

Amorphous and Co-Amorphous Olanzapine Stability in Formulations Intended for Wet Granulation and Pelletization




Abstract
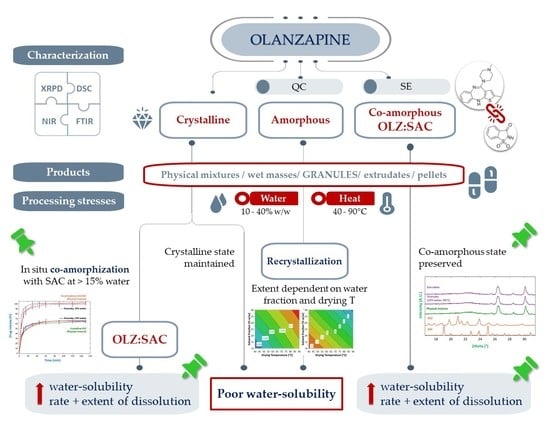
:
1. Introduction
2. Results and Discussion
2.1. Preparation of Partially Amorphous and Co-Amorphous Olanzapine
2.2. Preparation of Granules, Extrudates and Pellets Containing Crystalline, Partially Amorphous and Co-Amorphous Olanzapine
2.2.1. Diffractometric and Calorimetric Characterization of Materials
2.2.2. Fourier-Transformed Mid Infrared (FTIR) and Near Infrared (NIR) Spectroscopy
2.2.3. Dissolution Tests
3. Materials and Methods
3.1. Materials
3.2. Methods
3.2.1. Preparation of Amorphous and Co-Amorphous Olanzapine
3.2.2. Characterization of Amorphous and Co-Amorphous Olanzapine
3.2.3. Formulation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Component | Formulation | |
|---|---|---|
| A | B | |
| Olanzapine | 30 | 30 |
| Saccharin | 0 | 18 |
| Dibasic calcium phosphate anhydrous | 45 | 27 |
| Microcrystalline cellulose | 20 | 20 |
| Polyvinylpyrrolidone | 5 | 5 |
3.2.4. Preparation of Granules, Extrudates and Pellets
3.2.5. Characterization of Granules, Extrudates and Pellets
3.2.6. Statistical Analysis
3.2.7. Principal Component Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Attene-Ramos, M.S.; Austin, C.P.; Xia, M. High Throughput Screening. In Encyclopedia of Toxicology; Wexler, P., Ed.; Elsevier Inc.: Waltham, MA, USA, 2014; pp. 916–917. [Google Scholar]
- Aldewachi, H.; Al-Zidan, R.N.; Conner, M.T.; Salman, M.M. High-throughput screening platforms in the discovery of novel drugs for neurodegenerative diseases. Bioengineering 2021, 8, 30. [Google Scholar] [CrossRef]
- Owoyemi, B.C.D.; Da Silva, C.C.P.; Souza, M.S.; Diniz, L.F.; Ellena, J.; Carneiro, R.L. Fluconazole: Synthesis and Structural Characterization of Four New Pharmaceutical Cocrystal Forms. Cryst. Growth Des. 2019, 19, 648–657. [Google Scholar] [CrossRef]
- Caparica, R.; Júlio, A.; Fernandes, F.; Araújo, M.E.M.; Costa, J.G.; Santos de Almeida, T. Upgrading the topical delivery of poorly soluble drugs using ionic liquids as a versatile tool. Int. J. Mol. Sci. 2021, 22, 4338. [Google Scholar] [CrossRef] [PubMed]
- Alshehri, S.; Imam, S.S.; Hussain, A.; Altamimi, M.A.; Alruwaili, N.K.; Alotaibi, F.; Alanazi, A.; Shakeel, F. Potential of solid dispersions to enhance solubility, bioavailability, and therapeutic efficacy of poorly water-soluble drugs: Newer formulation techniques, current marketed scenario and patents. Drug Deliv. 2020, 27, 1625–1643. [Google Scholar] [CrossRef] [PubMed]
- Boyd, B.J.; Bergström, C.A.S.; Vinarov, Z.; Kuentz, M.; Brouwers, J.; Augustijns, P.; Brandl, M.; Bernkop-Schnürch, A.; Shrestha, N.; Préat, V.; et al. Successful oral delivery of poorly water-soluble drugs both depends on the intraluminal behavior of drugs and of appropriate advanced drug delivery systems. Eur. J. Pharm. Sci. 2019, 137, 104967. [Google Scholar] [CrossRef]
- Savjani, K.T.; Gajjar, A.K.; Savjani, J.K. Drug Solubility: Importance and Enhancement Techniques. ISRN Pharm. 2012, 2012, 195727. [Google Scholar] [CrossRef]
