
Target-Specific Machine Learning Scoring Function Improved Structure-Based Virtual Screening Performance for SARS-CoV-2 Drugs Development

 ,
,  and
and
Abstract
:1. Introduction
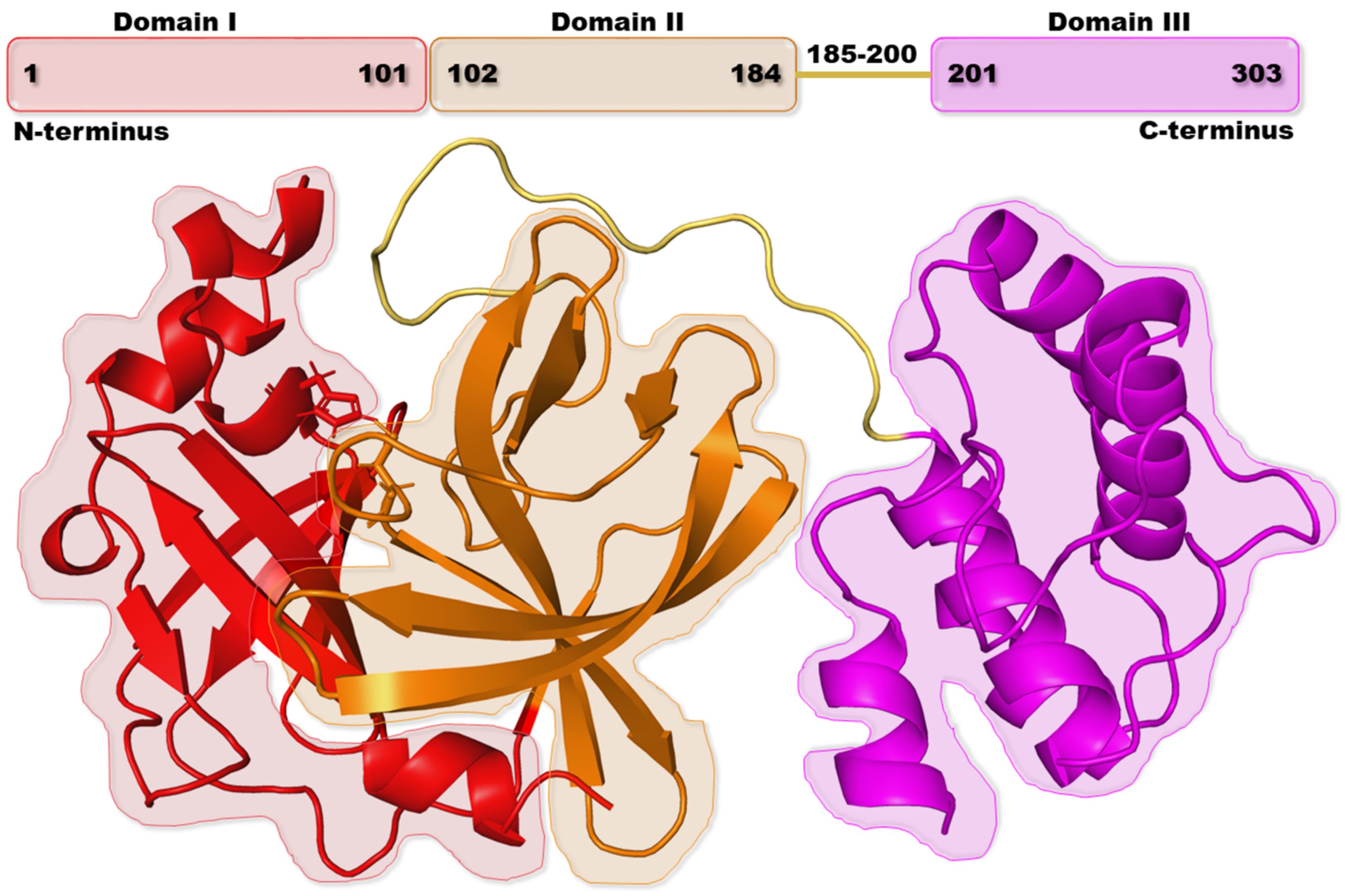
2. Results and Discussion
2.1. Chemical Space Analysis
2.2. CLpro-Specific Scoring Training
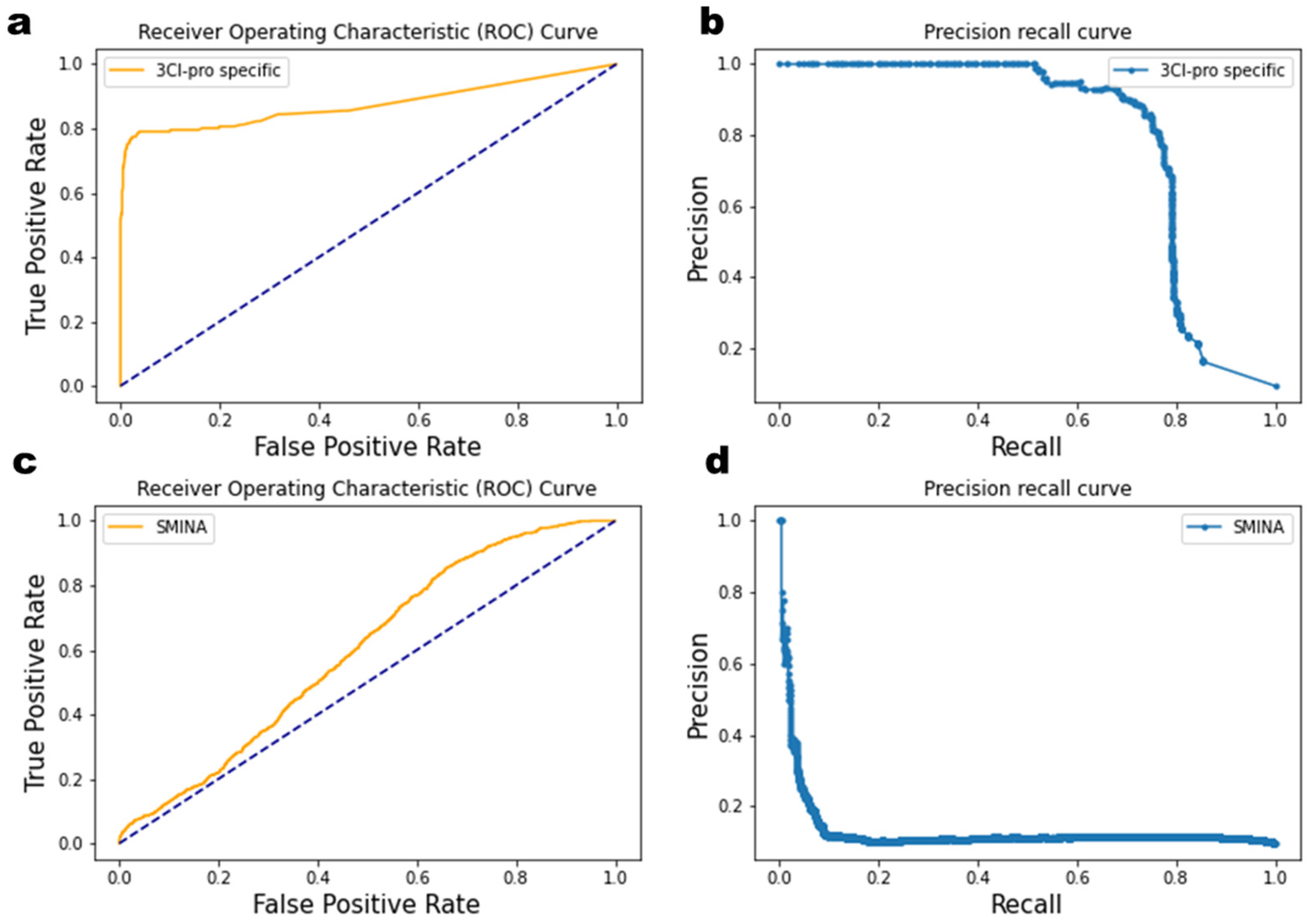
2.3. Enrichment Factor Analysis
2.4. Stability of Top-Ranked Molecules
3. Materials and Methods
3.1. Preparation of Actives
3.2. Preparation of Decoys
3.3. Generate 3D Coordinates for Actives and Decoys
3.4. Molecular Docking
3.5. Generation of Descriptors
3.6. Scoring Function
3.7. Model Evaluation
3.8. Molecular Dynamics (MD) Simulation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, Y.; Liu, Q.; Guo, D. Emerging coronaviruses: Genome structure, replication, and pathogenesis. J. Med. Virol. 2020, 92, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Christian, M.D.; Poutanen, S.M.; Loutfy, M.R.; Muller, M.P.; Low, D.E. Severe acute respiratory syndrome. Clin. Infect. Dis. 2004, 38, 1420–1427. [Google Scholar] [CrossRef]
- Zaki, A.M.; Van Boheemen, S.; Bestebroer, T.M.; Osterhaus, A.D.M.E.; Fouchier, R.A.M. Isolation of a Novel Coronavirus from a Man with Pneumonia in Saudi Arabia. N. Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- World Health Organization. Responding to Community Spread of COVID-19. WHO/COVID-19/Community_Transmission/2020.1; World Health Organization: Geneva, Switzerland, 2020.
