
Therapeutic Intervention with Anti-Complement Component 5 Antibody Does Not Reduce NASH but Does Attenuate Atherosclerosis and MIF Concentrations in Ldlr-/-.Leiden Mice

 , , , ,
, , , ,  , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
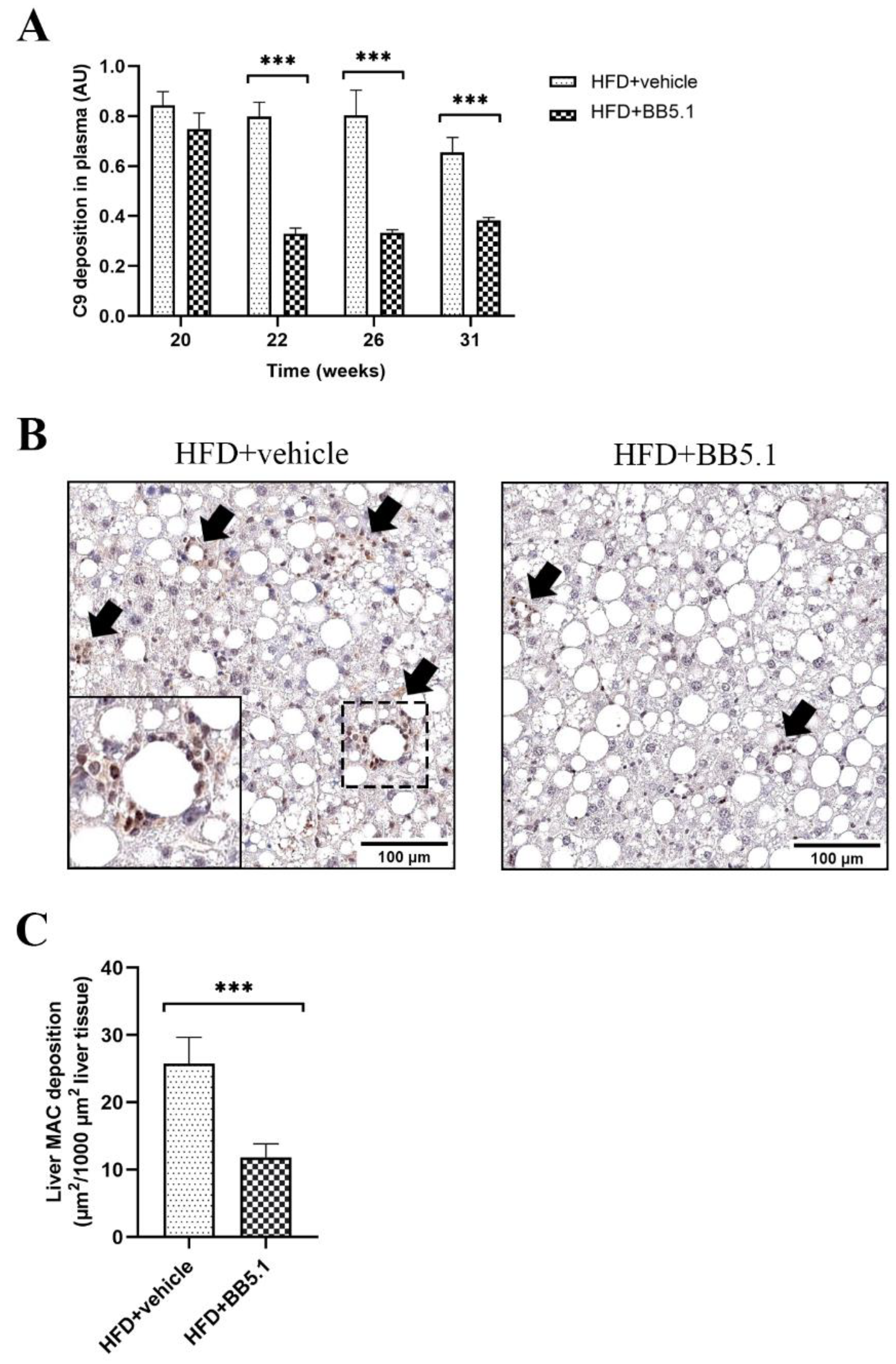
2.1. Anti-C5 Treatment Decreases Complement Activation
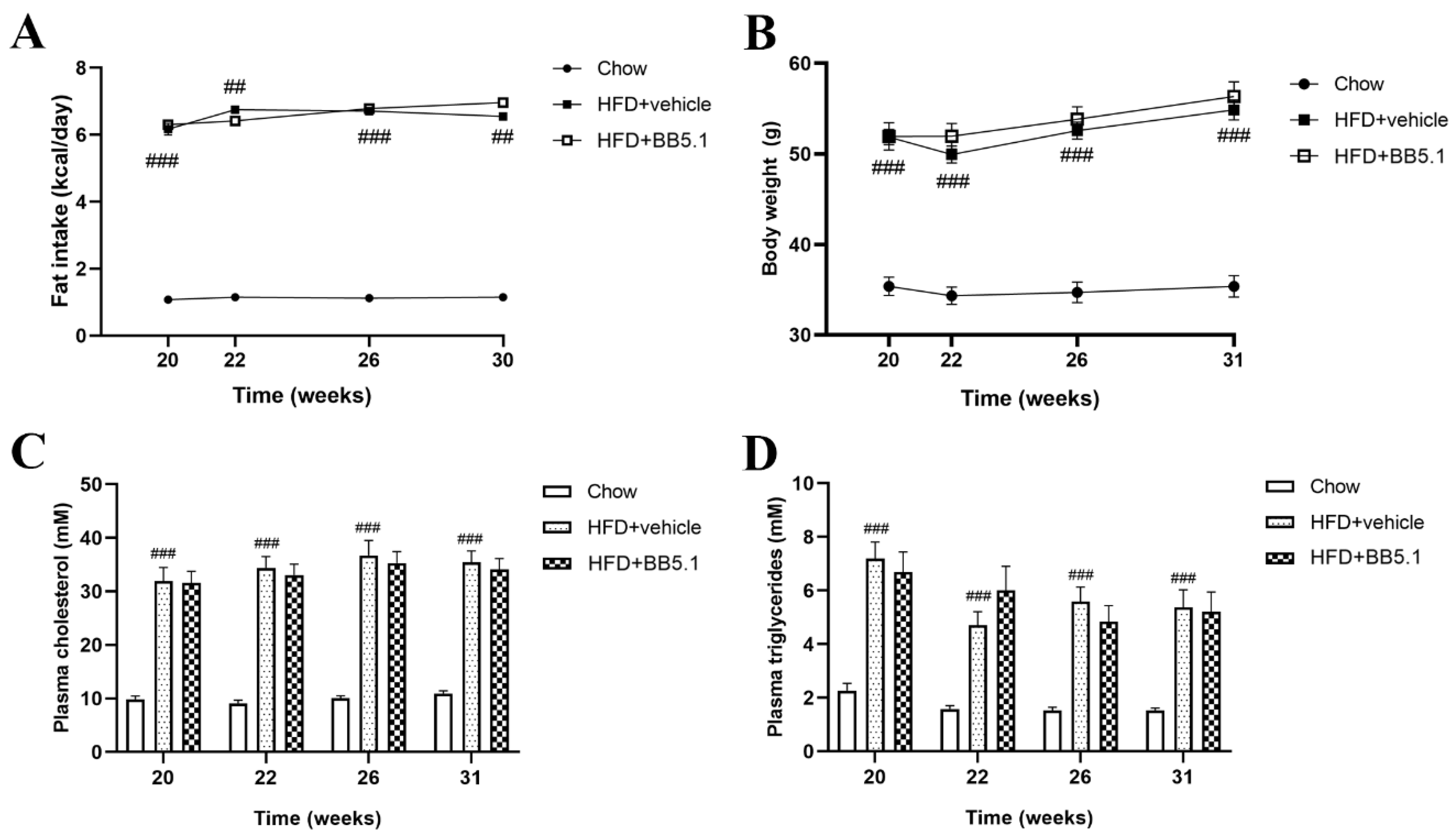
2.2. Anti-C5 Treatment Does Not Affect HFD-Induced Obesity and Its Metabolic Risk Factors
