
Long-Lived Individuals Show a Lower Burden of Variants Predisposing to Age-Related Diseases and a Higher Polygenic Longevity Score

 , , , , ,
, , , , , 
Abstract
:1. Introduction
2. Results
2.1. Association Analyses Do Not Reveal New Longevity Loci
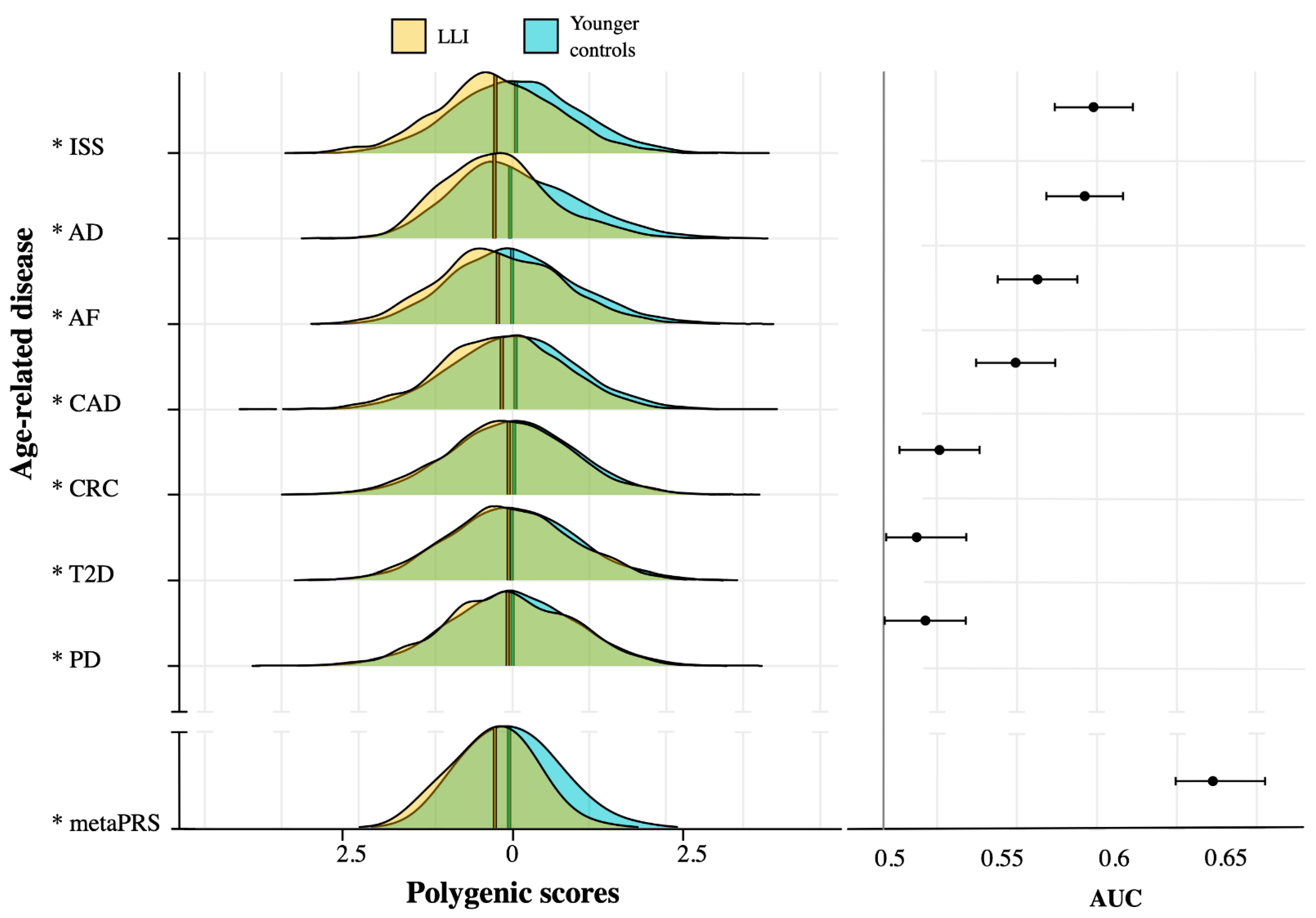
2.2. ARD-PRS Distributions Show Significant Differences between LLI and Controls
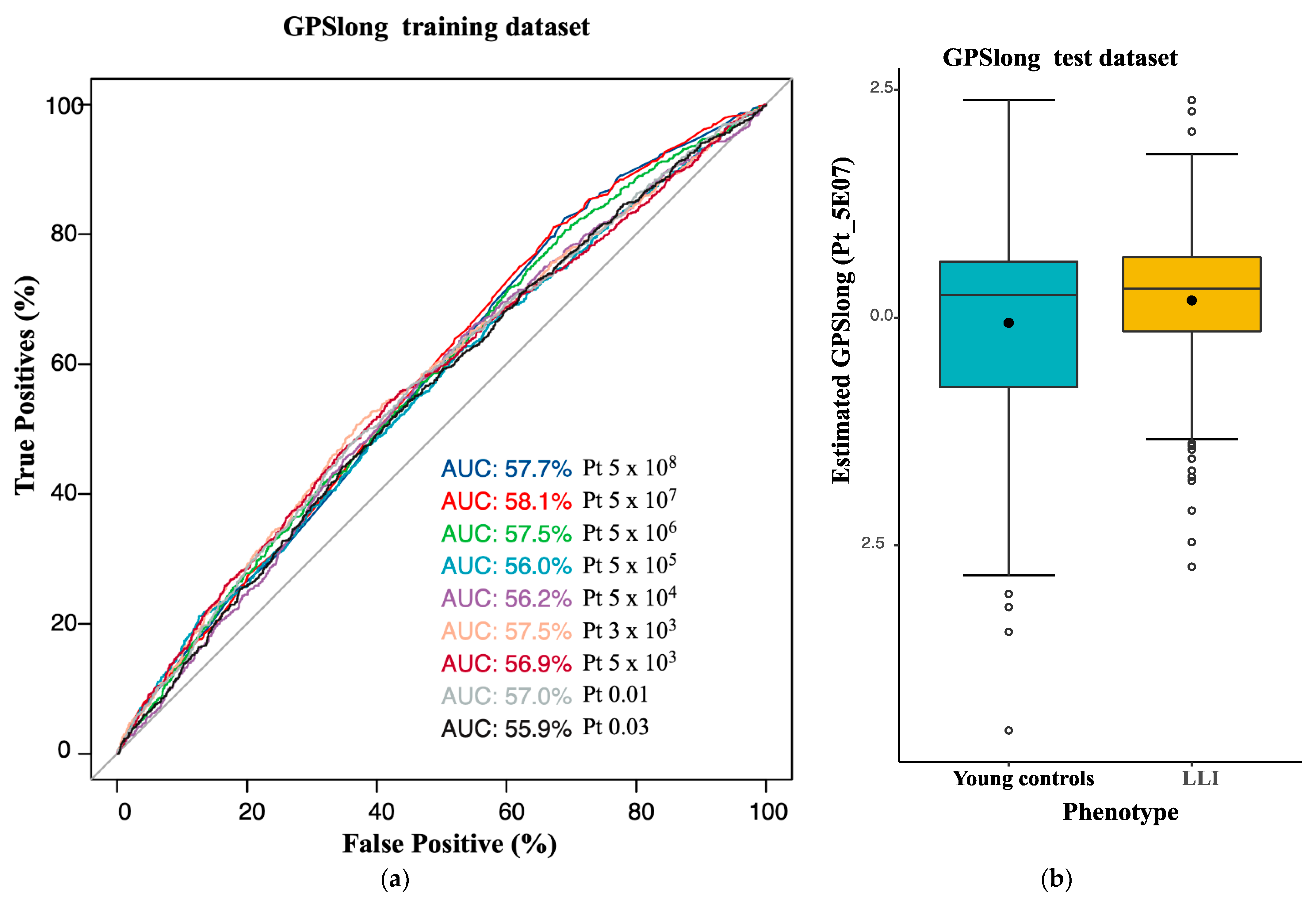
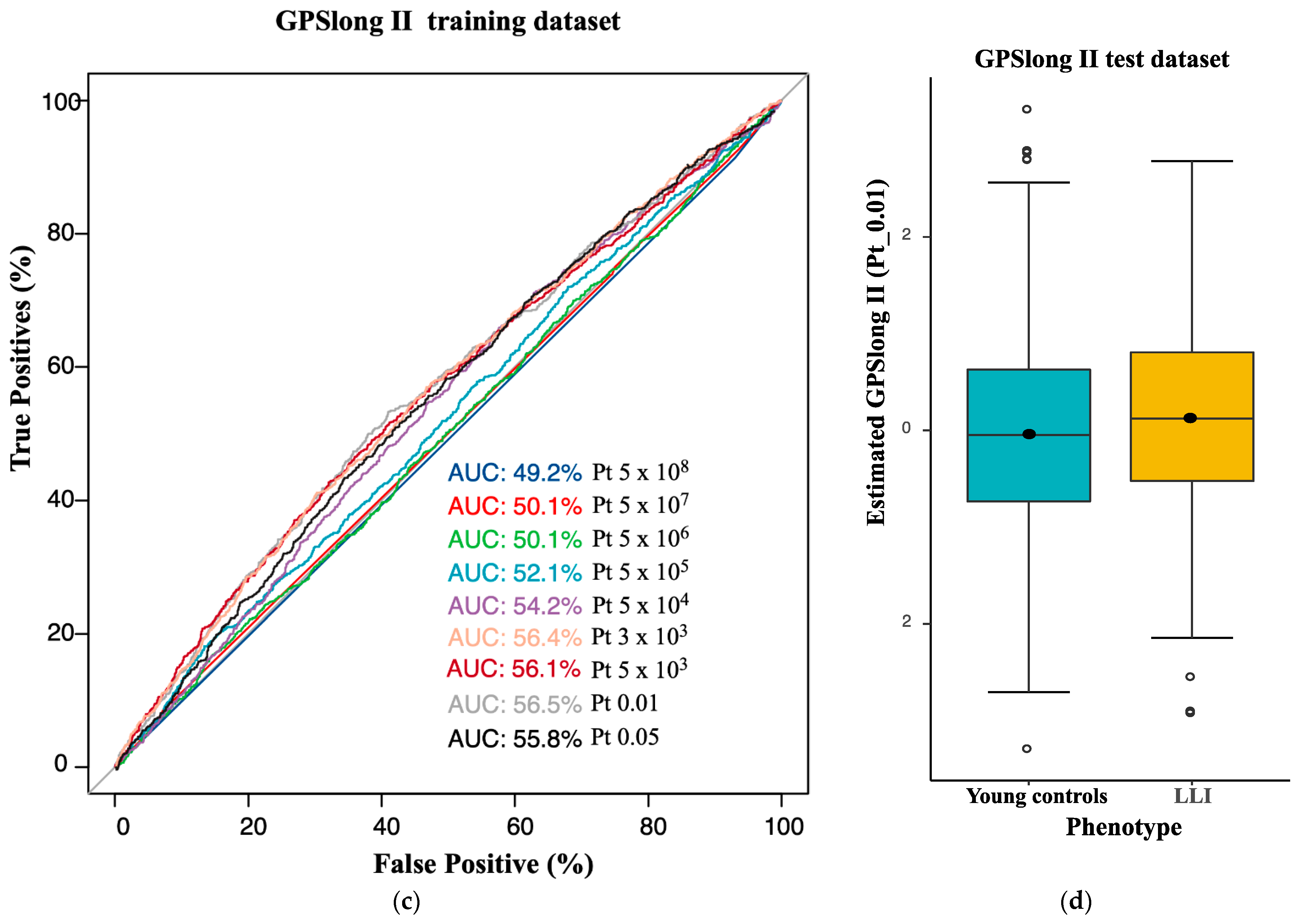
2.3. Longevity GPS Discriminates LLI and Controls with an AUC of 0.56
3. Discussion
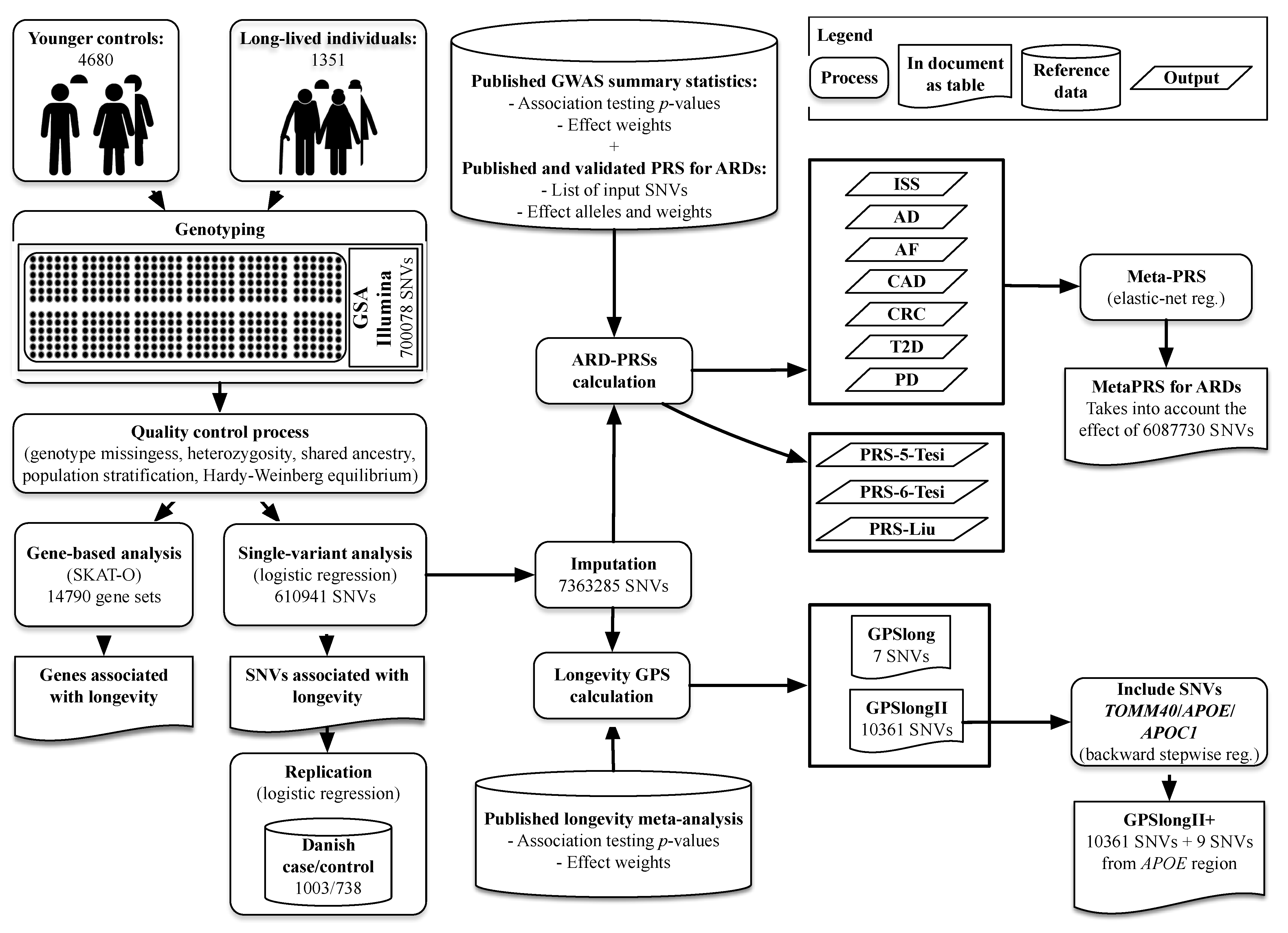
4. Materials and Methods
4.1. Study Populations
4.2. Variant Calling, Quality Control and Imputing for the German and Danish Study Populations
4.3. Longevity Association Analyses in the German and Danish Study Populations
4.4. Application of Published PRSs for Common ARDs in the German Longevity Study Population
4.5. Computation of a Longevity-GPS
4.6. MetaPRS Estimation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hjelmborg, J.V.; Iachine, I.; Skytthe, A.; Vaupel, J.W.; McGue, M.; Koskenvuo, M.; Kaprio, J.; Pedersen, N.L.; Christensen, K. Genetic influence on human lifespan and longevity. Qual. Life Res. 2006, 119, 312–321. [Google Scholar] [CrossRef]
- Flachsbart, F.; Caliebe, A.; Kleindorp, R.; Blanché, H.; von Eller-Eberstein, H.; Nikolaus, S.; Schreiber, S.; Nebel, A. Association of FOXO3A variation with human longevity confirmed in German centenarians. Proc. Natl. Acad. Sci. USA 2009, 106, 2700–2705. [Google Scholar] [CrossRef] [PubMed]
- Nebel, A.; Kleindorp, R.; Caliebe, A.; Nothnagel, M.; Blanché, H.; Junge, O.; Wittig, M.; Ellinghaus, D.; Flachsbart, F.; Wichmann, H.-E.; et al. A genome-wide association study confirms APOE as the major gene influencing survival in long-lived individuals. Mech. Ageing Dev. 2011, 132, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Sebastiani, P.; Solovieff, N.; DeWan, A.T.; Walsh, K.M.; Puca, A.; Hartley, S.W.; Melista, E.; Andersen, S.; Dworkis, D.A.; Wilk, J.B.; et al. Genetic Signatures of Exceptional Longevity in Humans. PLoS ONE 2012, 7, e29848. [Google Scholar] [CrossRef] [PubMed]
- Deelen, J.; Beekman, M.; Uh, H.-W.; Broer, L.; Ayers, K.L.; Tan, Q.; Kamatani, Y.; Bennet, A.M.; Tamm, R.; Trompet, S.; et al. Genome-wide association meta-analysis of human longevity identifies a novel locus conferring survival beyond 90 years of age. Hum. Mol. Genet. 2014, 23, 4420–4432. [Google Scholar] [CrossRef]
