural killer T cells in lipoprotein metabolism and atherosclerosis. Thromb. Haemost. 2011, 106, 814–819. [Google Scholar] [CrossRef] [PubMed]
- Majack, R.A.; Castle, C.K.; Goodman, L.V.; Weisgraber, K.H.; Mahley, R.W.; Shooter, E.M.; Gebicke-Haerter, P.J. Expression of apolipoprotein E by cultured vascular smooth muscle cells is controlled by growth state. J. Cell Biol. 1988, 107, 1207–1213. [Google Scholar] [CrossRef] [PubMed]
- Moore, Z.W.Q.; Zhu, B.; Kuhel, D.G.; Hui, D.Y. Vascular apolipoprotein E expression and recruitment from circulation to modulate smooth muscle cell response to endothelial denudation. Am. J. Pathol. 2004, 164, 2109–2116. [Google Scholar] [CrossRef]
- Hui, D.Y.; Basford, J.E. Distinct signaling mechanisms for apoE inhibition of cell migration and proliferation. Neurobiol. Aging 2005, 26, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, R.; Gordon, D.; Stanley, J.C.; Webb, R.C. Cell cycle effects of nitric oxide on vascular smooth muscle cells. Am. J. Physiol. 1997, 272, H1810–H1818. [Google Scholar] [CrossRef]
- Ji, Z.S.; Fazio, S.; Mahley, R.W. Variable heparan sulfate proteoglycan binding of apolipoprotein E variants may modulate the expression of Type III Hyperlipoproteinemia. J. Biol. Chem. 1994, 269, 13421–13428. [Google Scholar] [CrossRef]
- Kowal, R.C.; Herz, J.; Weisgraber, K.H.; Mahley, R.W.; Brown, M.S.; Goldstein, J.L. Opposing effects of apolipoproteins E and C on lipoprotein binding to low density lipoprotein receptor-related protein. J. Biol. Chem. 1990, 265, 10771–10779. [Google Scholar] [CrossRef]
- Moore, Z.W.Q.; Hui, D.Y. Apolipoprotein E inhibition of vascular hyperplasia and neointima formation requires inducible nitric oxide synthase. J. Lipid Res. 2005, 46, 2083–2090. [Google Scholar] [CrossRef] [PubMed]
- Tada, H. The E4 allele of apolipoprotein E is associated with increased restenosis after coronary angioplasty. Tokai J. Exp. Clin. Med. 2001, 26, 81–92. [Google Scholar] [PubMed]
- van Bockxmeer, F.M.; Mamotte, C.D.S.; Gibbons, F.R.; Taylor, R.R. Apolipoprotein E4 homozygosity—A determinant of restenosis after coronary angioplasty. Atherosclerosis 1994, 110, 195–202. [Google Scholar] [CrossRef]
- Kothapalli, D.; Liu, S.-L.; Bae, Y.H.; Monslow, J.; Xu, T.; Hawthorne, E.A.; Byfield, F.J.; Castagnino, P.; Rao, S.; Rader, D.J.; et al. Cardiovascular protection by apoE and apoE-HDL linked to suppression of ECM gene expression and arterial stiffening. Cell Rep. 2012, 2, 1259–1271. [Google Scholar] [CrossRef]
- Liu, F.; Mih, J.D.; Shea, B.S.; Kho, A.T.; Sharif, A.S.; Tager, A.M.; Tschumperlin, D.J. Feedback amplification of fibrosis through matrix siffening and COX-2 suppression. J. Cell Biol. 2010, 190, 693–706. [Google Scholar] [CrossRef] [PubMed]
- Alvim, R.O.; Freitas, S.R.S.; Ferreira, N.E.; Santos, P.C.J.L.; Cunha, R.S.; Mill, J.G.; Krieger, J.E.; Pereira, A.C. APOE polymphism is associated with lipid profile, but not with arterial stiffness in the general population. Lipids Health Dis. 2010, 9, 128. [Google Scholar] [CrossRef]
