
Multiple Timescale Dynamic Analysis of Functionally-Impairing Mutations in Human Ileal Bile Acid-Binding Protein



Abstract
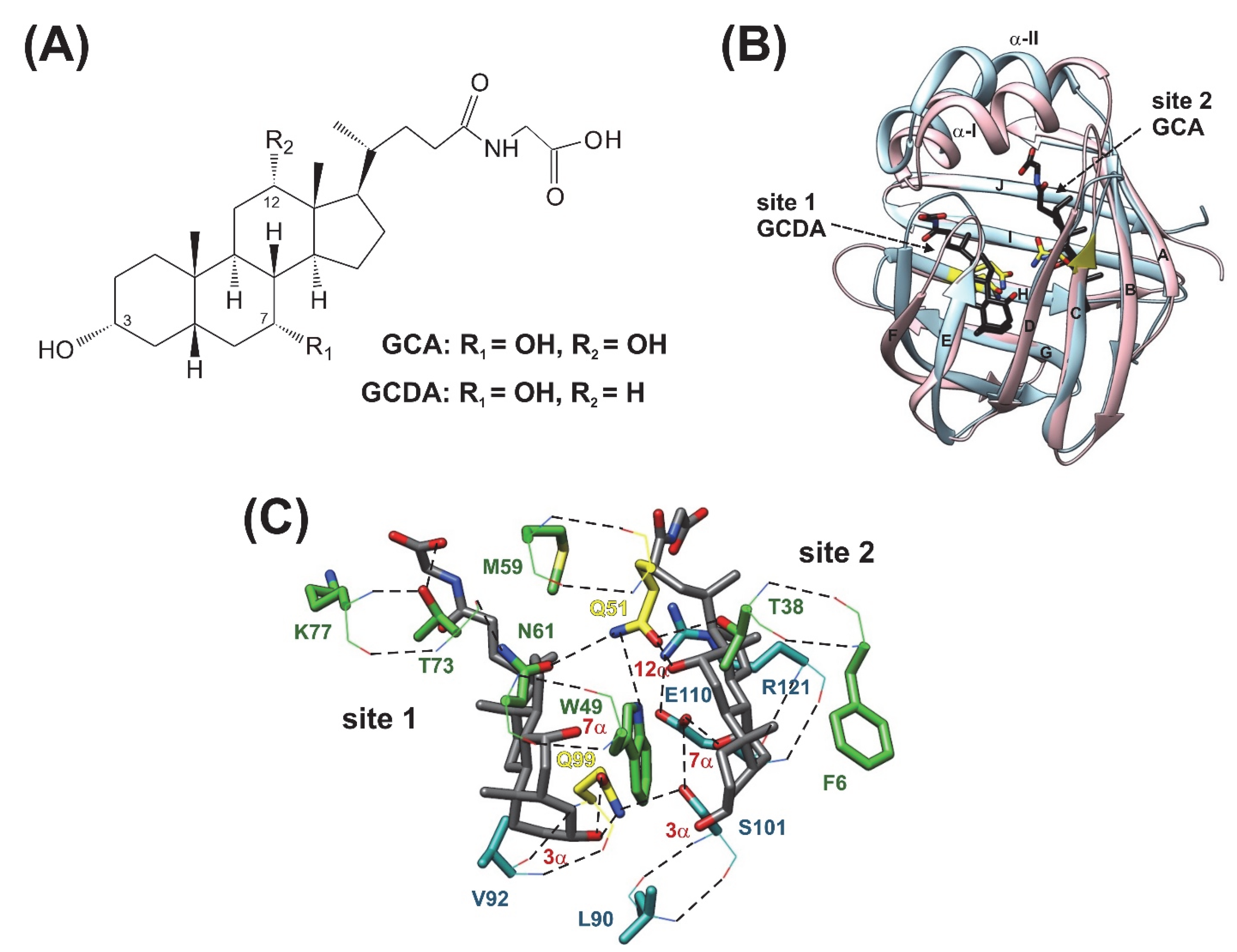
:1. Introduction
2. Results