- Torres, G.G.; Nygaard, M.; Caliebe, A.; Blanché, H.; Chantalat, S.; Galan, P.; Lieb, W.; Christiansen, L.; Deleuze, J.-F.; Christensen, K.; et al. Exome-Wide Association Study Identifies FN3KRP and PGP as New Candidate Longevity Genes. J. Gerontol. Ser. A 2021, 76, 786–795. [Google Scholar] [CrossRef]
- Andersen-Ranberg, K.; Schroll, M.; Jeune, B. Healthy centenarians do not exist, but autonomous centenarians do: A population-based study of morbidity among Danish centenarians. J. Am. Geriatr. Soc. 2001, 49, 900–908. [Google Scholar] [CrossRef]
- Evert, J.; Lawler, E.; Bogan, H.; Perls, T. Morbidity Profiles of Centenarians: Survivors, Delayers, and Escapers. J. Gerontol. Ser. A 2003, 58, M232–M237. [Google Scholar] [CrossRef]
- Ali, O. Genetics of type 2 diabetes. World J. Diabetes 2013, 4, 114. [Google Scholar] [CrossRef]
- Barber, R.C. The Genetics of Alzheimer’s Disease. Scientifica 2012, 2012, 1–14. [Google Scholar] [CrossRef] [Green Version]
- McPherson, R.; Tybjaerg-Hansen, A. Genetics of Coronary Artery Disease. Circ. Res. 2016, 118, 564–578. [Google Scholar] [CrossRef] [PubMed]
- Soerensen, M.; Nygaard, M.; Debrabant, B.; Mengel-From, J.; Dato, S.; Thinggaard, M.; Christensen, K.; Christiansen, L. No Association between Variation in Longevity Candidate Genes and Aging-related Phenotypes in Oldest-old Danes. Exp. Gerontol. 2016, 78, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, M.; Bae, H.; Schupf, N.; Andersen, S.; Zhang, Q.; Perls, T.; Sebastiani, P. Burden of disease variants in participants of the long life family Study. Aging 2015, 7, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Fortney, K.; Dobriban, E.; Garagnani, P.; Pirazzini, C.; Monti, D.; Mari, D.; Atzmon, G.; Barzilai, N.; Franceschi, C.; Owen, A.B.; et al. Genome-Wide Scan Informed by Age-Related Disease Identifies Loci for Exceptional Human Longevity. PLoS Genet. 2015, 11, e1005728. [Google Scholar] [CrossRef] [PubMed]
- Deelen, J.; Evans, D.S.; Arking, D.E.; Tesi, N.; Nygaard, M.; Liu, X.; Wojczynski, M.K.; Biggs, M.L.; van der Spek, A.; Atzmon, G.; et al. A meta-analysis of genome-wide association studies identifies multiple longevity genes. Nat. Commun. 2019, 10, 3669. [Google Scholar] [CrossRef]
- Melzer, D.; Pilling, L.C.; Ferrucci, L. The genetics of human ageing. Nat. Rev. Genet. 2019, 21, 88–101. [Google Scholar] [CrossRef]
- Liu, X.; Song, Z.; Li, Y.; Yao, Y.; Fang, M.; Bai, C.; An, P.; Chen, H.; Chen, Z.; Tang, B.; et al. Integrated genetic analyses revealed novel human longevity loci and reduced risks of multiple diseases in a cohort study of 15,651 Chinese individuals. Aging Cell 2021, 20, e13323. [Google Scholar] [CrossRef]