- Bernal, J.L.; Andrews, N.; Gower, C.; Gallagher, E.; Simmons, R.; Thelwall, S.; Stowe, J.; Tessier, E.; Groves, N.; Dabrera, G.; et al. Effectiveness of Covid-19 vaccines against the B. 1.617. 2 (Delta) variant. N. Engl. J. Med. 2021, 385, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Brian, D.A.; Baric, R.S. Coronavirus genome structure and replication. Coronavirus Replication Reverse Genet. 2005, 287, 1–30. [Google Scholar]
- Snijder, E.J.; Decroly, E.; Ziebuhr, J. The nonstructural proteins directing coronavirus RNA synthesis and processing. Adv. Virus Res. 2016, 96, 59–126. [Google Scholar]
- Chang, C.-k.; Hou, M.-H.; Chang, C.-F.; Hsiao, C.-D.; Huang, T.-h. The SARS coronavirus nucleocapsid protein–forms and functions. Antivir. Res. 2014, 103, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Morse, J.S.; Lalonde, T.; Xu, S.; Liu, W.R. Learning from the past: Possible urgent prevention and treatment options for severe acute respiratory infections caused by 2019-nCoV. Chembiochem 2020, 21, 730–738. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.-L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Zhang, B.; Jiang, X.-M.; Su, H.; Li, J.; Zhao, Y.; Xie, X.; Jin, Z.; Peng, J.; Liu, F.; et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 2020, 368, 1331–1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varnek, A.; Baskin, I. Machine learning methods for property prediction in chemoinformatics: Quo vadis? J. Chem. Inf. Modeling 2012, 52, 1413–1437. [Google Scholar] [CrossRef]
- Lo, Y.-C.; Rensi, S.E.; Torng, W.; Altman, R.B. Machine learning in chemoinformatics and drug discovery. Drug Discov. Today 2018, 23, 1538–1546. [Google Scholar] [CrossRef]
- Ali, S.M.; Hoemann, M.Z.; Aubé, J.; Georg, G.I.; Mitscher, L.A.; Jayasinghe, L.R. Butitaxel Analogues: Synthesis and Structure—Activity Relationships. J. Med. Chem. 1997, 40, 236–241. [Google Scholar] [CrossRef]
- Veerasamy, R. QSAR—An Important In-Silico Tool in Drug Design and Discovery. In Advances in Computational Modeling and Simulation; Springer: Berlin/Heidelberg, Germany, 2022; pp. 191–208. [Google Scholar]
- Priya, S.; Tripathi, G.; Singh, D.B.; Jain, P.; Kumar, A. Machine Learning Approaches and their Applications in Drug Discovery and Design. Chem. Biol. Drug Des. 2022, 100, 136–153. [Google Scholar] [CrossRef]
- Liu, Z.; Su, M.; Han, L.; Liu, J.; Yang, Q.; Li, Y.; Wang, R. Forging the basis for developing protein–ligand interaction scoring functions. Acc. Chem. Res. 2017, 50, 302–309. [Google Scholar] [CrossRef]
- Ragoza, M.; Hochuli, J.; Idrobo, E.; Sunseri, J.; Koes, D.R. Protein—ligand scoring with convolutional neural networks. J. Chem. Inf. Modeling 2017, 57, 942–957. [Google Scholar] [CrossRef]
- Stepniewska-Dziubinska, M.M.; Zielenkiewicz, P.; Siedlecki, P. Development and evaluation of a deep learning model for protein—ligand binding affinity prediction. Bioinformatics 2018, 34, 3666–3674. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, Y. Improving scoring-docking-screening powers of protein—ligand scoring functions using random forest. J. Comput. Chem. 2017, 38, 169–177. [Google Scholar] [CrossRef]
- Li, H.; Leung, K.-S.; Wong, M.-H.; Ballester, P.J. Substituting random forest for multiple linear regression improves binding affinity prediction of scoring functions: Cyscore as a case study. BMC Bioinform. 2014, 15, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Imrie, F.; Bradley, A.R.; Deane, C.M. Generating property-matched decoy molecules using deep learning. Bioinformatics 2021, 37, 2134–2141. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Morley, C.; Hutchison, G.R. Pybel: A Python wrapper for the OpenBabel cheminformatics toolkit. Chem. Cent. J. 2008, 2, 5. [Google Scholar] [CrossRef]
- Koes, D.R.; Baumgartner, M.P.; Camacho, C.J. Lessons learned in empirical scoring with smina from the CSAR 2011 benchmarking exercise. J. Chem. Inf. Modeling 2013, 53, 1893–1904. [Google Scholar] [CrossRef]