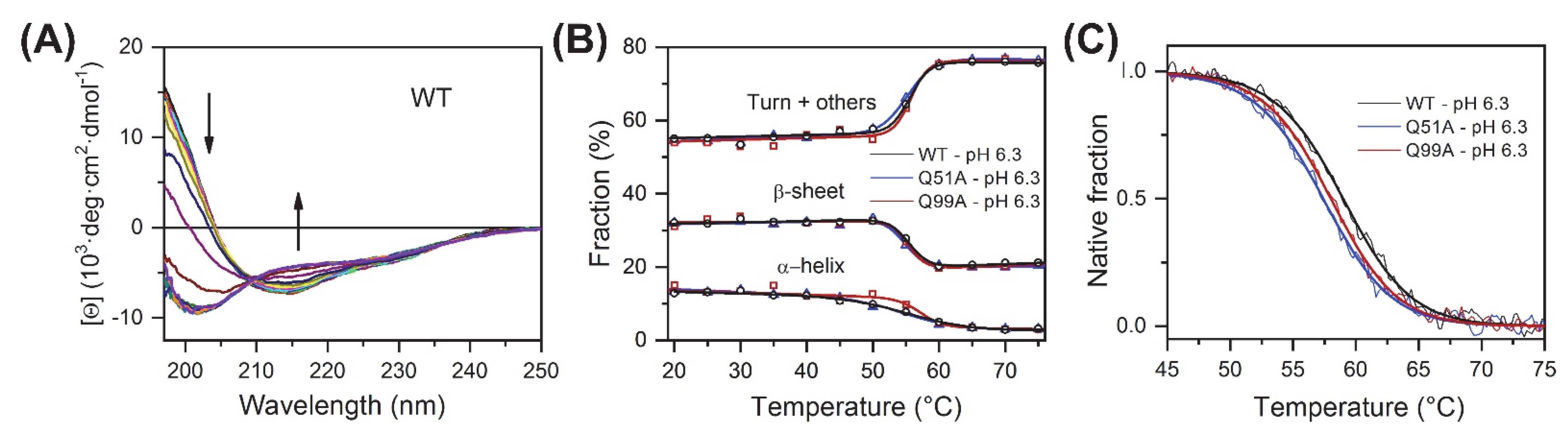
2.1. Overall Structure and Stability Studied by CD Spectroscopy