- Tesi, N.; Hulsman, M.; van der Lee, S.J.; Jansen, I.E.; Stringa, N.; van Schoor, N.M.; Scheltens, P.; van der Flier, W.M.; Huisman, M.; Reinders, M.J.T.; et al. The Effect of Alzheimer’s Disease-Associated Genetic Variants on Longevity. Front. Genet. 2021, 12, 748781. [Google Scholar] [CrossRef]
- Pilling, L.C.; Kuo, C.-L.; Sicinski, K.; Tamosauskaite, J.; Kuchel, G.A.; Harries, L.W.; Herd, P.; Wallace, R.; Ferrucci, L.; Melzer, D. Human longevity: 25 genetic loci associated in 389,166 UK biobank participants. Aging 2017, 9, 2504–2520. [Google Scholar] [CrossRef]
- Lin, J.-R.; Sin-Chan, P.; Napolioni, V.; Torres, G.G.; Mitra, J.; Zhang, Q.; Jabalameli, M.R.; Wang, Z.; Nguyen, N.; Gao, T.; et al. Rare genetic coding variants associated with human longevity and protection against age-related diseases. Nat. Aging 2021, 1, 783–794. [Google Scholar] [CrossRef]
- Mooijaart, S.P.; Van Heemst, D.; Noordam, R.; Rozing, M.P.; Wijsman, C.A.; De Craen, A.J.; Westendorp, R.G.; Beekman, M.; Slagboom, E.P. Polymorphisms associated with type 2 diabetes in familial longevity: The Leiden Longevity Study. Aging 2011, 3, 55–62. [Google Scholar] [CrossRef]
- Khera, A.V.; Chaffin, M.; Aragam, K.G.; Haas, M.E.; Roselli, C.; Choi, S.H.; Natarajan, P.; Lander, E.S.; Lubitz, S.A.; Ellinor, P.T.; et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet. 2018, 50, 1219–1224. [Google Scholar] [CrossRef] [PubMed]
- Tesi, N.; van der Lee, S.J.; Hulsman, M.; Jansen, I.E.; Stringa, N.; van Schoor, N.M.; Scheltens, P.; van der Flier, W.M.; Huisman, M.; Reinders, M.J.T.; et al. Polygenic Risk Score of Longevity Predicts Longer Survival Across an Age Continuum. J. Gerontol. Ser. A 2021, 76, 750–759. [Google Scholar] [CrossRef] [PubMed]
- Schächter, F.; Faure-Delanef, L.; Guénot, F.; Rouger, H.; Froguel, P.; Lesueur-Ginot, L.; Cohen, D. Genetic associations with human longevity at the APOE and ACE loci. Nat. Genet. 1994, 6, 29–32. [Google Scholar] [CrossRef] [PubMed]
- Szymczak, S.; Dose, J.; Torres, G.G.; Heinsen, F.-A.; Venkatesh, G.; Datlinger, P.; Nygaard, M.; Mengel-From, J.; Flachsbart, F.; Klapper, W.; et al. DNA methylation QTL analysis identifies new regulators of human longevity. Hum. Mol. Genet. 2020, 29, 1154–1167. [Google Scholar] [CrossRef] [PubMed]
- McDaid, A.F.; Joshi, P.K.; Porcu, E.; Komljenovic, A.; Li, H.; Sorrentino, V.; Litovchenko, M.; Bevers, R.P.J.; Rüeger, S.; Reymond, A.; et al. Bayesian association scan reveals loci associated with human lifespan and linked biomarkers. Nat. Commun. 2017, 8, 15842. [Google Scholar] [CrossRef]