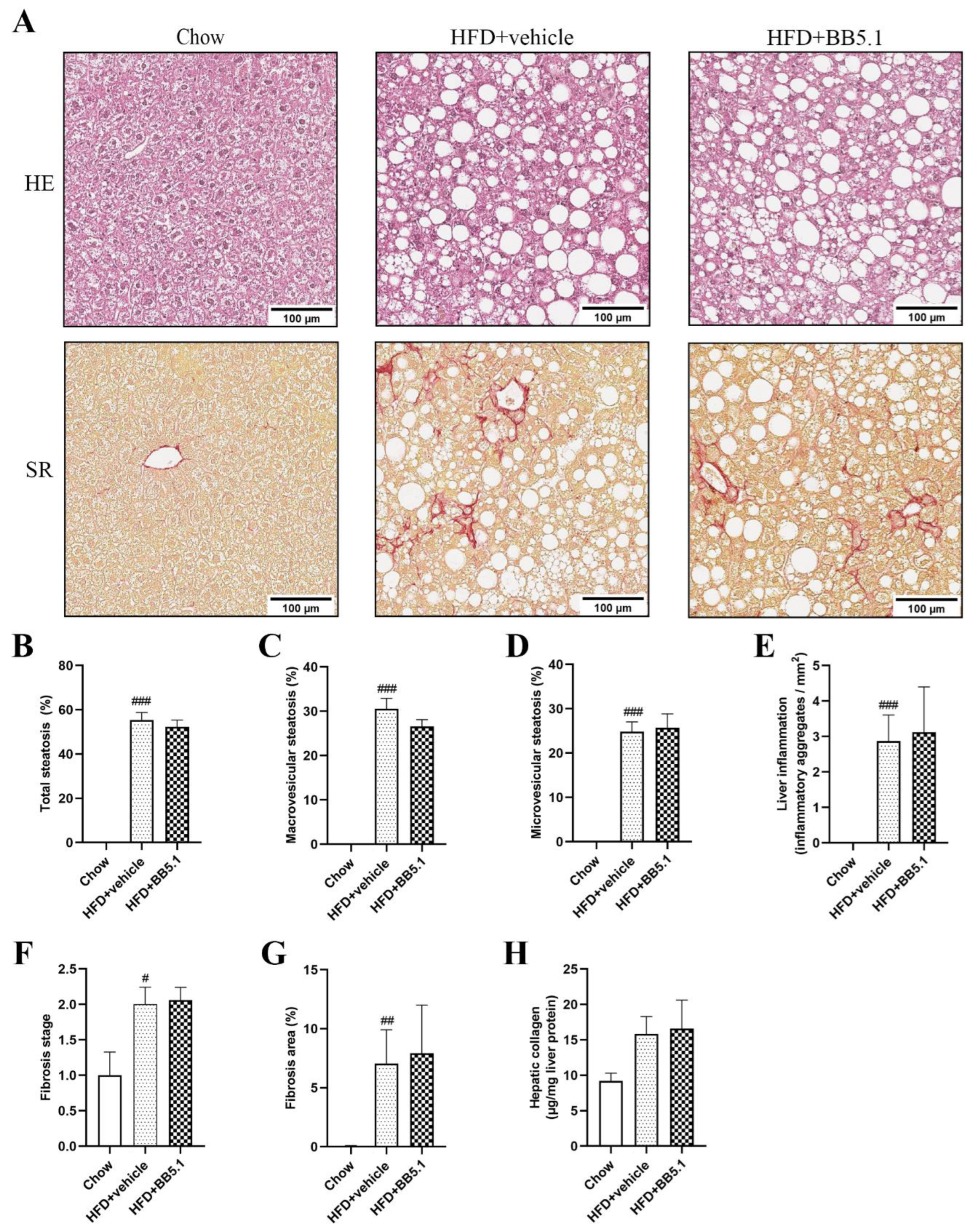
2.3. Anti-C5 Treatment Does Not Affect NASH Development
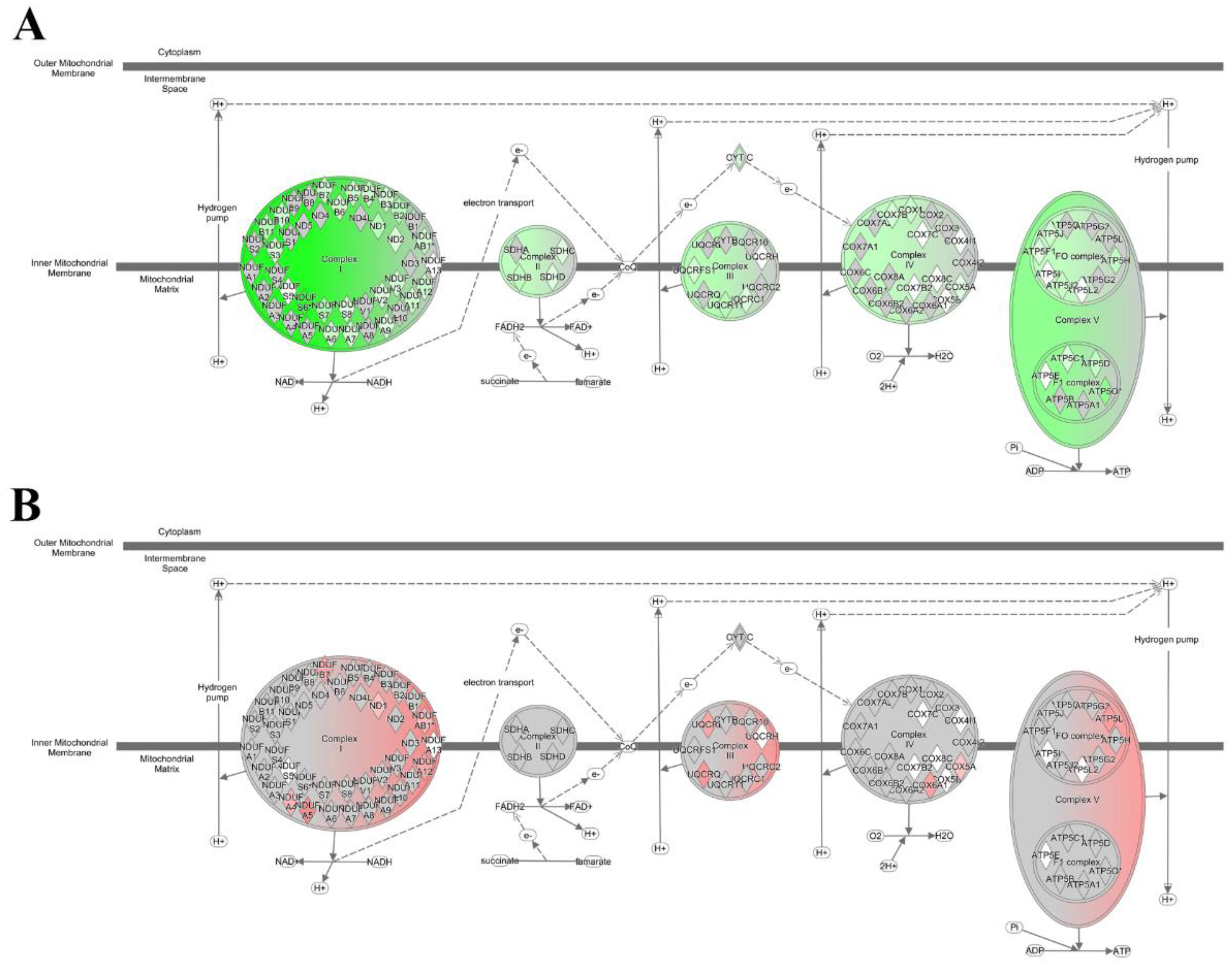
2.4. Anti-C5 Treatment Reverses HFD-Induced Effects on Oxidative Phosphorylation Genes
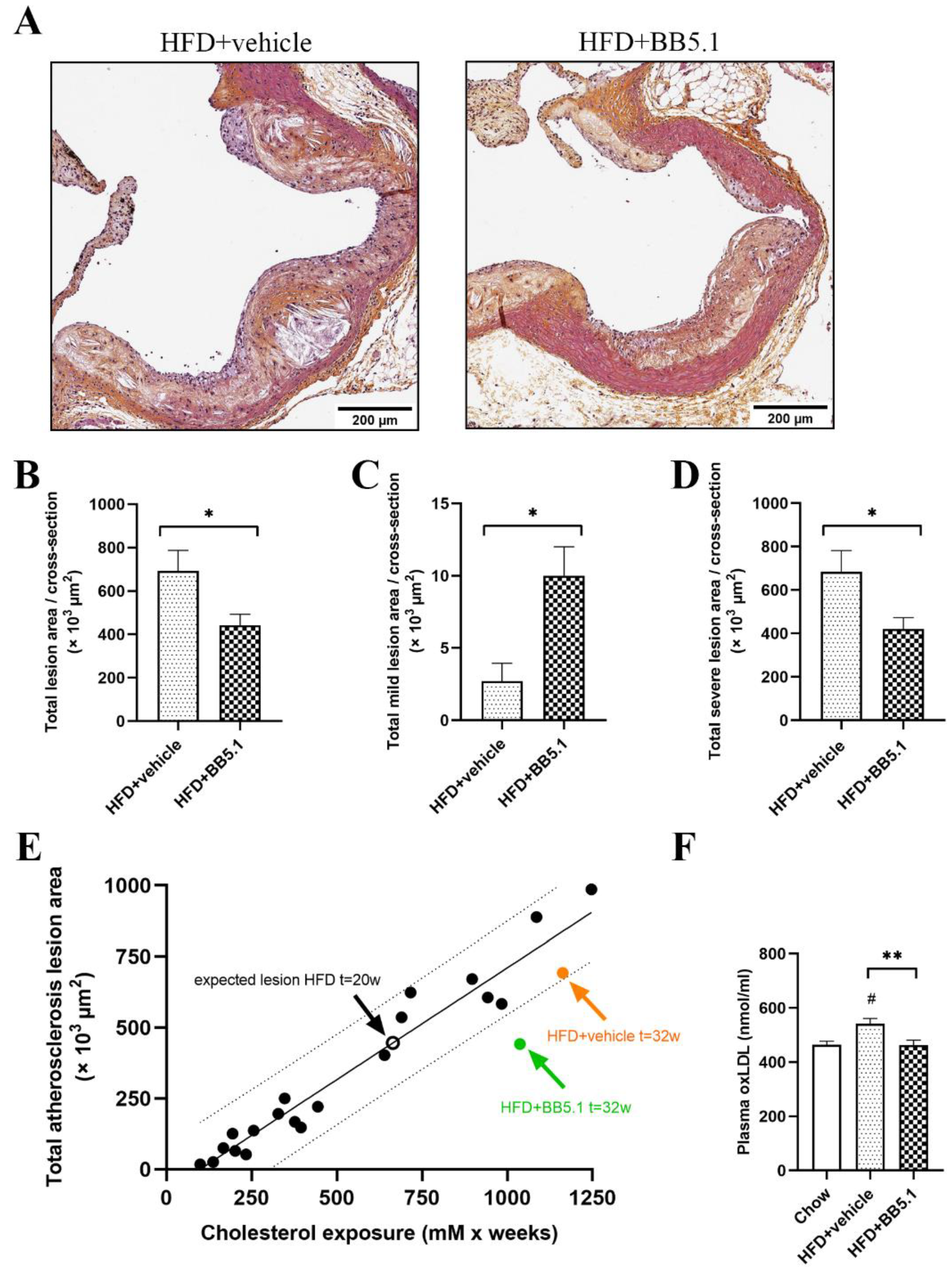
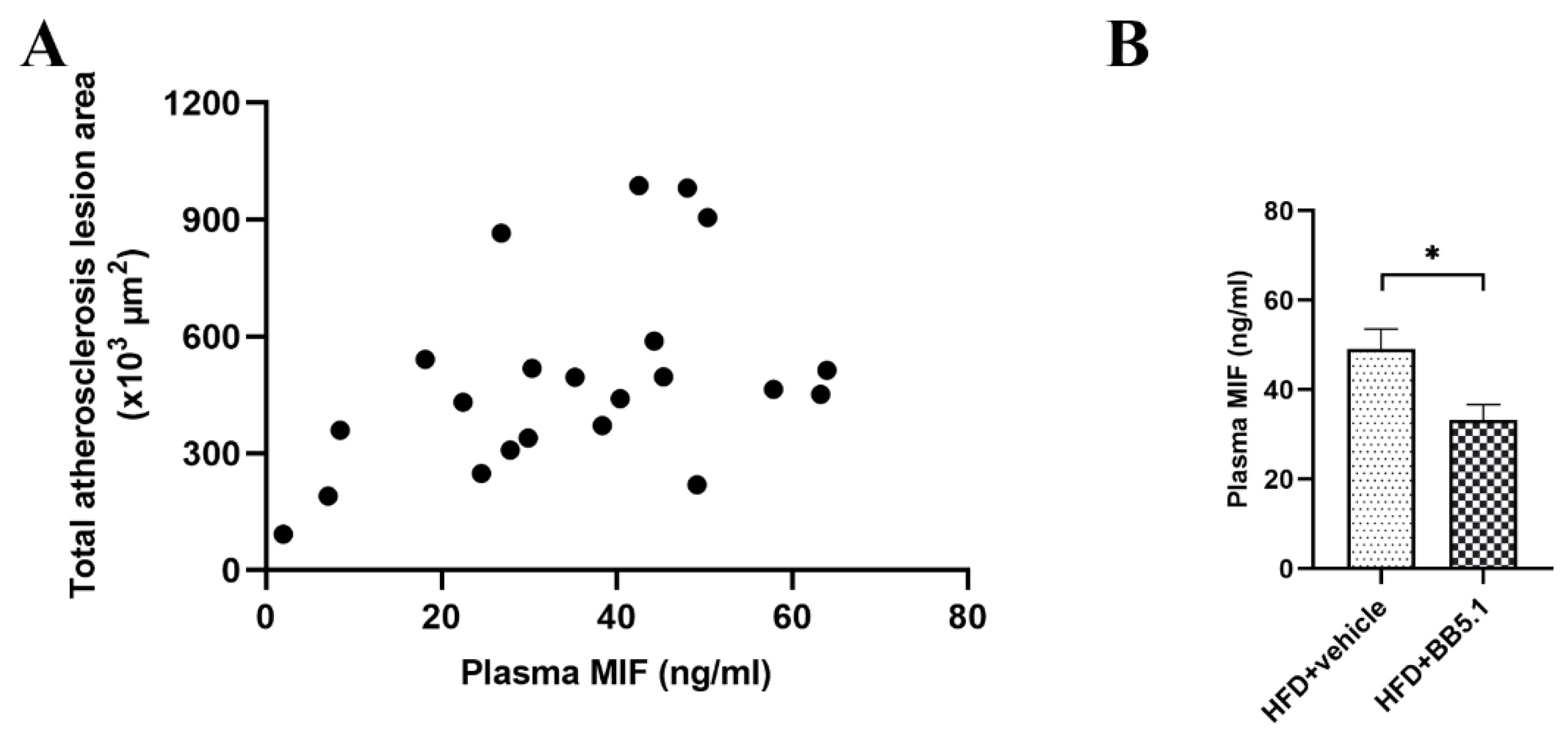
2.5. Anti-C5 Treatment Reduces Atherosclerotic Plaque Growth and Severity Independently of Circulating Cholesterol
3. Discussion
4. Materials and Methods
4.1. Animal Studies
4.2. BB5.1 Antibody Production
4.3. Plasma and Liver Biochemical Analyses
4.3.1. Blood Glucose, Plasma Triglycerides and Cholesterol
4.3.2. Complement Component 9 (C9) Functional Complement Enzyme-Linked Immunoassay (ELISA)
4.3.3. Hepatic Collagen Content
4.3.4. Plasma oxLDL, Chemokines and Adhesion Molecules
4.4. Histological Analyses of the Livers and Aortic Roots
4.4.1. NAFLD/NASH and Atherosclerosis
4.4.2. MAC Immunostaining
4.5. Liver Gene Expression and Pathway Analysis
4.6. Relationship between Cholesterol Exposure and Atherosclerotic Lesion Size
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Development Initiatives Global Nutrition Report 2017: Nourishing the SDGs. Available online: https://globalnutritionreport.org/reports/2017-global-nutrition-report/ (accessed on 18 October 2021).