- Volcik, K.A.; Barkley, R.A.; Hutchinson, R.G.; Mosley, T.H.; Heiss, G.; Sharrett, A.R.; Ballantyne, C.M.; Boerwinkle, E. Apolipoprotein E polymorphisms predict low density lipoprotein cholesterol levels and carotid artery wall thickness but not incident coronary heart disease in 12,491 ARIC study participants. Am. J. Epidemiol. 2006, 164, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Boulenouar, H.; Benchekor, S.M.; Djellouli, H.O.; Hetraf, S.A.L.; Houti, L.; Hammani-Medjauui, I. Association study of APOE gene polymorphisms with diabetes and the main cardiometabolic risk factors in the Algerian population. Egypt. J. Med. Hum. Genet. 2019, 20, 5. [Google Scholar] [CrossRef]
- Galal, A.A.; Abd Elmajeed, A.A.; Elbaz, R.A.; Wafa, A.M.; Elshazli, R.M. Association of apolipoprotein E gene polymorphism with the risk of T2DM and obesity among Egyptian subjects. Gene 2021, 769, 145223. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, X.; Fan, P.; Liu, R.; Huang, Y.; Liang, S.; Liu, Y.; Wu, Y.; Bai, H. Distribution and effect of apoE genotype on plasma lipid and apolipoprotein profiles in overweight/obese and nonobese Chinese subjects. J. Clin. Lab. Anal. 2012, 26, 200–205. [Google Scholar] [CrossRef]
- Zarkesh, M.; Daneshpour, M.S.; Faam, B.; Hedayati, M.; Azizi, F. Is there any association of apolipoprotein E gene polymorphism with obesity status and lipid profiles. Tehran lipid and gucose study (TLGS). Gene 2012, 509, 282–285. [Google Scholar] [CrossRef] [PubMed]
- Kuhel, D.G.; Konaniah, E.S.; Basford, J.E.; McVey, C.; Goodin, C.T.; Chatterjee, T.K.; Weintraub, N.L.; Hui, D.Y. Apolipoprotein E2 accentuates postprandial inflammation and diet-induced obesity to promote hyperinsulinemia in mice. Diabetes 2013, 62, 382–391. [Google Scholar] [CrossRef]
- Utermann, G. Apolipoprotein E polymorphism in health and disease. Am. Heart J. 1987, 113, 433–440. [Google Scholar] [CrossRef]
- Hofmann, S.M.; Perez-Tilve, D.; Greer, T.M.; Coburn, B.A.; Grant, E.; Basford, J.E.; Tschop, M.H.; Hui, D.Y. Defective lipid delivery modulates glucose tolerance and metabolic response to diet in apolipoprotein E-deficient mice. Diabetes 2008, 57, 5–12. [Google Scholar] [CrossRef]
- Huang, Z.H.; Gu, D.; Mazzone, T. Role of adipocyte-derived apoE in modulating adipocyte size, lipid metabolism, and gene expression in vivo. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E1110–E1119. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.H.; Reardon, C.A.; Mazzone, T. Endogenous apoE expression modulates adipocyte triglyceride content and turnover. Diabetes 2006, 55, 3394–3402. [Google Scholar] [CrossRef]
- Oh, J.-Y.; Barrett-Conner, E. Apolipoprotein E polymorphism and lipid levels differ by gender and family history of diabetes: The Rancho Bernardo Study. Clin. Genet. 2001, 60, 132–137. [Google Scholar] [CrossRef]
- Lumsden, A.L.; Mulugeta, A.; Zhou, A.; Hypponen, E. Apolipoprotein E (APOE) genotype-assoicated disease risks: A phenome-wide, registry-bsed, case-control study utilising the UK biobank. EBioMedicine 2020, 59, 102954. [Google Scholar] [CrossRef] [PubMed]