- Timmers, P.R.; Mounier, N.; Lall, K.; Fischer, K.; Ning, Z.; Feng, X.; Bretherick, A.D.; Clark, D.W.; Agbessi, M.; Ahsan, H.; et al. Genomics of 1 million parent lifespans implicates novel pathways and common diseases and distinguishes survival chances. eLife 2019, 8, e39856. [Google Scholar] [CrossRef]
- Maruszak, A.; Pepłońska, B.; Safranow, K.; Chodakowska-Żebrowska, M.; Barcikowska, M.; Żekanowski, C. TOMM40 rs10524523 Polymorphism’s Role in Late-Onset Alzheimer’s Disease and in Longevity. J. Alzheimer’s Dis. 2012, 28, 309–322. [Google Scholar] [CrossRef]
- Pavlidis, N.; Stanta, G.; Audisio, R.A. Cancer prevalence and mortality in centenarians: A systematic review. Crit. Rev. Oncol. 2012, 83, 145–152. [Google Scholar] [CrossRef]
- Nolen, S.C.; Evans, M.A.; Fischer, A.; Corrada, M.M.; Kawas, C.H.; Bota, D.A. Cancer—Incidence, prevalence and mortality in the oldest-old. A comprehensive review. Mech. Ageing Dev. 2017, 164, 113–126. [Google Scholar] [CrossRef] [Green Version]
- von Campenhausen, S.; Bornschein, B.; Wick, R.; Bötzel, K.; Sampaio, C.; Poewe, W.; Oertel, W.; Siebert, U.; Berger, K.; Dodel, R. Prevalence and incidence of Parkinson’s disease in Europe. Eur. Neuropsychopharmacol. 2005, 15, 473–490. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Simon, D.K. No evidence for heritability of Parkinson disease in Swedish twins. Neurology 2005, 64, 932. [Google Scholar] [CrossRef] [PubMed]
- Marcon, G.; Manganotti, P.; Tettamanti, M. Is Parkinson’s Disease a Very Rare Pathology in Centenarians? A Clinical Study in a Cohort of Subjects. J. Alzheimer’s Dis. 2020, 73, 73–76. [Google Scholar] [CrossRef]
- Kerminen, S.; Martin, A.R.; Koskela, J.; Ruotsalainen, S.E.; Havulinna, A.S.; Surakka, I.; Palotie, A.; Perola, M.; Salomaa, V.; Daly, M.J.; et al. Geographic Variation and Bias in the Polygenic Scores of Complex Diseases and Traits in Finland. Am. J. Hum. Genet. 2019, 104, 1169–1181. [Google Scholar] [CrossRef]
- Janssens, A.C.J.W. Validity of polygenic risk scores: Are we measuring what we think we are? Hum. Mol. Genet. 2019, 28, R143–R150. [Google Scholar] [CrossRef]
- Marnetto, D.; Pärna, K.; Läll, K.; Molinaro, L.; Montinaro, F.; Haller, T.; Metspalu, M.; Mägi, R.; Fischer, K.; Pagani, L. Ancestry deconvolution and partial polygenic score can improve susceptibility predictions in recently admixed individuals. Nat. Commun. 2020, 11, 1628. [Google Scholar] [CrossRef]