- Bahetibieke, S.; Moinuddin, S.M.; Baiyisaiti, A.; Liu, X.; Zhang, J.; Liu, G.; Shi, Q.; Peng, A.; Tao, J.; Di, C.; et al. Co-Amorphous Formation of Simvastatin-Ezetimibe: Enhanced Physical Stability, Bioavailability and Cholesterol-Lowering Effects in LDLr−/−Mice. Pharmaceutics 2022, 14, 1258. [Google Scholar] [CrossRef]
- Blagden, N.; de Matas, M.; Gavan, P.T.; York, P. Crystal engineering of active pharmaceutical ingredients to improve solubility and dissolution rates. Adv. Drug Deliv. Rev. 2007, 59, 617–630. [Google Scholar] [CrossRef]
- Almarsson, Ö.; Zaworotko, M.J. Crystal engineering of the composition of pharmaceutical phases. Do pharmaceutical co-crystals represent a new path to improved medicines? Chem. Commun. 2004, 35, 1889–1896. [Google Scholar] [CrossRef]
- Varshosaz, J.; Ghassami, E.; Ahmadipour, S. Crystal Engineering for Enhanced Solubility and Bioavailability of Poorly Soluble Drugs. Curr. Pharm. Des. 2018, 24, 2473–2496. [Google Scholar] [CrossRef]
- Kasten, G.; Löbmann, K.; Grohganz, H.; Rades, T. Co-former selection for co-amorphous drug-amino acid formulations. Int. J. Pharm. 2019, 557, 366–373. [Google Scholar] [CrossRef] [PubMed]
- Qian, K.; Stella, L.; Jones, D.S.; Andrews, G.P.; Du, H.; Tian, Y. Drug-rich phases induced by amorphous solid dispersion: Arbitrary or intentional goal in oral drug delivery? Pharmaceutics 2021, 13, 889. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, Y.; Cheng, J.; Chen, H.; Xu, J.; Liu, Z.; Shi, Q.; Zhang, C. Recent Advances in the Application of Characterization Techniques for Studying Physical Stability of Amorphous Pharmaceutical Solids. Crystals 2021, 11, 1440. [Google Scholar] [CrossRef]
- Graeser, K.A.; Patterson, J.E.; Zeitler, J.A.; Rades, T. The role of configurational entropy in amorphous systems. Pharmaceutics 2010, 2, 224–244. [Google Scholar] [CrossRef] [PubMed]
- Babu, N.J.; Nangia, A. Solubility advantage of amorphous drugs and pharmaceutical cocrystals. Cryst. Growth Des. 2011, 11, 2662–2679. [Google Scholar] [CrossRef]
- Su, M.; Xia, Y.; Shen, Y.; Heng, W.; Wei, Y.; Zhang, L.; Gao, Y.; Zhang, J.; Qian, S. A novel drug-drug coamorphous system without molecular interactions: Improve the physicochemical properties of tadalafil and repaglinide. RSC Adv. 2019, 10, 565–583. [Google Scholar] [CrossRef]
- Wu, W.; Löbmann, K.; Schnitzkewitz, J.; Knuhtsen, A.; Pedersen, D.S.; Grohganz, H.; Rades, T. Aspartame as a co-former in co-amorphous systems. Int. J. Pharm. 2018, 549, 380–387. [Google Scholar] [CrossRef]
- D’Angelo, A.; Edgar, B.; Hurt, A.P.; Antonijević, M.D. Physico-chemical characterisation of three-component co-amorphous systems generated by a melt-quench method. J. Therm. Anal. Calorim. 2018, 134, 381–390. [Google Scholar] [CrossRef]
- Fael, H.; Demirel, A.L. Indomethacin co-amorphous drug-drug systems with improved solubility, supersaturation, dissolution rate and physical stability. Int. J. Pharm. 2021, 600, 120448. [Google Scholar] [CrossRef]
- Ruponen, M.; Kettunen, K.; Pires, M.S.; Laitinen, R. Co-amorphous formulations of furosemide with arginine and p-glycoprotein inhibitor drugs. Pharmaceutics 2021, 13, 171. [Google Scholar] [CrossRef]