- Csige, I.; Ujvárosy, D.; Szabó, Z.; Lőrincz, I.; Paragh, G.; Harangi, M.; Somodi, S. The Impact of Obesity on the Cardiovascular System. J. Diabetes Res. 2018, 2018, 3407306. [Google Scholar] [CrossRef] [PubMed]
- Pang, Q.; Zhang, J.-Y.; Song, S.-D.; Qu, K.; Xu, X.-S.; Liu, S.-S.; Liu, C. Central Obesity and Nonalcoholic Fatty Liver Disease Risk after Adjusting for Body Mass Index. World J. Gastroenterol. 2015, 21, 1650–1662. [Google Scholar] [CrossRef] [PubMed]
- Farrell, G.C.; Van Rooyen, D.; Gan, L.; Chitturi, S. NASH Is an Inflammatory Disorder: Pathogenic, Prognostic and Therapeutic Implications. Gut Liver 2012, 6, 149–171. [Google Scholar] [CrossRef] [PubMed]
- Geovanini, G.R.; Libby, P. Atherosclerosis and Inflammation: Overview and Updates. Clin. Sci. 2018, 132, 1243–1252. [Google Scholar] [CrossRef] [PubMed]
- Zipfel, P.F.; Skerka, C. Complement Regulators and Inhibitory Proteins. Nat. Rev. Immunol. 2009, 9, 729–740. [Google Scholar] [CrossRef]
- Wlazlo, N.; Van Greevenbroek, M.M.J.; Ferreira, I.; Jansen, E.H.J.M.; Feskens, E.J.M.; Van der Kallen, C.J.H.; Schalkwijk, C.G.; Bravenboer, B.; Stehouwer, C.D.A. Activated Complement Factor 3 Is Associated with Liver Fat and Liver Enzymes: The CODAM Study. Eur. J. Clin. Investig. 2013, 43, 679–688. [Google Scholar] [CrossRef]
- Jia, Q.; Li, C.; Xia, Y.; Zhang, Q.; Wu, H.; Du, H.; Liu, L.; Wang, C.; Shi, H.; Guo, X.; et al. Association between Complement C3 and Prevalence of Fatty Liver Disease in an Adult Population: A Cross-Sectional Study from the Tianjin Chronic Low-Grade Systemic Inflammation and Health (TCLSIHealth) Cohort Study. PLoS ONE 2015, 10, e0122026. [Google Scholar] [CrossRef]
- Xu, C.; Chen, Y.; Xu, L.; Miao, M.; Li, Y.; Yu, C. Serum Complement C3 Levels Are Associated with Nonalcoholic Fatty Liver Disease Independently of Metabolic Features in Chinese Population. Sci. Rep. 2016, 6, 6–11. [Google Scholar] [CrossRef]
- Rensen, S.S.; Slaats, Y.; Driessen, A.; Peutz-Kootstra, C.J.; Nijhuis, J.; Steffensen, R.; Greve, J.W.; Buurman, W.A. Activation of the Complement System in Human Nonalcoholic Fatty Liver Disease. Hepatology 2009, 50, 1809–1817. [Google Scholar] [CrossRef]
- Guo, R.F.; Ward, P.A. Role of C5a in Inflammatory Responses. Annu. Rev. Immunol. 2005, 23, 821–852. [Google Scholar] [CrossRef]
- Hillebrandt, S.; Wasmuth, H.E.; Weiskirchen, R.; Hellerbrand, C.; Keppeler, H.; Werth, A.; Schirin-Sokhan, R.; Wilkens, G.; Geier, A.; Lorenzen, J.; et al. Complement Factor 5 Is a Quantitative Trait Gene That Modifies Liver Fibrogenesis in Mice and Humans. Nat. Genet. 2005, 37, 835–843. [Google Scholar] [CrossRef] [PubMed]
- Bavia, L.; Cogliati, B.; Dettoni, J.B.; Ferreira Alves, V.A.; Isaac, L. The Complement Component C5 Promotes Liver Steatosis and Inflammation in Murine Non-Alcoholic Liver Disease Model. Immunol. Lett. 2016, 177, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Onat, A.; Can, G.; Rezvani, R.; Cianflone, K. Complement C3 and Cleavage Products in Cardiometabolic Risk. Clin. Chim. Acta 2011, 412, 1171–1179. [Google Scholar] [CrossRef] [PubMed]
- Si, W.; He, P.; Wang, Y.; Fu, Y.; Li, X.; Lin, X.; Chen, F.; Cao, G.; Zhang, H. Complement Complex C5b-9 Levels Are Associated with the Clinical Outcomes of Acute Ischemic Stroke and Carotid Plaque Stability. Transl. Stroke Res. 2019, 10, 279–286. [Google Scholar] [CrossRef]
- Niculescu, F.; Hugo, F.; Rus, H.G.; Vlaicu, R.; Bhakdi, S. Quantitative Evaluation of the Terminal C5b-9 Complement Complex by ELISA in Human Atherosclerotic Arteries. Clin. Exp. Immunol. 1987, 69, 477–483. [Google Scholar]
- Martínez-López, D.; Roldan-Montero, R.; García-Marqués, F.; Nuñez, E.; Jorge, I.; Camafeita, E.; Minguez, P.; Rodriguez de Cordoba, S.; López-Melgar, B.; Lara-Pezzi, E.; et al. Complement C5 Protein as a Marker of Subclinical Atherosclerosis. J. Am. Coll. Cardiol. 2020, 75, 1926–1941. [Google Scholar] [CrossRef]
- Oksjoki, R.; Kovanen, P.T.; Mäyränpää, M.I.; Laine, P.; Blom, A.M.; Meri, S.; Pentikäinen, M.O. Complement Regulation in Human Atherosclerotic Coronary Lesions. Immunohistochemical Evidence That C4b-Binding Protein Negatively Regulates the Classical Complement Pathway, and That C5b-9 Is Formed via the Alternative Complement Pathway. Atherosclerosis 2007, 192, 40–48. [Google Scholar] [CrossRef]
- Niculescu, F.; Rus, H. The Role of Complement Activation in Atherosclerosis. Immunol. Res. 2004, 30, 73–80. [Google Scholar] [CrossRef]
- Vlaicu, S.I.; Tatomir, A.; Rus, V.; Mekala, A.P.; Mircea, P.A.; Niculescu, F.; Rus, H. The Role of Complement Activation in Atherogenesis: The First 40 Years. Immunol. Res. 2016, 64, 1–13. [Google Scholar] [CrossRef]
- Schmiedt, W.; Kinscherf, R.; Deigner, H.P.; Kamencic, H.; Nauen, O.; Kilo, J.; Oelert, H.; Metz, J.; Bhakdi, S. Complement C6 Deficiency Protects against Diet-Induced Atherosclerosis in Rabbits. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 1790–1795. [Google Scholar] [CrossRef]
- Yun, S.; Leung, V.W.Y.; Botto, M.; Boyle, J.J.; Haskard, D.O. Brief Report: Accelerated Atherosclerosis in Low-Density Lipoprotein Receptor-Deficient Mice Lacking the Membrane-Bound Complement Regulator CD59. Arter. Thromb. Vasc. Biol. 2008, 28, 1714–1716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, R.D.; Jackson, C.L.; Morgan, B.P.; Hughes, T.R. The Membrane Attack Complex of Complement Drives the Progression of Atherosclerosis in Apolipoprotein E Knockout Mice. Mol. Immunol. 2010, 47, 1098–1105. [Google Scholar] [CrossRef] [PubMed]