- Rudolph, A.; Song, M.; Brook, M.; Milne, R.L.; Mavaddat, N.; Michailidou, K.; Bolla, M.K.; Wang, Q.; Dennis, J.; Wilcox, A.; et al. Joint associations of a polygenic risk score and environmental risk factors for breast cancer in the Breast Cancer Association Consortium. Int. J. Epidemiol. 2018, 47, 526–536. [Google Scholar] [CrossRef] [PubMed]
- Barnes, D.R.; Rookus, M.A.; McGuffog, L.; Leslie, G.; Mooij, T.M.; Dennis, J.; Mavaddat, N.; Adlard, J.; Ahmed, M.; Aittomäki, K.; et al. Polygenic risk scores and breast and epithelial ovarian cancer risks for carriers of BRCA1 and BRCA2 pathogenic variants. Genet. Med. 2020, 22, 1653–1666. [Google Scholar] [CrossRef] [PubMed]
- Polygenic Risk Score Task Force of the International Common Disease Alliance; Adeyemo, A.; Balaconis, M.K.; Darnes, D.R.; Fatumo, S.; Moreno, P.G.; Hodonsky, C.J.; Inouye, M.; Kanai, M.; Kato, K.; et al. Responsible use of polygenic risk scores in the clinic: Potential benefits, risks and gaps. Nat. Med. 2021, 27, 1876–1884. [Google Scholar] [CrossRef]
- Nebel, A.; Croucher, P.J.P.; Stiegeler, R.; Nikolaus, S.; Krawczak, M.; Schreiber, S. No association between microsomal triglyceride transfer protein (MTP) haplotype and longevity in humans. Proc. Natl. Acad. Sci. USA 2005, 102, 7906–7909. [Google Scholar] [CrossRef] [Green Version]
- Müller, N.; Schulte, D.M.; Türk, K.; Freitag-Wolf, S.; Hampe, J.; Zeuner, R.; Schröder, J.O.; Gouni-Berthold, I.; Berthold, H.K.; Krone, W.; et al. IL-6 blockade by monoclonal antibodies inhibits apolipoprotein (a) expression and lipoprotein (a) synthesis in humans. J. Lipid Res. 2015, 56, 1034–1042. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, S.H.; Andersen-Ranberg, K.; Thinggaard, M.; Jeune, B.; Skytthe, A.; Christiansen, L.; Vaupel, J.W.; McGue, M.; Christensen, K. Cohort Profile: The 1895, 1905, 1910 and 1915 Danish Birth Cohort Studies-secular trends in the health and functioning of the very old. Int. J. Epidemiol. 2017, 46, 1746-1746j. [Google Scholar] [CrossRef] [PubMed]
- Robine, J.-M.; Cheung, S.L.K.; Saito, Y.; Jeune, B.; Parker, M.G.; Herrmann, F.R. Centenarians Today: New Insights on Selection from the 5-COOP Study. Curr. Gerontol. Geriatr. Res. 2010, 2010, 120354. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, D.A.; Larsen, L.A.; Nygaard, M.; Mengel-From, J.; McGue, M.; Dalgård, C.; Hvidberg, L.; Hjelmborg, J.; Skytthe, A.; Holm, N.V.; et al. The Danish Twin Registry: An Updated Overview. Twin Res. Hum. Genet. 2019, 22, 499–507. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Breunig, M.M.; Kriegel, H.-P.; Ng, R.T.; Sander, J. LOF: Identifying density-based local outliers. In Proceedings of the 2000 ACM SIGMOD international conference on Management of data, Dallas, TX, USA, 16–18 May 2000; pp. 93–104. [Google Scholar]