- Tejedor, M.T.; Garcia-Sobreviela, M.P.; Ledesma, M.; Arbones-Mainar, J.M. The apolipoprotein E polymorphism rs7412 associates with body fatness independently of plasma lipids in middle aged men. PLoS ONE 2014, 9, e108605. [Google Scholar] [CrossRef]
- Arbones-Mainar, J.M.; Johnson, L.A.; Altenburg, M.K.; Kim, H.-S.; Maeda, N. Impaired adipogenic response to thiazolidinediones in mice expressing human apolipoprotein E4. FASEB J. 2010, 24, 3809–3818. [Google Scholar] [CrossRef]
- Arbones-Mainar, J.M.; Johnson, L.A.; Torres-Perez, E.; Garcia, A.E.; Perez-Diaz, S.; Raber, J.; Maeda, N. Metabolic shifts toward fatty acid usage and increased thermogenesis are associated with impaired adipogenesis in mice expressing human APOE4. Int. J. Obes. 2016, 40, 1574–1581. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Martinez, A.B.; Torres-Perez, E.; Devannehy, N.; Del Moral, R.; Johnson, L.A.; Arbones-Mainar, J.M. Beyond the CNS: The many peripheral roles of APOE. Neurobiol. Dis 2020, 138, 104809. [Google Scholar] [CrossRef] [PubMed]
- Elosua, R.; Demissie, S.; Cupples, L.A.; Meigs, J.B.; Wilson, P.W.F.; Schaefer, E.J.; Corella, D.; Ordovas, J.M. Obesity modulates the association among APOE genotype, insulin, and glucose in men. Obes. Res. 2003, 11, 1502–1508. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Liu, J.; Weng, R.; Gu, X.; Zhong, Z. Apolipoprotein E gene polymorphism and the risk of cardiovascular disease and type 2 diabetes. BMC Cardiovasc. Disord. 2019, 19, 213. [Google Scholar] [CrossRef] [PubMed]
- Keeney, J.T.-R.; Ibrahimi, S.; Zhao, L. Human apoE isoforms differentially modulate glucose and amyloid metabolic pathways in female brain: Evidence of the mechanism of neuroprotection by apOE2 and implications for Alzheimer’s prevention and early intervention. J. Alzheimer’s Dis. 2015, 48, 411–424. [Google Scholar] [CrossRef]
- Arbones-Mainar, J.M.; Johnson, L.A.; Altenburg, M.K.; Maeda, N. Differential modulation of diet-induced obesity and adipocyte functionality by human apolipoprotein E3 and E4 in mice. Int. J. Obes. 2008, 32, 1595–1605. [Google Scholar] [CrossRef] [PubMed]
- Apostolova, L.G.; Risacher, S.L.; Duran, T.; Stage, E.C.; Goukasian, N.; West, J.D.; Do, T.M.; Grotts, J.; Wilhalme, H.; Nho, K.; et al. Associations of the Top 20 Alzheimer Disease Risk Variants with Brain Amyloidosis. JAMA Neurol. 2018, 75, 328–341. [Google Scholar] [CrossRef] [PubMed]
- Naj, A.C.; Schellenberg, G.D.; Alzheimer’s Disease Genetics Consortium. Genomic variants, genes, and pathways of Alzheimer’s disease: An overview. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2017, 174, 5–26. [Google Scholar] [CrossRef]
- Nagy, Z.S.; Esiri, M.M.; Jobst, K.A.; Johnston, C.; Litchfield, S.; Sim, E.; Smith, A.D. Influence of the apolipoprotein E genotype on amyloid deposition and neurofibrillary tangle formation in Alzheimer’s disease. Neuroscience 1995, 69, 757–761. [Google Scholar] [CrossRef]