- Cruz-Angeles, J.; Videa, M.; Martínez, L.M. Highly Soluble Glimepiride and Irbesartan Co-amorphous Formulation with Potential Application in Combination Therapy. AAPS PharmSciTech 2019, 20, 144. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Moinuddin, S.M.; Cai, T. Advances in coamorphous drug delivery systems. Acta Pharm. Sin. B 2019, 9, 19–35. [Google Scholar] [CrossRef] [PubMed]
- Khanfar, M.; Al-Remawi, M.; Al-Akayleh, F.; Hmouze, S. Preparation and Evaluation of Co-amorphous Formulations of Telmisartan—Amino Acids as a Potential Method for Solubility and Dissolution Enhancement. AAPS PharmSciTech 2021, 22, 112. [Google Scholar] [CrossRef]
- Chen, X.; Li, D.; Zhang, H.; Duan, Y.; Huang, Y. Co-amorphous systems of sinomenine with nonsteroidal anti-inflammatory drugs: A strategy for solubility improvement, sustained release, and drug combination therapy against rheumatoid arthritis. Int. J. Pharm. 2021, 606, 120894. [Google Scholar] [CrossRef] [PubMed]
- Dengale, S.J.; Grohganz, H.; Rades, T.; Löbmann, K. Recent advances in co-amorphous drug formulations. Adv. Drug Deliv. Rev. 2016, 100, 116–125. [Google Scholar] [CrossRef]
- Ueda, H.; Wu, W.; Löbmann, K.; Grohganz, H.; Müllertz, A.; Rades, T. Application of a Salt Coformer in a Co-Amorphous Drug System Dramatically Enhances the Glass Transition Temperature: A Case Study of the Ternary System Carbamazepine, Citric Acid, and l -Arginine. Mol. Pharm. 2018, 15, 2036–2044. [Google Scholar] [CrossRef]
- Martínez-Jiménez, C.; Cruz-Angeles, J.; Videa, M.; Martínez, L.M. Co-amorphous simvastatin-nifedipine with enhanced solubility for possible use in combination therapy of hypertension and hypercholesterolemia. Molecules 2018, 23, 2161. [Google Scholar] [CrossRef]
- Pandi, P.; Bulusu, R.; Kommineni, N.; Khan, W.; Singh, M. Amorphous solid dispersions: An update for preparation, characterization, mechanism on bioavailability, stability, regulatory considerations and marketed products. Int. J. Pharm. 2020, 586, 119560. [Google Scholar] [CrossRef]
- Ayala, A.P.; Siesler, H.W.; Boese, R.; Hoffmann, G.G.; Polla, G.I.; Vega, D.R. Solid state characterization of olanzapine polymorphs using vibrational spectroscopy. Int. J. Pharm. 2006, 326, 69–79. [Google Scholar] [CrossRef]
- Reutzel-Edens, S.M.; Bush, J.K.; Magee, P.A.; Stephenson, G.A.; Byrn, S.R. Anhydrates and Hydrates of Olanzapine: Crystallization, Solid-State Characterization, and Structural Relationships. Cryst. Growth Des. 2003, 3, 897–907. [Google Scholar] [CrossRef]
- Mauri, M.C.; Volonteri, L.S.; Colasanti, A.; Fiorentini, A.; De Gaspari, I.F.; Bareggi, S.R. Clinical Pharmacokinetics of Atypical Antipsychotics. Clin. Pharmacokinet. 2007, 46, 359–388. [Google Scholar] [CrossRef] [PubMed]
- Balbão, M.S.; Hallak, J.E.C.; Nunes, E.A.; de Mello, M.H.; Triffoni-Melo, A.T.; Ferreira, F.I.S.; Chaves, C.; Durão, A.M.S.; Ramos, A.P.P.; Crippa, J.A.S.; et al. Olanzapine, weight change and metabolic effects: A naturalistic 12-month follow up. Ther. Adv. Psychopharmacol. 2014, 4, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Da Costa, N.F.; Santos, I.A.; Fernandes, A.I.; Pinto, J.F. Sulfonic acid derivatives in the production of stable co-amorphous systems for solubility enhancement. J. Pharm. Sci. 2022, in press. [CrossRef]