- Das, S.; Forer, L.; Schönherr, S.; Sidore, C.; Locke, A.E.; Kwong, A.; Vrieze, S.I.; Chew, E.Y.; Levy, S.; McGue, M.; et al. Next-generation genotype imputation service and methods. Nat. Genet. 2016, 48, 1284–1287. [Google Scholar] [CrossRef]
- Loh, P.-R.; Danecek, P.; Palamara, P.F.; Fuchsberger, C.; Reshef, Y.A.; Finucane, H.K.; Schoenherr, S.; Forer, L.; McCarthy, S.; Abecasis, C.F.G.R.; et al. Reference-based phasing using the Haplotype Reference Consortium panel. Nat. Genet. 2016, 48, 1443–1448. [Google Scholar] [CrossRef]
- Howie, B.N.; Donnelly, P.; Marchini, J. A Flexible and Accurate Genotype Imputation Method for the Next Generation of Genome-Wide Association Studies. PLoS Genet. 2009, 5, e1000529. [Google Scholar] [CrossRef]
- Newman, A.B.; Walter, S.; Lunetta, K.; Garcia, M.E.; Slagboom, P.; Christensen, K.; Arnold, A.M.; Aspelund, T.; Aulchenko, Y.; Benjamin, E.; et al. A Meta-analysis of Four Genome-Wide Association Studies of Survival to Age 90 Years or Older: The Cohorts for Heart and Aging Research in Genomic Epidemiology Consortium. J. Gerontol. Ser. A 2010, 65, 478–487. [Google Scholar] [CrossRef]
- Ionita-Laza, I.; Lee, S.; Makarov, V.; Buxbaum, J.D.; Lin, X. Sequence Kernel Association Tests for the Combined Effect of Rare and Common Variants. Am. J. Hum. Genet. 2013, 92, 841–853. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Irizar, P.A.; Schäuble, S.; Esser, D.; Groth, M.; Frahm, C.; Priebe, S.; Baumgart, M.; Hartmann, N.; Marthandan, S.; Menzel, U.; et al. Transcriptomic alterations during ageing reflect the shift from cancer to degenerative diseases in the elderly. Nat. Commun. 2018, 9, 327. [Google Scholar] [CrossRef] [PubMed]
- Abraham, G.; Malik, R.; Yonova-Doing, E.; Salim, A.; Wang, T.; Danesh, J.; Butterworth, A.S.; Howson, J.M.M.; Inouye, M.; Dichgans, M. Genomic risk score offers predictive performance comparable to clinical risk factors for ischaemic stroke. Nat. Commun. 2019, 10, 5819. [Google Scholar] [CrossRef] [PubMed]
- Chaudhury, S.; Brookes, K.J.; Patel, T.; Fallows, A.; Guetta-Baranes, T.; Turton, J.C.; Guerreiro, R.; Bras, J.; Hardy, J.; Francis, P.T.; et al. Alzheimer’s disease polygenic risk score as a predictor of conversion from mild-cognitive impairment. Transl. Psychiatry 2019, 9, 154. [Google Scholar] [CrossRef]