- Genin, E.; Hannequin, D.; Wallon, D.; Sleegers, K.; Hiltunen, M.; Combarros, O.; Bullido, M.J.; Engelborghs, S.; De Deyn, P.; Berr, C.; et al. APOE and Alzheimer disease: A major gene with semi-dominant inheritance. Mol. Psychiatr. 2011, 16, 903–907. [Google Scholar] [CrossRef] [PubMed]
- Farrer, L.A.; Cupples, L.A.; Haines, J.L.; Hyman, B.; Kukull, W.A.; Mayeux, R.; Myers, R.H.; Pericak-Vance, M.A.; Risch, N.; van Duijn, C.M. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 1997, 278, 1349–1356. [Google Scholar] [CrossRef]
- Martins, I.J.; Hone, E.; Foster, J.K.; Sünram-Lea, S.I.; Gnjec, A.; Fuller, S.J.; Nolan, D.; Gandy, S.E.; Martins, R.N. Apolipoprotein E, cholesterol metabolism, diabetes, and the convergence of risk factors for Alzheimer’s disease and cardiovascular disease. Mol. Psychiatr. 2006, 11, 721–736. [Google Scholar] [CrossRef] [PubMed]
- Post, S.G. Future scenarios for the prevention and delay of Alzheimer disease onset in high-risk groups. An ethical perspective. Am. J. Prev. Med. 1999, 16, 105–110. [Google Scholar] [CrossRef]
- Stojakovic, T.; Scharnagl, H.; März, W. ApoE: Crossroads between Alzheimer’s Disease and Atherosclerosis. Semin. Vasc. Med. 2004, 4, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, M.; Rockenstein, E.; Crews, L.; Masliah, E. Role of Protein Aggregation in Mitochondrial Dysfunction and Neurodegeneration in Alzheimer’s and Parkinson’s Diseases. NeuroMol. Med. 2003, 4, 21–35. [Google Scholar] [CrossRef]
- Koutsodendris, N.; Nelson, M.R.; Rao, A.; Huang, Y. Apolipoprotien E and Alzheimer’s disease: Findings, hypotheses, and potential mechanisms. Ann. Rev. Pathol. Mech. Dis. 2022, 17, 73–99. [Google Scholar] [CrossRef]
- Raulin, A.-C.; Martens, Y.A.; Bu, G. Lipoproteins in the central nervous system: From biology to pathobiology. Ann. Rev. Biochem. 2022, 91, 731–759. [Google Scholar] [CrossRef]
- Huang, Y.-W.A.; Zhou, B.; Wernig, M.; Südhof, T.C. ApoE2, ApoE3, and ApoE4 Differentially Stimulate APP Transcription and Aβ Secretion. Cell 2017, 168, 427–441.e421. [Google Scholar] [CrossRef]
- Safieh, M.; Korczyn, A.D.; Michaelson, D.M. ApoE4: An emerging therapeutic target for Alzheimer’s disease. BMC Med. 2019, 17, 64. [Google Scholar] [CrossRef]
- Tachibana, M.; Holm, M.-L.; Liu, C.-C.; Shinohara, M.; Aikawa, T.; Oue, H.; Yamazaki, Y.; Martens, Y.A.; Murray, M.E.; Sullivan, P.M.; et al. APOE4-mediated amyloid-β pathology depends on its neuronal receptor LRP1. J. Clin. Investig. 2019, 129, 1272–1277. [Google Scholar] [CrossRef]
- LaDu, M.; Falduto, M.; Manelli, A.; Reardon, C.; Getz, G.; Frail, D. Isoform-specific binding of apolipoprotein E to beta-amyloid. J. Biol. Chem. 1994, 269, 23403–23406. [Google Scholar] [CrossRef]
- Deane, R.; Sagare, A.; Hamm, K.; Parisi, M.; Lane, S.; Finn, M.B.; Holtzman, D.M.; Zlokovic, B.V. ApoE isoform-specific disruption of amyloid beta peptide clearance from mouse brain. J. Clin. Investig. 2008, 118, 4002–4013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deane, R.; Sagare, A.; Zlokovic, B.V. The role of the cell surface LRP and soluble LRP in blood-brain barrier Abeta clearance in Alzheimer’s disease. Curr. Pharm. Des. 2008, 14, 1601–1605. [Google Scholar] [CrossRef] [PubMed]