- Da Costa, N.F.; Fernandes, A.I.; Pinto, J.F. Measurement of the amorphous fraction of olanzapine incorporated in a co-amorphous formulation. Int. J. Pharm. 2020, 588, 119716. [Google Scholar] [CrossRef] [PubMed]
- Da Costa, N.F.; Pinto, J.F.; Fernandes, A.I. Cohesiveness of Powdered Co-Amorphous Olanzapine and Impact on Tablet Production. Med. Sci. Forum 2021, 5, 2. [Google Scholar] [CrossRef]
- Da Costa, N.F.; Daniels, R.; Fernandes, A.I.; Pinto, J.F. Downstream Processing of Amorphous and Co-Amorphous Olanzapine Powder Blends. Pharmaceutics 2022, 14, 1535. [Google Scholar] [CrossRef]
- Sirisha, V.R.; Sri, K.V.; Suresh, K.; Reddy, G.K.; Devanna, N. A Review of Pellets and Pelletization Process—A Multiparticulate Drug Delivery System. Int. J. Pharm. Sci. Res. 2013, 4, 2145–2158. [Google Scholar] [CrossRef]
- Muley, S.; Nandgude, T.; Poddar, S. Extrusion–spheronization a promising pelletization technique: In-depth review. Asian J. Pharm. Sci. 2016, 11, 684–699. [Google Scholar] [CrossRef]
- Pinto, J.F.; Podczeck, F.; Newton, J.M. The Use of Statistical Moment Analysis to Elucidate the Mechanism of Release of a Model Drug from Pellets Produced by Extrusion and Spheronisation. Chem. Pharm. Bull. 1997, 45, 171–180. [Google Scholar] [CrossRef]
- Theismann, E.M.; Keppler, J.K.; Owen, M.; Schwarz, K.; Schlindwein, W. Modelling the effect of process parameters on the wet extrusion and spheronisation of high-loaded nicotinamide pellets using a quality by design approach. Pharmaceutics 2019, 11, 154. [Google Scholar] [CrossRef] [Green Version]
- Greco, S.; Authelin, J.R.; Leveder, C.; Segalini, A. A practical method to predict physical stability of amorphous solid dispersions. Pharm. Res. 2012, 29, 2792–2805. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.A.; Zografi, G.; Gao, P.; Gong, Y.; Zhang, G.G.Z. Modeling Physical Stability of Amorphous Solids Based on Temperature and Moisture Stresses. J. Pharm. Sci. 2016, 105, 2932–2939. [Google Scholar] [CrossRef]
- Bastos, A.C.; Santos, I.A.; Pinto, J.F.; Fernandes, A.I. Co-formability, solubility enhancement and stability of olanzapine co-amorphous systems produced with different co-formers. Ann. Med. 2021, 53, S112–S113. [Google Scholar] [CrossRef]
- Démuth, B.; Farkas, A.; Balogh, A.; Bartosiewicz, K.; Kállai-Szabó, B.; Bertels, J.; Vigh, T.; Mensch, J.; Verreck, G.; Van Assche, I.; et al. Lubricant-Induced Crystallization of Itraconazole From Tablets Made of Electrospun Amorphous Solid Dispersion. J. Pharm. Sci. 2016, 105, 2982–2988. [Google Scholar] [CrossRef]
- Kondo, K.; Miyamoto, K.; Miura, S.; Niwa, T. Solventless granulation and spheronization of indomethacin crystals using a mechanical powder processor: Effects of mechanically induced amorphization on particle formation. Eur. J. Pharm. Biopharm. 2020, 154, 348–358. [Google Scholar] [CrossRef] [PubMed]
- Szafraniec-Szczęsny, J.; Antosik-Rogóż, A.; Knapik-Kowalczuk, J.; Kurek, M.; Szefer, E.; Gawlak, K.; Chmiel, K.; Peralta, S.; Niwiński, K.; Pielichowski, K.; et al. Compression-induced phase transitions of bicalutamide. Pharmaceutics 2020, 12, 438. [Google Scholar] [CrossRef]
- Karagianni, A.; Kachrimanis, K.; Nikolakakis, I. Co-Amorphous Solid Dispersions for Solubility and Absorption Improvement of Drugs: Composition, Preparation, Characterization and Formulations for Oral Delivery. Pharmaceutics 2018, 10, 98. [Google Scholar] [CrossRef]