- An, G.; Li, B.; Liu, X.; Zhang, M.; Gao, F.; Zhao, Y.; An, F.; Zhang, Y.; Zhang, C. Overexpression of Complement Component C5a Accelerates the Development of Atherosclerosis in ApoE-Knockout Mice. Oncotarget 2016, 7, 56060–56070. [Google Scholar] [CrossRef] [PubMed]
- Shagdarsuren, E.; Bidzhekov, K.; Mause, S.F.; Simsekyilmaz, S.; Polakowski, T.; Hawlisch, H.; Gessner, J.E.; Zernecke, A.; Weber, C. C5a Receptor Targeting in Neointima Formation after Arterial Injury in Atherosclerosis-Prone Mice. Circulation 2010, 122, 1026–1036. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.; Liu, F.; Ren, M.; Qin, Z.; Rout, N.; Yang, X.-F.; Wang, H.; Tomlinson, S.; Qin, X. Complement Inhibition Targeted to Injury Specific Neoepitopes Attenuates Atherogenesis in Mice. Front. Cardiovasc. Med. 2021, 8, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Wu, L.; Wu, G.; Wang, C.; Zhang, L.; Tomlinson, S.; Qin, X. Targeted Mouse Complement Inhibitor CR2-Crry Protects against the Development of Atherosclerosis in Mice. Atherosclerosis 2014, 234, 237–243. [Google Scholar] [CrossRef]
- Manthey, H.D.; Thomas, A.C.; Shiels, I.A.; Zernecke, A.; Woodruff, T.M.; Rolfe, B.; Taylor, S.M. Complement C5a Inhibition Reduces Atherosclerosis in ApoE –/– Mice. FASEB J. 2011, 25, 2447–2455. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Hu, W.; Shahsafaei, A.; Song, W.; Dobarro, M.; Sukhova, G.K.; Bronson, R.R.; Shi, G.P.; Rother, R.P.; Halperin, J.A.; et al. Complement Regulator Cd59 Protects against Atherosclerosis by Restricting the Formation of Complement Membrane Attack Complex. Circ. Res. 2009, 104, 550–558. [Google Scholar] [CrossRef] [PubMed]
- Zelek, W.M.; Menzies, G.E.; Brancale, A.; Stockinger, B.; Morgan, B.P. Characterizing the Original Anti-C5 Function-Blocking Antibody, BB5.1, for Species Specificity, Mode of Action and Interactions with C5. Immunology 2020, 161, 103–113. [Google Scholar] [CrossRef]
- Guti, J.; Santos, A.; Armendariz-borunda, J. Pathophysiological Molecular Mechanisms of Obesity: A Link between MAFLD and NASH with Cardiovascular Diseases. Int. J. Mol. Sci. 2021, 22, 11629. [Google Scholar]
- Van den Hoek, A.M.; Verschuren, L.; Worms, N.; van Nieuwkoop, A.; de Ruiter, C.; Attema, J.; Menke, A.L.; Caspers, M.P.M.; Radhakrishnan, S.; Salic, K.; et al. A Translational Mouse Model for NASH with Advanced Fibrosis and Atherosclerosis Expressing Key Pathways of Human Pathology. Cells 2020, 9, 2014. [Google Scholar] [CrossRef] [PubMed]
- Morrison, M.C.; Verschuren, L.; Salic, K.; Verheij, J.; Menke, A.; Wielinga, P.Y.; Iruarrizaga-Lejarreta, M.; Gole, L.; Yu, W.M.; Turner, S.; et al. Obeticholic Acid Modulates Serum Metabolites and Gene Signatures Characteristic of Human NASH and Attenuates Inflammation and Fibrosis Progression in Ldlr-/-.Leiden Mice. Hepatol. Commun. 2018, 2, 1513–1532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santhekadur, P.K.; Kumar, D.P.; Sanyal, A.J. Preclinical Models of Non-Alcoholic Fatty Liver Disease. J. Hepatol. 2018, 68, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Morrison, M.C.; Kleemann, R.; van Koppen, A.; Hanemaaijer, R.; Verschuren, L. Key Inflammatory Processes in Human NASH Are Reflected in Ldlr−/−.Leiden Mice: A Translational Gene Profiling Study. Front. Physiol. 2018, 9, 132. [Google Scholar] [CrossRef] [PubMed]
- Van Koppen, A.; Verschuren, L.; van den Hoek, A.M.; Verheij, J.; Morrison, M.C.; Li, K.; Nagabukuro, H.; Costessi, A.; Caspers, M.P.M.; van den Broek, T.J.; et al. Uncovering a Predictive Molecular Signature for the Onset of NASH-Related Fibrosis in a Translational NASH Mouse Model. Cell. Mol. Gastroenterol. Hepatol. 2017, 5, 83–98.e10. [Google Scholar] [CrossRef]
- Martínez-Arranz, I.; Bruzzone, C.; Noureddin, M.; Gil-Redondo, R.; Mincholé, I.; Bizkarguenaga, M.; Arretxe, E.; Iruarrizaga-Lejarreta, M.; Fernández-Ramos, D.; Lopitz-Otsoa, F.; et al. Metabolic Subtypes of Patients with NAFLD Exhibit Distinctive Cardiovascular Risk Profiles. Hepatology 2022. [Google Scholar] [CrossRef]
- Wu, L.; Gao, X.; Guo, Q.; Li, J.; Yao, J.; Yan, K.; Xu, Y.; Jiang, X.; Ye, D.; Guo, J. The Role of Neutrophils in Innate Immunity-Driven Nonalcoholic Steatohepatitis: Lessons Learned and Future Promise. Hepatol. Int. 2020, 14, 652–666. [Google Scholar] [CrossRef]
- Perianayagam, M.C.; Balakrishnan, V.S.; Pereira, B.J.G.; Jaber, B.L. C5a Delays Apoptosis of Human Neutrophils via an Extracellular Signal-Regulated Kinase and Bad-Mediated Signalling Pathway. Eur. J. Clin. Investig. 2004, 34, 50–56. [Google Scholar] [CrossRef]
- Lee, Y.A.; Friedman, S.L. Inflammatory and Fibrotic Mechanisms in NAFLD—Implications for New Treatment Strategies. J. Intern. Med. 2021, 291, 11–31. [Google Scholar] [CrossRef]
- Parthasarathy, G.; Revelo, X.; Malhi, H. Pathogenesis of Nonalcoholic Steatohepatitis: An Overview. Hepatol. Commun. 2020, 4, 478–492. [Google Scholar] [CrossRef]
- Martinus, R.D.; Cook, C.J. The Effect of Complement C5a on Mitochondrial Functions of PC12 Cells. Neuroreport 2011, 22, 581–585. [Google Scholar] [CrossRef] [PubMed]