- Mahley, R.W.; Weisgraber, K.H.; Huang, Y. Apolipoprotein E4: A causative factor and therapeutic target in neuropathology, including Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2006, 103, 5644–5651. [Google Scholar] [CrossRef] [PubMed]
- Mann, D.M.; Iwatsubo, T.; Pickering-Brown, S.M.; Owen, F.; Saido, T.C.; Perry, R.H. Preferential deposition of amyloid beta protein (Abeta) in the form Abeta40 in Alzheimer’s disease is associated with a gene dosage effect of the apolipoprotein E E4 allele. Neurosci. Lett. 1997, 221, 81–84. [Google Scholar] [CrossRef]
- Holtzman, D.M.; Bales, K.R.; Tenkova, T.; Fagan, A.M.; Parsadanian, M.; Sartorius, L.J.; Mackey, B.; Olney, J.; McKeel, D.; Wozniak, D.; et al. Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2000, 97, 2892–2897. [Google Scholar] [CrossRef]
- Bien-Ly, N.; Gillespie, A.K.; Walker, D.W.; Yoon, S.Y.; Huang, Y. Reducing human apolipoprotein E levels attenuates age-dependent Ab accumulation in mutant human amyloid precursor protein transgenic mice. J. Neurosci. 2012, 32, 4803–4811. [Google Scholar] [CrossRef]
- Bales, K.R.; Verina, T.; Dodel, R.C.; Du, Y.; Altstiel, L.; Bender, M.; Hyslop, P.; Johnstone, E.M.; Little, S.P.; Cummins, D.J.; et al. Lack of apolipoprotein E dramatically reduces amyloid beta-peptide deposition. Nat. Genet. 1997, 17, 263–264. [Google Scholar] [CrossRef]
- Cao, J.; Gaamouch, F.E.; Meabon, J.S.; Meeker, K.D.; Zhu, L.; Zhong, M.B.; Bendik, J.; Elder, G.; Jing, P.; Xia, J. ApoE4-associated phospholipid dysregulation contributes to development of Tau hyper-phosphorylation after traumatic brain injury. Sci. Rep. 2017, 7, 11372. [Google Scholar] [CrossRef]
- Vasilevskaya, A.; Taghdiri, F.; Burke, C.; Tarazi, A.; Naeimi, S.A.; Khodadadi, M.; Goswami, R.; Sato, C.; Grinberg, M.; Moreno, D. Interaction of APOE4 alleles and PET tau imaging in former contact sport athletes. NeuroImage Clin. 2020, 26, 102212. [Google Scholar] [CrossRef]
- Wang, C.; Najm, R.; Xu, Q.; Jeong, D.-E.; Walker, D.; Balestra, M.E.; Yoon, S.Y.; Yuan, H.; Li, G.; Miller, Z.A. Gain of toxic apolipoprotein E4 effects in human iPSC-derived neurons is ameliorated by a small-molecule structure corrector. Nat. Med. 2018, 24, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Yamada, K.; Liddelow, S.A.; Smith, S.T.; Zhao, L.; Luo, W.; Tsai, R.M.; Spina, S.; Grinberg, L.T.; Rojas, J.C. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 2017, 549, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Harris, F.M.; Brecht, W.J.; Xu, Q.; Mahley, R.W.; Huang, Y. Increased tau Phosphorylation in Apolipoprotein E4 Transgenic Mice Is Associated with Activation of Extracellular Signal-regulated Kinase: Modulation by zinc. J. Biol. Chem. 2004, 279, 44795–44801. [Google Scholar] [CrossRef] [PubMed]