- Feinstein, W.P.; Brylinski, M. Calculating an optimal box size for ligand docking and virtual screening against experimental and predicted binding pockets. J. Cheminform. 2015, 7, 18. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Wójcikowski, M.; Zielenkiewicz, P.; Siedlecki, P. Open Drug Discovery Toolkit (ODDT): A new open-source player in the drug discovery field. J. Cheminform. 2015, 7, 26. [Google Scholar] [CrossRef]
- Cereto-Massagué, A.; Ojeda, M.J.; Valls, C.; Mulero, M.; Garcia-Vallvé, S.; Pujadas, G. Molecular fingerprint similarity search in virtual screening. Methods 2015, 71, 58–63. [Google Scholar] [CrossRef]
- Morgan, H. The generation of a unique machine description for chemical structures-a technique developed at chemical abstracts service. J. Chem. Doc. 1965, 5, 107–113. [Google Scholar] [CrossRef]
- Nicholls, A. What do we know and when do we know it? J. Comput.-Aided Mol. Des. 2008, 22, 239–255. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Alnammi, M.; Ericksen, S.S.; Voter, A.F.; Ananiev, G.E.; Keck, J.L.; Hoffmann, F.M.; Wildman, S.A.; Gitter, A. Practical model selection for prospective virtual screening. J. Chem. Inf. Modeling 2018, 59, 282–293. [Google Scholar] [CrossRef]
- Fresnais, L.; Ballester, P.J. The impact of compound library size on the performance of scoring functions for structure-based virtual screening. Briefings. Bioinform. 2021, 22, bbaa095. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An overview of the Amber biomolecular simulation package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- Zwanzig, R. Nonlinear generalized Langevin equations. J. Stat. Phys. 1973, 9, 215–220. [Google Scholar] [CrossRef]
- Zhou, R.; Harder, E.; Xu, H.; Berne, B. Efficient multiple time step method for use with Ewald and particle mesh Ewald for large biomolecular systems. J. Chem. Phys. 2001, 115, 2348–2358. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Gotz, A.W.; Williamson, M.J.; Xu, D.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born. J. Chem. Theory Comput. 2012, 8, 1542–1555. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Top 5 Molecules Scored by Smina | Top 5 Molecules Scored by Smina 3CLpro-Specific Machine Learning Model | ||||
|---|---|---|---|---|---|
| Molecules Top 5 | Smina Score | Actual pIC50 | Molecules Top 5 | 3CLpro-Specific Score | Actual pIC50 |
| Mol_1514 | −10.80 | 4.79 | Mol_336 | 6.95 | 7.10 |
| Mol_890 | −10.64 | 4.79 | Mol_821 | 6.67 | 7.01 |
| Mol_1170 | −10.43 | 2 | Mol_522 | 6.62 | 7.08 |
| Mol_1112 | −10.35 | 2 | Mol_1355 | 6.47 | 7.27 |
| Mol_280 | −10.25 | 4.49 | Mol_819 | 6.39 | 6.66 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tahir ul Qamar, M.; Zhu, X.-T.; Chen, L.-L.; Alhussain, L.; Alshiekheid, M.A.; Theyab, A.; Algahtani, M. Target-Specific Machine Learning Scoring Function Improved Structure-Based Virtual Screening Performance for SARS-CoV-2 Drugs Development. Int. J. Mol. Sci. 2022, 23, 11003. https://doi.org/10.3390/ijms231911003
Tahir ul Qamar M, Zhu X-T, Chen L-L, Alhussain L, Alshiekheid MA, Theyab A, Algahtani M. Target-Specific Machine Learning Scoring Function Improved Structure-Based Virtual Screening Performance for SARS-CoV-2 Drugs Development. International Journal of Molecular Sciences. 2022; 23(19):11003. https://doi.org/10.3390/ijms231911003
Chicago/Turabian StyleTahir ul Qamar, Muhammad, Xi-Tong Zhu, Ling-Ling Chen, Laila Alhussain, Maha A. Alshiekheid, Abdulrahman Theyab, and Mohammad Algahtani. 2022. "Target-Specific Machine Learning Scoring Function Improved Structure-Based Virtual Screening Performance for SARS-CoV-2 Drugs Development" International Journal of Molecular Sciences 23, no. 19: 11003. https://doi.org/10.3390/ijms231911003
APA StyleTahir ul Qamar, M., Zhu, X. -T., Chen, L. -L., Alhussain, L., Alshiekheid, M. A., Theyab, A., & Algahtani, M. (2022). Target-Specific Machine Learning Scoring Function Improved Structure-Based Virtual Screening Performance for SARS-CoV-2 Drugs Development. International Journal of Molecular Sciences, 23(19), 11003. https://doi.org/10.3390/ijms231911003