- Triantafilou, K.; Hughes, T.R.; Triantafilou, M.; Morgan, P.P. The Complement Membrane Attack Complex Triggers Intracellular Ca2+ Fluxes Leading to NLRP3 Inflammasome Activation. J. Cell Sci. 2013, 126, 2903–2913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Begriche, K.; Massart, J.; Robin, M.A.; Bonnet, F.; Fromenty, B. Mitochondrial Adaptations and Dysfunctions in Nonalcoholic Fatty Liver Disease. Hepatology 2013, 58, 1497–1507. [Google Scholar] [CrossRef]
- An, G.; Miwa, T.; Song, W.L.; Lawson, J.A.; Rader, D.J.; Zhang, Y.; Song, W.C. CD59 but Not DAF Deficiency Accelerates Atherosclerosis in Female ApoE Knockout Mice. Mol. Immunol. 2009, 46, 1702–1709. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Thelander, E.M.; Hernandez, M.; Montenegro, J.; Hassing, H.; Burton, C.; Mundt, S.; Hermanowski-Vosatka, A.; Wright, S.D.; Chao, Y.S.; et al. ApoE−/− Mice Develop Atherosclerosis in the Absence of Complement Component C5. Biochem. Biophys. Res. Commun. 2001, 286, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Yin, C.; Ackermann, S.; Ma, Z.; Mohanta, S.K.; Zhang, C.; Li, Y.; Nietzsche, S.; Westermann, M.; Peng, L.; Hu, D.; et al. ApoE Attenuates Unresolvable Inflammation by Complex Formation with Activated C1q. Nat. Med. 2019, 25, 496–506. [Google Scholar] [CrossRef] [PubMed]
- Ji, E.; Lee, S. Antibody-Based Therapeutics for Atherosclerosis and Cardiovascular Diseases. Int. J. Mol. Sci. 2021, 22, 5770. [Google Scholar] [CrossRef] [PubMed]
- Khatana, C.; Saini, N.K.; Chakrabarti, S.; Saini, V.; Sharma, A.; Saini, R.V.; Saini, A.K. Mechanistic Insights into the Oxidized Low-Density Lipoprotein-Induced Atherosclerosis. Oxid. Med. Cell. Longev. 2020, 2020, 5245308. [Google Scholar] [CrossRef]
- Hoebinger, C.; Rajcic, D.; Hendrikx, T. Oxidized Lipids: Common Immunogenic Drivers of Non-Alcoholic Fatty Liver Disease and Atherosclerosis. Front. Cardiovasc. Med. 2022, 8, 1–12. [Google Scholar] [CrossRef]
- Kilgore, K.S.; Schmidt, E.; Shanley, T.P.; Flory, C.M.; Maheswari, V.; Tramontini, N.L.; Cohen, H.; Ward, P.A.; Friedl, H.P.; Warren, J.S. Sublytic Concentrations of the Membrane Attack Complex of Complement Induce Endothelial Interleukin-8 and Monocyte Chemoattractant Protein-1 through Nuclear Factor-ΚB Activation. Am. J. Pathol. 1997, 150, 2019–2031. [Google Scholar]
- Speidl, W.S.; Kastl, S.P.; Huber, K.; Wojta, J. Complement in Atherosclerosis: Friend or Foe? J. Thromb. Haemost. 2011, 9, 428–440. [Google Scholar] [CrossRef] [PubMed]
- Flynn, M.C.; Pernes, G.; Lee, M.K.S.; Nagareddy, P.R.; Murphy, A.J. Monocytes, Macrophages, and Metabolic Disease in Atherosclerosis. Front. Pharmacol. 2019, 10, 1–13. [Google Scholar] [CrossRef]
- Szmitko, P.E.; Wang, C.H.; Weisel, R.D.; De Almeida, J.R.; Anderson, T.J.; Verma, S. New Markers of Inflammation and Endothelial Cell Activation Part I. Circulation 2003, 108, 1917–1923. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, K.; Leonard, W.J. Cytokine and Cytokine Receptor Pleiotropy and Redundancy. J. Biol. Chem. 2002, 277, 29355–29358. [Google Scholar] [CrossRef] [PubMed]
- Verschuren, L.; Kooistra, T.; Bernhagen, J.; Voshol, P.J.; Ouwens, D.M.; van Erk, M.; de Vries-van der Weij, J.; Leng, L.; van Bockel, J.H.; van Dijk, K.W.; et al. MIF Deficiency Reduces Chronic Inflammation in White Adipose Tissue and Impairs the Development of Insulin Resistance, Glucose Intolerance, and Associated Atherosclerotic Disease. Circ. Res. 2009, 105, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Burger-Kentischer, A.; Goebel, H.; Seiler, R.; Fraedrich, G.; Schaefer, H.E.; Dimmeler, S.; Kleemann, R.; Bernhagen, J.; Ihling, C. Expression of Macrophage Migration Inhibitory Factor in Different Stages of Human Atherosclerosis. Circulation 2002, 105, 1561–1566. [Google Scholar] [CrossRef]
- Bernhagen, J.; Krohn, R.; Lue, H.; Gregory, J.L.; Zernecke, A.; Koenen, R.R.; Dewor, M.; Georgiev, I.; Schober, A.; Leng, L.; et al. MIF Is a Noncognate Ligand of CXC Chemokine Receptors in Inflammatory and Atherogenic Cell Recruitment. Nat. Med. 2007, 13, 587–596. [Google Scholar] [CrossRef]
- Pan, J.H.; Sukhova, G.K.; Yang, J.T.; Wang, B.; Xie, T.; Fu, H.; Zhang, Y.; Satoskar, A.R.; David, J.R.; Metz, C.N.; et al. Macrophage Migration Inhibitory Factor Deficiency Impairs Atherosclerosis in Low-Density Lipoprotein Receptor-Deficient Mice. Circulation 2004, 109, 3149–3153. [Google Scholar] [CrossRef]
- Sendler, M.; Beyer, G.; Mahajan, U.M.; Kauschke, V.; Maertin, S.; Schurmann, C.; Homuth, G.; Völker, U.; Völzke, H.; Halangk, W.; et al. Complement Component 5 Mediates Development of Fibrosis, via Activation of Stellate Cells, in 2 Mouse Models of Chronic Pancreatitis. Gastroenterology 2015, 149, e10–e776. [Google Scholar] [CrossRef]
- Chen, L.; Yang, G.; Zhang, X.; Wu, J.; Gu, Q.; Wei, M.; Yang, J.; Zhu, Y.; Wang, N.; Guan, Y. Induction of MIF Expression by Oxidized LDL via Activation of NF-ΚB in Vascular Smooth Muscle Cells. Atherosclerosis 2009, 207, 428–433. [Google Scholar] [CrossRef]
- Gart, E.; Salic, K.; Morrison, M.C.; Caspers, M.; van Duyvenvoorde, W.; Heijnk, M.; Giera, M.; Bobeldijk-Pastorova, I.; Keijer, J.; Storsve, A.B.; et al. Krill Oil Treatment Increases Distinct Pufas and Oxylipins in Adipose Tissue and Liver and Attenuates Obesity-Associated Inflammation via Direct and Indirect Mechanisms. Nutrients 2021, 13, 2836. [Google Scholar] [CrossRef] [PubMed]