- Dubnikov, T.; Cohen, E. The Emerging Roles of Early Protein Folding Events in the Secretory Pathway in the Development of Neurodegenerative Maladies. Front. Neurosci. 2017, 11, 48. [Google Scholar] [CrossRef]
- Richey, P.L.; Siedlak, S.L.; Smith, M.A.; Perry, G. Apolipoprotein E interaction with the neurofibrillary tangles and senile plaques in Alzheimer disease: Implications for disease pathogenesis. Biochem. Biophys. Res. Commun. 1995, 208, 657–663. [Google Scholar] [CrossRef]
- Theendakara, V.; Bredesen, d.E.; Rao, R.V. Downregulation of protein phosphatase 2A by apolipoprotein E: Implications for Alzheimer’s disease. Mol. Cell Neurosci. 2017, 83, 83–91. [Google Scholar] [CrossRef]
- Brecht, W.J.; Harris, F.M.; Chang, S.; Tesseur, I.; Yu, G.-Q.; Xu, Q.; Fish, J.D.; Wyss-Coray, T.; Buttini, M.; Mucke, L.; et al. Neuron-specific apolipoprotein E4 proteolysis is associated with increased tau phosphorylation in brains of transgenic mice. J. Neurosci. 2004, 24, 2527–2534. [Google Scholar] [CrossRef]
- Harris, F.M.; Brecht, W.J.; Xu, Q.; Tesseur, I.; Kekonius, L.; Wyss-Coray, T.; Fish, J.D.; Masliah, E.; Hopkins, P.C.; Scearce-Levie, K.; et al. Carboxyl-terminal-truncated apolipoprotein E4 causes Alzheimer’s diease-like neurodegeneration and behavioral deficits in transgenic mice. Proc. Natl. Acad. Sci. USA 2003, 100, 10966–10971. [Google Scholar] [CrossRef]
- Yu, Y.; Yang, M.; Zhuang, X.; Pan, J.; Zhao, Y.; Yu, Y. Effects of toxic apolipoprotein E fragments on Tau phosphorylation and cognitive impairment in neonatal mice under sevoflurane anesthesia. Brain Behav. 2022, 12, e2702. [Google Scholar] [CrossRef]
- Medway, C.W.; Abdul-Hay, S.; Mims, T.; Ma, L.; Bisceglio, G.; Zou, F.; Pankratz, S.; Sando, S.B.; Aasly, J.O.; Barcikowska, M.; et al. ApoE variant p.V236E is associated wtih markedly reduced risk of Alzheimer’s disease. Mol. Neurodegener 2014, 9, 11. [Google Scholar] [CrossRef]
- Le Guen, Y.; Belloy, M.E.; Grenier-Boley, B.; de Rojas, I.; Castillo-Morales, A.; Jansen, I.; Nicolas, A.; Bellenguez, C.; Dalmasso, C.; Küçükali, F.; et al. Association of rare APOE missense variants V236E and R251G with risk of Alzheimer’s disease. JAMA Neurol. 2022, 79, 652–663. [Google Scholar] [CrossRef]
- Williams, B.; Convertino, M.; Das, J.; Dokholyan, N.V. ApoE4-specific misfolded intermediate identified by molecular dynamics simulations. PLoS Comput. Biol. 2015, 11, e1004359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, W.S.; Verghese, P.B.; Chakraborty, C.; Joung, J.K.; Hyman, B.T.; Ulrich, J.D.; Holtzman, D.M.; Barres, B.A. Novel allele-dependent role for APOE in controlling the reate of synapse pruning by astrocytes. Proc. Nat. Acad. Sci. USA 2016, 113, 10186–10191. [Google Scholar] [CrossRef]
- Najm, R.; Zalocusky, K.A.; Zilberer, M.; Yoon, S.Y.; Hao, Y.; Koutsodendris, N.; Nelson, M.R.; Rao, A.; Taubes, A.; Jones, E.A.; et al. In vivo chimeric Alzheimer’s disease modeling of apolipoprotein E4 toxicity in human neurons. Cell Rep. 2020, 32, 107962. [Google Scholar] [CrossRef]