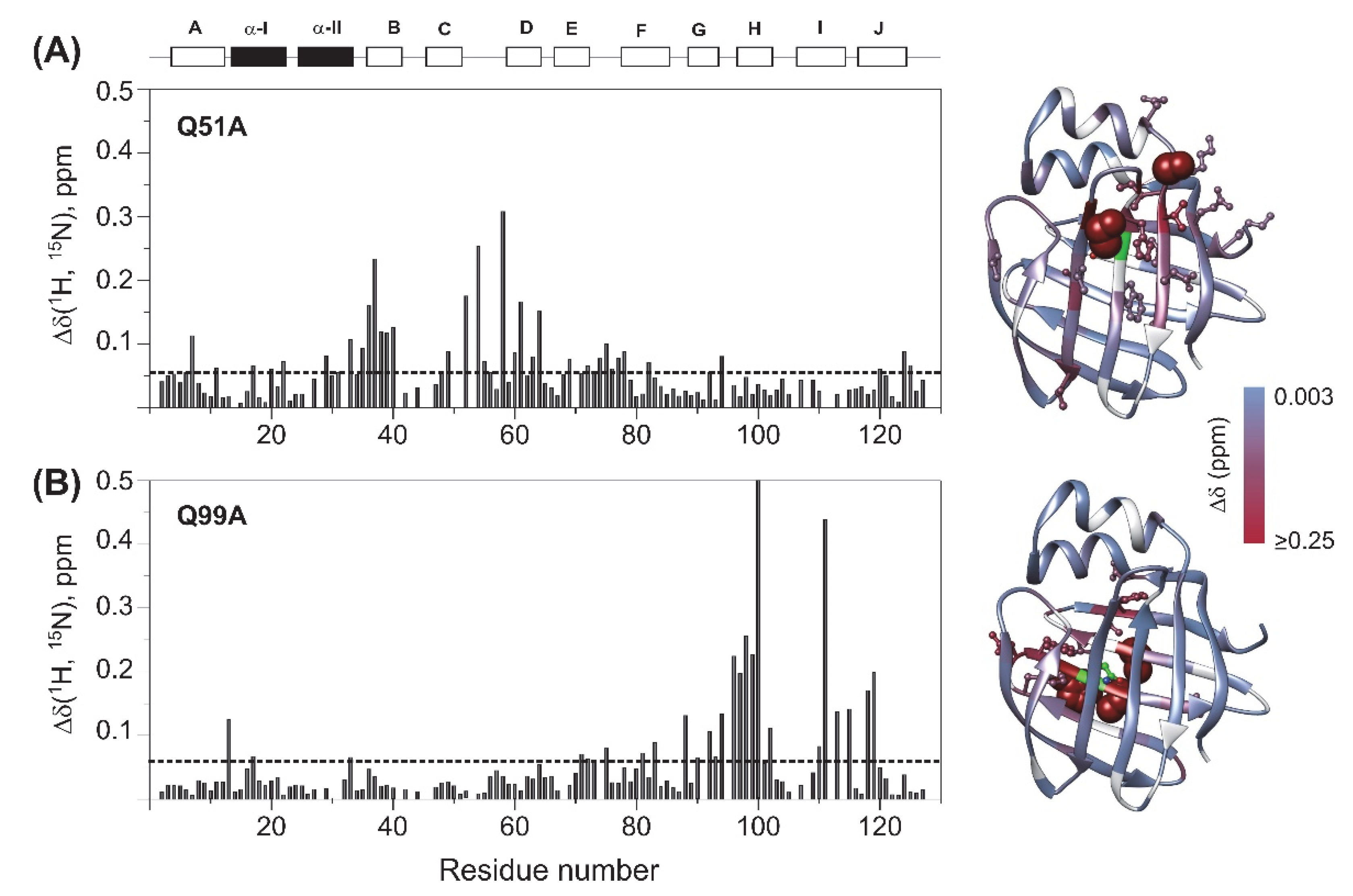
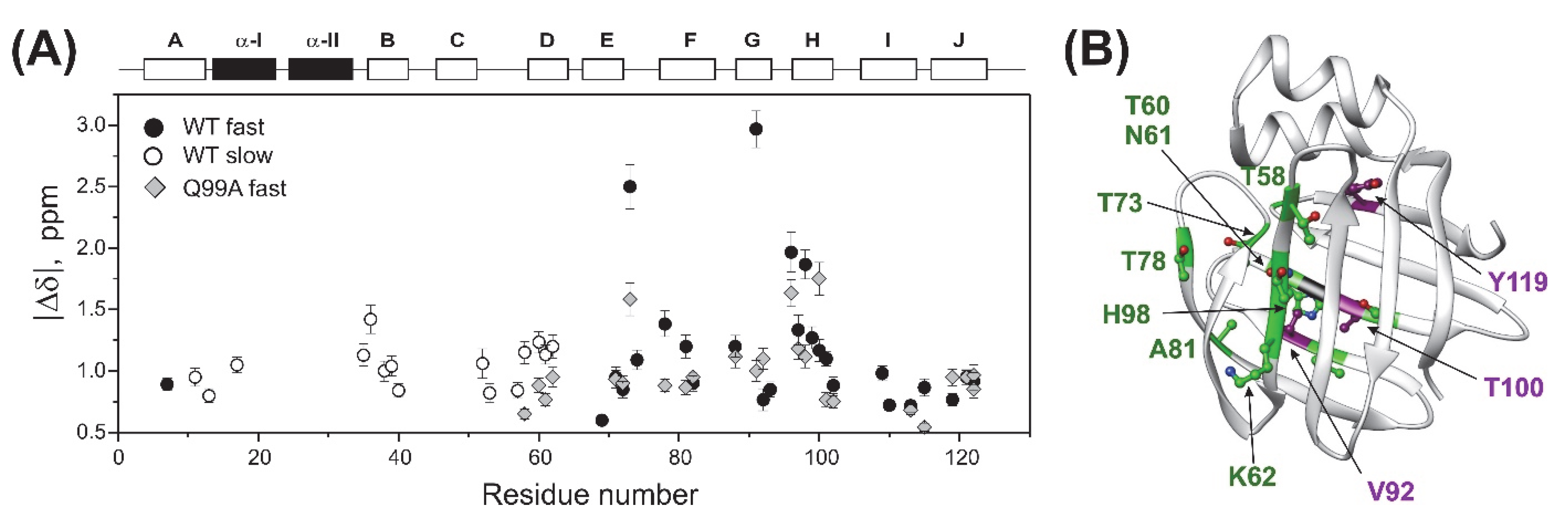