- Nalls, M.A.; Blauwendraat, C.; Vallerga, C.L.; Heilbron, K.; Bandres-Ciga, S.; Chang, D.; Tan, M.; Kia, D.A.; Noyce, A.J.; Xue, A.; et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: A meta-analysis of genome-wide association studies. Lancet Neurol. 2019, 18, 1091–1102. [Google Scholar] [CrossRef]
- Jia, G.; Lu, Y.; Wen, W.; Long, J.; Liu, Y.; Tao, R.; Li, B.; Denny, J.C.; Shu, X.-O.; Zheng, W. Evaluating the Utility of Polygenic Risk Scores in Identifying High-Risk Individuals for Eight Common Cancers. JNCI Cancer Spectr. 2020, 4, pkaa021. [Google Scholar] [CrossRef]
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.-C.; Müller, M. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 2011, 12, 77. [Google Scholar] [CrossRef]
- Friedman, J.H.; Hastie, T.; Tibshirani, R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J. Stat. Softw. 2010, 33, 1–22. [Google Scholar] [CrossRef]
- Choi, S.W.; O’Reilly, P.F. PRSice-2: Polygenic Risk Score software for biobank-scale data. GigaScience 2019, 8, giz082. [Google Scholar] [CrossRef]
- Mangiafico, S. rcompanion: Functions to support extension education program evaluation. Cran Repos 2017, 20, 1–71. [Google Scholar]
- Zou, H.; Hastie, T. Regularization and variable selection via the elastic net. J. R. Stat. Soc. Stat. Methodol. Ser. B 2005, 67, 301–320. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, M. Building predictive models in R using the caret package. J. Stat. Softw. 2008, 28, 1–26. [Google Scholar] [CrossRef]
- Menardi, G.; Torelli, N. Training and assessing classification rules with imbalanced data. Data Min. Knowl. Discov. 2014, 28, 92–122. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Age-Related Disease | AUC 1 | AUC_L95 | AUC_U95 | Beta 3 | OR 4 | p-Value 2 | PRS-Input-SNVs 5(No.) | Input-SNVs Covered 6(No. (%)) |
|---|---|---|---|---|---|---|---|---|
| PD | 0.52 | 0.50 | 0.53 | −0.142 | 0.87 | 1.91 × 10−3 | 1805 | 1715 (95.01) |
| T2D | 0.51 | 0.50 | 0.53 | −0.056 | 0.95 | 2.61 × 10−3 | 6,917,436 | 6,024,432 (87.09) |
| CRC | 0.52 | 0.50 | 0.54 | −0.097 | 0.91 | 5.85 × 10−3 | 95 | 92 (96.84) |
| CAD | 0.56 | 0.54 | 0.57 | −0.159 | 0.85 | 1.88 × 10−12 | 6,630,150 | 5,920,526 (89.30) |
| AF | 0.57 | 0.55 | 0.58 | −0.170 | 0.84 | 1.07 × 10−16 | 6,730,541 | 5,973,364 (88.75) |
| ISS | 0.59 | 0.57 | 0.61 | −0.283 | 0.75 | 2.84 × 10−35 | 2,759,740 | 2,573,737 (93.26) |
| AD | 0.59 | 0.57 | 0.60 | −0.219 | 0.80 | 3.16 × 10−25 | 167 | 161 (96.41) |