- Iannucci, J.; Sen, A.; Grammas, P. Isoform-specific effects of apolipoprotein E on markers of inflammation and toxicity in brain glia and neuronal cells in vitro. Curr. Issues Mol. Biol. 2021, 43, 215–225. [Google Scholar] [CrossRef]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581 e569. [Google Scholar] [CrossRef]
- Li, J.-T.; Zhang, Y. TREM2 regulates innate immunity in Alzheimer’s disease. J. Neuroinflamm. 2018, 15, 107. [Google Scholar] [CrossRef] [PubMed]
- Maezawa, I.; Nivison, M.; Montine, K.S.; Maeda, N.; Montine, T.J. Neurotoxicity from innate immune response is greatest with targeted replacement of E4 allele of apolipoproein E gene and is mediated by microglial p38MAPK. FASEB J. 2006, 20, 797–799. [Google Scholar] [CrossRef] [PubMed]
- Lanfranco, M.F.; Sepulveda, J.; Kopetsky, G.; Rebeck, G.W. Expression and secretion of apoE isoforms in astrocytes and microglia during inflammation. Glia 2021, 69, 1478–1493. [Google Scholar] [CrossRef] [PubMed]
- Buttini, M.; Masliah, E.; Yu, G.-Q.; Palop, J.J.; Chang, S.; Bernardo, A.; Lin, C.; Wyss-Coray, T.; Huang, Y.; Mucke, L. Cellular source of apolipoprotein E4 determines neuronal susceptibility to excitotoxic injury in transgenic mice. Am. J. Pathol. 2010, 177, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Knoferle, J.; Yoon, S.Y.; Walker, D.W.; Leung, L.; Gillespie, A.K.; Tong, L.M.; Bien-Ly, N.; Huang, Y. Apolipoprotein E4 produced in GABAergic interneurons causes learning and memory deficits in mice. J. Neurosci. 2014, 34, 14069–14078. [Google Scholar] [CrossRef]
- Chang, S.; Ma, T.R.; Miranda, R.D.; Balestra, M.E.; Mahley, R.W.; Huang, Y. Lipid- and receptor-binding regions of apolipoprotein E4 fragments act in concert to cause mitochondrial dysfunction and neurotoxicity. Proc. Natl. Acad. Sci. USA 2005, 102, 18694–18699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, T.; Watanabe, A.; Fujino, T.; Hosono, T.; Michikawa, M. Apolipoprotein E4 (1-272) fragment is associated with mitochondrial proteins and affects mitochondrial function in neuronal cells. Mol. Neurodegener 2009, 4, 35. [Google Scholar] [CrossRef]
- Pang, S.; Li, J.; Zhang, Y.; Chen, J. Meta-Analysis of the Relationship between the APOE Gene and the Onset of Parkinson’s Disease Dementia. Parkinsons Dis. 2018, 2018, 9497147. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.-Y.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Gallardo, G.; Schluter, O.M.; Sudhof, T.C. A molecular pathway of neurodegeneration linking alpha-synuclein to apoE and Abeta peptides. Nat. Neurosci. 2008, 11, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Ogaki, K.; Martens, Y.A.; Heckman, M.G.; Koga, S.; Labbe, C.; Lorenzo-Betancor, O.; Wernick, A.I.; Walton, R.L.; Soto, A.I.; Vargas, E.R.; et al. Multiple system atrophy and apolipoprotein E. Mov. Disord. 2018, 33, 647–650. [Google Scholar] [CrossRef]
- Li, Y.; Macyczko, J.R.; Liu, C.-C.; Bu, G. ApoE4 reduction: An emerging and promising therapeutic strategy for Alzheimer’s disease. Neurobiol. Aging 2022, 115, 20–28. [Google Scholar] [CrossRef]