- Marsac, P.J.; Rumondor, A.C.F.; Nivens, D.E.; Kestur, U.S.; Stanciu, L.; Taylor, L.S. Effect of Temperature and Moisture on the Miscibility of Amorphous Dispersions of Felodipine and Poly(vinyl pyrrolidone). J. Pharm. Sci. 2010, 99, 169–185. [Google Scholar] [CrossRef]
- Xie, T.; Taylor, L.S. Effect of Temperature and Moisture on the Physical Stability of Binary and Ternary Amorphous Solid Dispersions of Celecoxib. J. Pharm. Sci. 2017, 106, 100–110. [Google Scholar] [CrossRef]
- Paisana, M.; Wahl, M.; Pinto, J. Role of polymeric excipients in the stabilization of olanzapine when exposed to aqueous environments. Molecules 2015, 20, 22364–22382. [Google Scholar] [CrossRef] [Green Version]
- Bastin, R.J.; Bowker, M.J.; Slater, B.J. Salt selection and optimisation procedures for pharmaceutical new chemical entities. Org. Process Res. Dev. 2000, 4, 427–435. [Google Scholar] [CrossRef]
- Petry, I.; Löbmann, K.; Grohganz, H.; Rades, T.; Leopold, C.S. Undesired co-amorphisation of indomethacin and arginine during combined storage at high humidity conditions. Int. J. Pharm. 2018, 544, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Petry, I.; Löbmann, K.; Grohganz, H.; Rades, T.; Leopold, C.S. In situ co-amorphisation of arginine with indomethacin or furosemide during immersion in an acidic medium—A proof of concept study. Eur. J. Pharm. Biopharm. 2018, 133, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Petry, I.; Löbmann, K.; Grohganz, H.; Rades, T.; Leopold, C.S. Solid state properties and drug release behavior of co-amorphous indomethacin-arginine tablets coated with Kollicoat® Protect. Eur. J. Pharm. Biopharm. 2017, 119, 150–160. [Google Scholar] [CrossRef] [PubMed]
- Priemel, P.A.; Laitinen, R.; Grohganz, H.; Rades, T.; Strachan, C.J. In situ amorphisation of indomethacin with Eudragit® E during dissolution. Eur. J. Pharm. Biopharm. 2013, 85, 1259–1265. [Google Scholar] [CrossRef]
- Marsac, P.J.; Romary, D.P.; Shamblin, S.L.; Baird, J.A.; Taylor, L.S. Spontaneous Crystallinity Loss of Drugs in the Disordered Regions of Poly(Ethylene Oxide) in the Presence of Water. J. Pharm. Sci. 2008, 97, 3182–3194. [Google Scholar] [CrossRef]
- Tanabe, S.; Higashi, K.; Umino, M.; Limwikrant, W.; Yamamoto, K.; Moribe, K. Yellow coloration phenomena of incorporated indomethacin into folded sheet mesoporous materials. Int. J. Pharm. 2012, 429, 38–45. [Google Scholar] [CrossRef]
- Wu, Y.; Lv, S.; Wang, C.; Gao, X.; Li, J.; Meng, Q. Comparative analysis of volatiles difference of Yunnan sun-dried Pu-erh green tea from different tea mountains: Jingmai and Wuliang mountain by chemical fingerprint similarity combined with principal component analysis and cluster analysis. Chem. Cent. J. 2016, 10, 1–11. [Google Scholar] [CrossRef]
- Combes, C.; Azema, J. Clustering using principal component analysis applied to autonomy-disability of elderly people. Decis. Support Syst. 2013, 55, 578–586. [Google Scholar] [CrossRef]
- FDA. Guidance for Industry Dissolution Testing of Immediate Release Solid Oral Dosage Forms; FDA: Rockville, MD, USA, 1997.