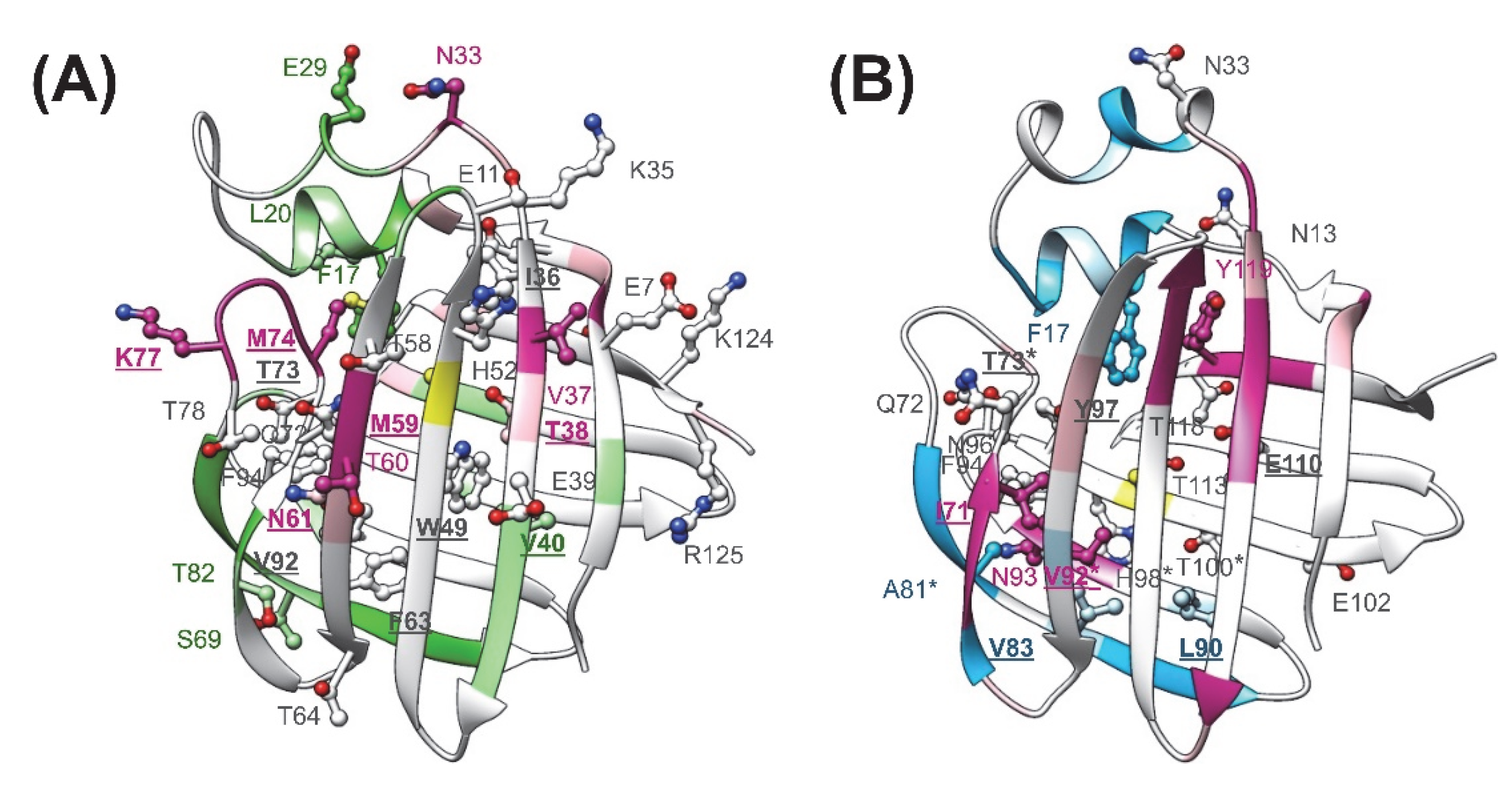
2.2. Chemical Shift Perturbation Mapping