- Mahley, R.W. Apolipoprotein E: From cardiovascular disease to neurodegenerative disorders. J. Mol. Med. 2016, 94, 739–746. [Google Scholar] [CrossRef]
- Lynch, J.R.; Tang, W.; Wang, H.; Vitek, M.P.; Bennett, E.R.; Sullivan, P.M.; Warner, D.S.; Laskowitz, D.T. APOE Genotype and an ApoE-mimetic Peptide Modify the Systemic and Central Nervous System Inflammatory Response. J. Biol. Chem. 2003, 278, 48529–48533. [Google Scholar] [CrossRef]
- Kockx, M.; Jessup, W.; Kritharides, L. Regulation of endogenous apolipoprotein E secretion by macrophages. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1060–1067. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Kantor, B.; Chiba-Falek, O. APOE: The new frontier in the development of a therapeutic target towards precision medicine in late-onset Alzheimer’s. Int. J. Mol. Sci. 2021, 22, 1244. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.D.; Evans, V.; Owen, J.S. ApoE gene therapy to treat hyperlipidemia and atherosclerosis. Curr. Opin. Mol. Ther. 2006, 8, 275–287. [Google Scholar] [PubMed]
- Angelopoulou, E.; Paudel, Y.N.; Papageorgiou, S.G.; Piperi, C. APOE genotype and Alzheimer’s disease: The influence of lifestyle and environmental factors. ACS Chem. Neurosci. 2021, 12, 2749–2764. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Conditions | ApoE−/− Mice | APOE2 Mice | APOE2 Humans | APOE4 Mice | APOE4 Humans |
|---|---|---|---|---|---|
| Cardiovascular | |||||
| Atherosclerosis | ↑↑ | ↑ | ↑↓ depends on plasma lipids | ↑ | ↑ |
| Plasma lipids | ↑↑ | ↑ | ↑↓ with confounding factors | ↑ | ↑ |
| Inflammation | ↑↑ | ↑ | ↑↓ depends on plasma lipids | ↑ | ↑ |
| Metabolic diseases | |||||
| Obesity | ↓ | ↑ | ↑↓ with confounding factors | ↓ | ↑↓ controversial |
| Diabetes | ↑ | ↑ | ↑ | ↑ | ↑ |
| Neurodegenerative diseases | |||||
| Cognition | ↓ | ↑ | ↑ | ↓ | ↓ |
| Aβ accumulation | ↓ | ↓ | ↓ | ↑ | ↑ |
| Tau phosphorylation | ↓ | ↓ | ↓ | ↑ | ↑ |
| Neuroinflammation | ↑ | ↓ | ↓ | ↑ | ↑ |
| Neuronal toxicity | - | - | - | ↑ | ↑ |
| Parkinson’s disease | ↓ | - | - | ↑ | ↑ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alagarsamy, J.; Jaeschke, A.; Hui, D.Y. Apolipoprotein E in Cardiometabolic and Neurological Health and Diseases. Int. J. Mol. Sci. 2022, 23, 9892. https://doi.org/10.3390/ijms23179892
Alagarsamy J, Jaeschke A, Hui DY. Apolipoprotein E in Cardiometabolic and Neurological Health and Diseases. International Journal of Molecular Sciences. 2022; 23(17):9892. https://doi.org/10.3390/ijms23179892
Chicago/Turabian StyleAlagarsamy, Jeyashree, Anja Jaeschke, and David Y. Hui. 2022. "Apolipoprotein E in Cardiometabolic and Neurological Health and Diseases" International Journal of Molecular Sciences 23, no. 17: 9892. https://doi.org/10.3390/ijms23179892
APA StyleAlagarsamy, J., Jaeschke, A., & Hui, D. Y. (2022). Apolipoprotein E in Cardiometabolic and Neurological Health and Diseases. International Journal of Molecular Sciences, 23(17), 9892. https://doi.org/10.3390/ijms23179892