- Kotimaa, J.P.; van Werkhoven, M.B.; O’Flynn, J.; Klar-Mohamad, N.; van Groningen, J.; Schilders, G.; Rutjes, H.; Daha, M.R.; Seelen, M.A.; van Kooten, C. Functional Assessment of Mouse Complement Pathway Activities and Quantification of C3b/C3c/IC3b in an Experimental Model of Mouse Renal Ischaemia/Reperfusion Injury. J. Immunol. Methods 2015, 419, 25–34. [Google Scholar] [CrossRef]
- Salic, K.; Morrison, M.C.; Verschuren, L.; Wielinga, P.Y.; Wu, L.; Kleemann, R.; Gjorstrup, P.; Kooistra, T. Resolvin E1 Attenuates Atherosclerosis in Absence of Cholesterol-Lowering Effects and on Top of Atorvastatin. Atherosclerosis 2016, 250, 158–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, W.; Menke, A.L.; Driessen, A.; Koek, G.H.; Lindeman, J.H.; Stoop, R.; Havekes, L.M.; Kleemann, R.; Van Den Hoek, A.M. Establishment of a General NAFLD Scoring System for Rodent Models and Comparison to Human Liver Pathology. PLoS ONE 2014, 9, e115922. [Google Scholar] [CrossRef] [PubMed]
- Morrison, M.C.; Mulder, P.; Stavro, P.M.; Suárez, M.; Arola-Arnal, A.; Van Duyvenvoorde, W.; Kooistra, T.; Wielinga, P.Y.; Kleemann, R. Replacement of Dietary Saturated Fat by PUFA-Rich Pumpkin Seed Oil Attenuates Non-Alcoholic Fatty Liver Disease and Atherosclerosis Development, with Additional Health Effects of Virgin over Refined Oil. PLoS ONE 2015, 10, e0139196. [Google Scholar] [CrossRef] [PubMed]
- Schoemaker, M.H.; Kleemann, R.; Morrison, M.C.; Verheij, J.; Salic, K.; Van Tol, E.A.F.; Kooistra, T.; Wielinga, P.Y. A Casein Hydrolysate Based Formulation Attenuates Obesity and Associated Nonalcoholic Fatty Liver Disease and Atherosclerosis in LDLr-/-.Leiden Mice. PLoS ONE 2017, 12, e0180648. [Google Scholar] [CrossRef]
- Stary, H.; Chandler, A.; Glagov, S.; Jr, G.; Insull, W.; Rosenfeld, M.J.; Schaffer, S.; Schwartz, C.; Wagner, W.; Wissler, R. AHA Medical / Scientific Statement Special Report A Definition of Initial, Fatty Streak, and Intermediate Lesions of Atherosclerosis. Circulation 1994, 89, 2462–2478. [Google Scholar] [CrossRef]
- Stary, H.C.; Chandler, A.B.; Dinsmore, R.E.; Fuster, V.; Glagov, S.; Insull, W.; Rosenfeld, M.E.; Schwartz, C.J.; Wagner, W.D.; Wissler, R.W. A Definition of Advanced Types of Atherosclerotic Lesions and a Histological Classification of Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 1512–1531. [Google Scholar] [CrossRef]
- Salic, K.; Gart, E.; Seidel, F.; Verschuren, L.; Caspers, M.; van Duyvenvoorde, W.; Wong, K.E.; Keijer, J.; Bobeldijk-Pastorova, I.; Wielinga, P.Y.; et al. Combined Treatment with L-Carnitine and Nicotinamide Riboside Improves Hepatic Metabolism and Attenuates Obesity and Liver Steatosis. Int. J. Mol. Sci. 2019, 20, 4359. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Mueller, A.M.; Kleemann, R.; Gart, E.; van Duyvenvoorde, W.; Verschuren, L.; Caspers, M.; Menke, A.; Krömmelbein, N.; Salic, K.; Burmeister, Y.; et al. Cholesterol Accumulation as a Driver of Hepatic Inflammation Under Translational Dietary Conditions Can Be Attenuated by a Multicomponent Medicine. Front. Endocrinol. 2021, 12, 601160. [Google Scholar] [CrossRef] [PubMed]
- Krämer, A.; Green, J.; Pollard, J.; Tugendreich, S. Causal Analysis Approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef] [PubMed]
- van den Hoek, A.M.; de Jong, J.C.B.C.; Worms, N.; van Nieuwkoop, A.; Voskuilen, M.; Menke, A.L.; Lek, S.; Caspers, M.P.M.; Verschuren, L.; Kleemann, R. Diet and Exercise Reduce Pre-Existing NASH and Fibrosis and Have Additional Beneficial Effects on the Vasculature, Adipose Tissue and Skeletal Muscle via Organ-Crosstalk. Metabolism 2021, 124, 154873. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seidel, F.; Kleemann, R.; van Duyvenvoorde, W.; van Trigt, N.; Keijzer, N.; van der Kooij, S.; van Kooten, C.; Verschuren, L.; Menke, A.; Kiliaan, A.J.; et al. Therapeutic Intervention with Anti-Complement Component 5 Antibody Does Not Reduce NASH but Does Attenuate Atherosclerosis and MIF Concentrations in Ldlr-/-.Leiden Mice. Int. J. Mol. Sci. 2022, 23, 10736. https://doi.org/10.3390/ijms231810736
Seidel F, Kleemann R, van Duyvenvoorde W, van Trigt N, Keijzer N, van der Kooij S, van Kooten C, Verschuren L, Menke A, Kiliaan AJ, et al. Therapeutic Intervention with Anti-Complement Component 5 Antibody Does Not Reduce NASH but Does Attenuate Atherosclerosis and MIF Concentrations in Ldlr-/-.Leiden Mice. International Journal of Molecular Sciences. 2022; 23(18):10736. https://doi.org/10.3390/ijms231810736
Chicago/Turabian StyleSeidel, Florine, Robert Kleemann, Wim van Duyvenvoorde, Nikki van Trigt, Nanda Keijzer, Sandra van der Kooij, Cees van Kooten, Lars Verschuren, Aswin Menke, Amanda J. Kiliaan, and et al. 2022. "Therapeutic Intervention with Anti-Complement Component 5 Antibody Does Not Reduce NASH but Does Attenuate Atherosclerosis and MIF Concentrations in Ldlr-/-.Leiden Mice" International Journal of Molecular Sciences 23, no. 18: 10736. https://doi.org/10.3390/ijms231810736
APA StyleSeidel, F., Kleemann, R., van Duyvenvoorde, W., van Trigt, N., Keijzer, N., van der Kooij, S., van Kooten, C., Verschuren, L., Menke, A., Kiliaan, A. J., Winter, J., Hughes, T. R., Morgan, B. P., Baas, F., Fluiter, K., & Morrison, M. C. (2022). Therapeutic Intervention with Anti-Complement Component 5 Antibody Does Not Reduce NASH but Does Attenuate Atherosclerosis and MIF Concentrations in Ldlr-/-.Leiden Mice. International Journal of Molecular Sciences, 23(18), 10736. https://doi.org/10.3390/ijms231810736