- Rinnan, Å.; van den Berg, F.; Engelsen, S.B. Review of the most common pre-processing techniques for near-infrared spectra. TrAC-Trends Anal. Chem. 2009, 28, 1201–1222. [Google Scholar] [CrossRef]
- Maleki, M.R.; Mouazen, A.M.; Ramon, H.; De Baerdemaeker, J. Multiplicative Scatter Correction during On-line Measurement with Near Infrared Spectroscopy. Biosyst. Eng. 2007, 96, 427–433. [Google Scholar] [CrossRef]










| Sample | Drying Temperature (°C) | Area under the Peak (Intensity.°2θ) | Enthalpy of Recrystallization (J/g) | |
|---|---|---|---|---|
| OLZ QC (amorphous) | Physical Mixture | 0.079 ± 0.002 ## | 12.61 ± 0.11 ## | |
| Granules (10% water) | 40 | 0.079 ± 0.002 ## | 11.80 ± 0.61 ## | |
| 65 | 0.084 ± 0.003 ## | 10.82 ± 0.31 *,## | ||
| 90 | 0.099 ± 0.003 **,# | 7.78 ± 0.88 **,## | ||
| Granules (25% water) | 40 | 0.081 ± 0.001 ## | 10.91 ± 0.53 *,## | |
| 65 | 0.095 ± 0.002 **,# | 9.40 ± 0.25 **,## | ||
| 90 | 0.102 ± 0.002 ** | 5.09 ± 0.63 **,## | ||
| Granules (40% water) | 40 | 0.091 ± 0.003 *,## | 8.49 ± 0.09 **,## | |
| 65 | 0.101 ± 0.002 ** | 5.62 ± 0.20 **,## | ||
| 90 | 0.107 ± 0.001 ** | 4.16 ± 0.10 **,## | ||
| Wet Masses (24 h storage) (40% water) | 40 | 0.105 ± 0.001 ** | 3.63 ± 0.20 **,## | |
| 65 | 0.104 ± 0.003 ** | 2.15 ± 0.25 **,# | ||
| 90 | 0.107 ± 0.001 ** | 2.00 ± 0.20 **,# | ||
| Extrudate | 40 | 0.107 ± 0.002 ** | 1.26 ± 0.21 ** | |
| Pellet | 40 | 0.108 ± 0.001 ** | 0.00 ± 0.00 (a),** | |
| OLZ RM (crystalline) | Physical Mixture | 0.107 ± 0.003 ** | 0.00 ± 0.00 (a),** | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
da Costa, N.F.; Daniels, R.; Fernandes, A.I.; Pinto, J.F. Amorphous and Co-Amorphous Olanzapine Stability in Formulations Intended for Wet Granulation and Pelletization. Int. J. Mol. Sci. 2022, 23, 10234. https://doi.org/10.3390/ijms231810234
da Costa NF, Daniels R, Fernandes AI, Pinto JF. Amorphous and Co-Amorphous Olanzapine Stability in Formulations Intended for Wet Granulation and Pelletization. International Journal of Molecular Sciences. 2022; 23(18):10234. https://doi.org/10.3390/ijms231810234
Chicago/Turabian Styleda Costa, Nuno F., Rolf Daniels, Ana I. Fernandes, and João F. Pinto. 2022. "Amorphous and Co-Amorphous Olanzapine Stability in Formulations Intended for Wet Granulation and Pelletization" International Journal of Molecular Sciences 23, no. 18: 10234. https://doi.org/10.3390/ijms231810234
APA Styleda Costa, N. F., Daniels, R., Fernandes, A. I., & Pinto, J. F. (2022). Amorphous and Co-Amorphous Olanzapine Stability in Formulations Intended for Wet Granulation and Pelletization. International Journal of Molecular Sciences, 23(18), 10234. https://doi.org/10.3390/ijms231810234