| Meta-PRS 7 | 0.64 | 0.63 | 0.67 | −0.403 | 0.67 | 6.45 × 10−15 | 6,087,730 | 6,087,730 (100) |
| GPS | AUC 1 | AUC_L95 | AUC_U95 | Beta 2 | OR 3 | p-Value 4 | Number of SNVs 5 |
|---|---|---|---|---|---|---|---|
| GPSlong (Germans) | 0.56 | 0.53 | 0.58 | 0.281 | 1.32 | 1.45 × 10−5 | 7 |
| GPSlongII (Germans) | 0.55 | 0.52 | 0.55 | 0.158 | 1.17 | 9.85 × 10−3 | 10,361 |
| PRS-5 (Germans) | 0.56 | 0.54 | 0.57 | 0.223 | 1.25 | 5.33 × 10−11 | 324 |
| PRS-5 [23] | NA | NA | NA | 0.149 | 1.41 | 3.50 × 10−9 | 334 |
| PRS-6 (Germans) | 0.58 | 0.56 | 0.59 | 0.329 | 1.39 | 1.37 × 10−20 | 91 |
| PRS-6 [23] | NA | 0.57 | 0.61 | 0.158 | 1.44 | 7.30 × 10−10 | 96 |
| PRS_Liu (Germans) | 0.53 | 0.51 | 0.54 | 0.102 | 1.11 | 2.22 × 10−3 | 3414 |
| PRS_Liu [17] | 0.76 | 1.90 × 10−5 | 3966 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torres, G.G.; Dose, J.; Hasenbein, T.P.; Nygaard, M.; Krause-Kyora, B.; Mengel-From, J.; Christensen, K.; Andersen-Ranberg, K.; Kolbe, D.; Lieb, W.; et al. Long-Lived Individuals Show a Lower Burden of Variants Predisposing to Age-Related Diseases and a Higher Polygenic Longevity Score. Int. J. Mol. Sci. 2022, 23, 10949. https://doi.org/10.3390/ijms231810949
Torres GG, Dose J, Hasenbein TP, Nygaard M, Krause-Kyora B, Mengel-From J, Christensen K, Andersen-Ranberg K, Kolbe D, Lieb W, et al. Long-Lived Individuals Show a Lower Burden of Variants Predisposing to Age-Related Diseases and a Higher Polygenic Longevity Score. International Journal of Molecular Sciences. 2022; 23(18):10949. https://doi.org/10.3390/ijms231810949
Chicago/Turabian StyleTorres, Guillermo G., Janina Dose, Tim P. Hasenbein, Marianne Nygaard, Ben Krause-Kyora, Jonas Mengel-From, Kaare Christensen, Karen Andersen-Ranberg, Daniel Kolbe, Wolfgang Lieb, and et al. 2022. "Long-Lived Individuals Show a Lower Burden of Variants Predisposing to Age-Related Diseases and a Higher Polygenic Longevity Score" International Journal of Molecular Sciences 23, no. 18: 10949. https://doi.org/10.3390/ijms231810949
APA StyleTorres, G. G., Dose, J., Hasenbein, T. P., Nygaard, M., Krause-Kyora, B., Mengel-From, J., Christensen, K., Andersen-Ranberg, K., Kolbe, D., Lieb, W., Laudes, M., Görg, S., Schreiber, S., Franke, A., Caliebe, A., Kuhlenbäumer, G., & Nebel, A. (2022). Long-Lived Individuals Show a Lower Burden of Variants Predisposing to Age-Related Diseases and a Higher Polygenic Longevity Score. International Journal of Molecular Sciences, 23(18), 10949. https://doi.org/10.3390/ijms231810949