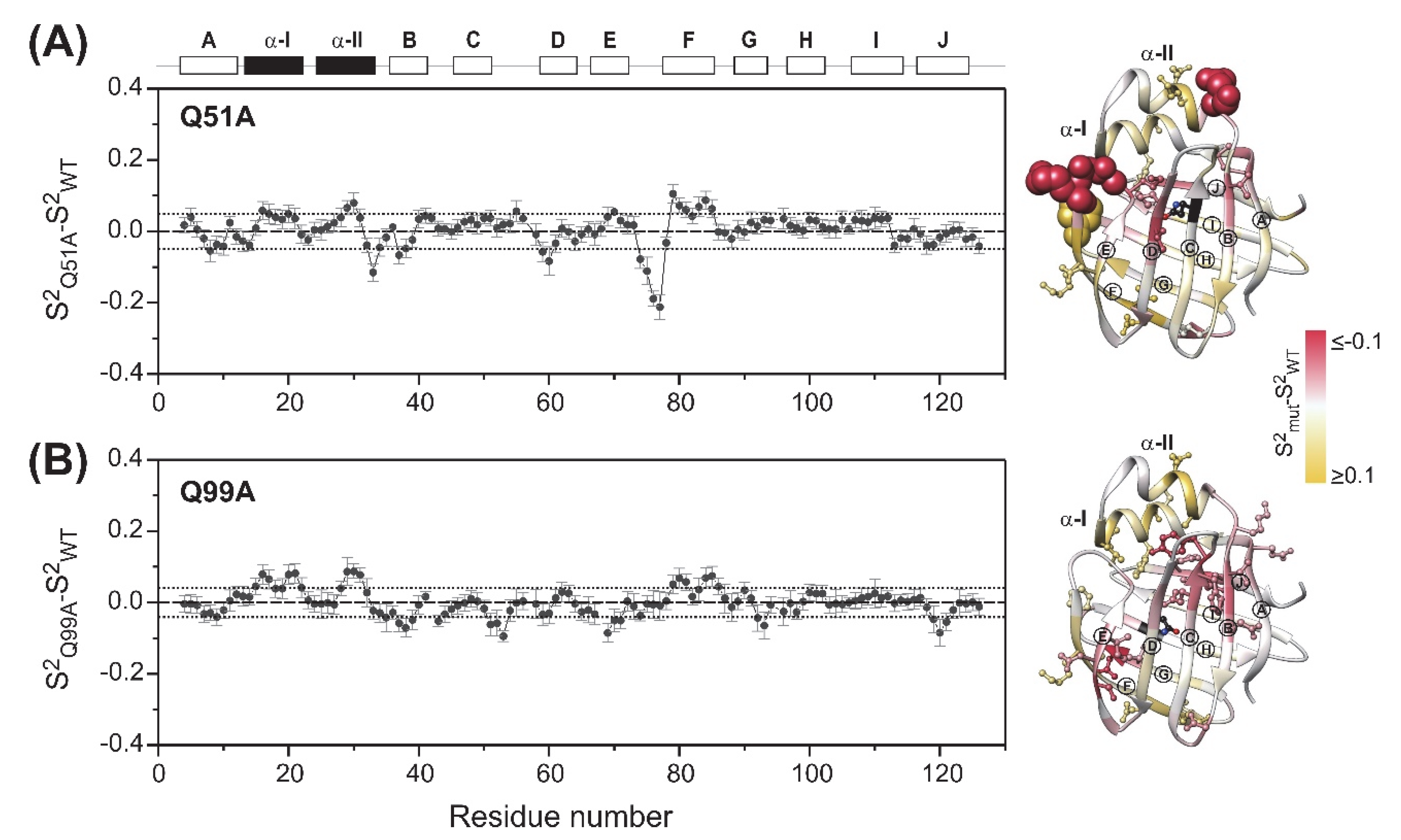
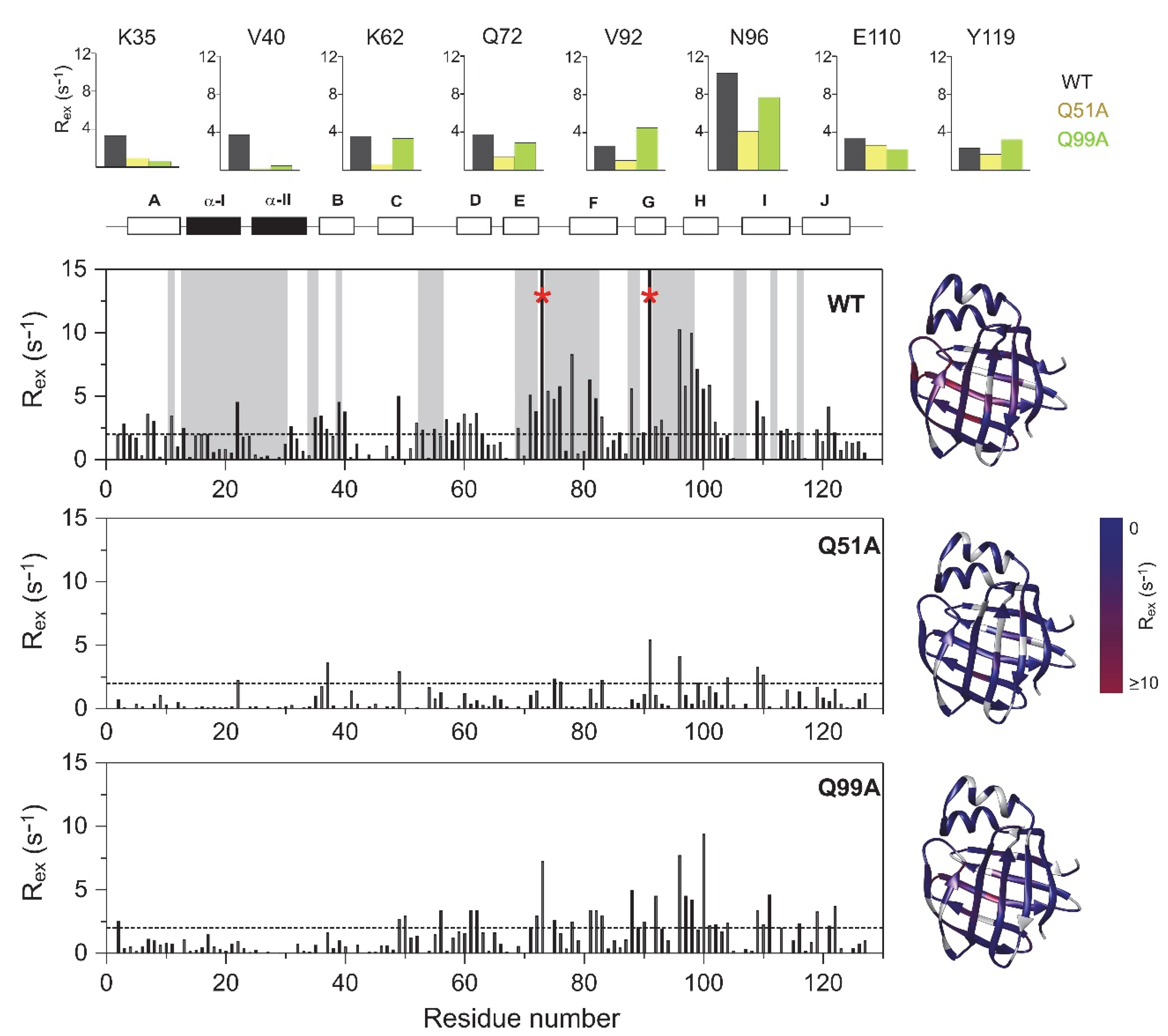
2.3. Fast Protein Motions by NMR
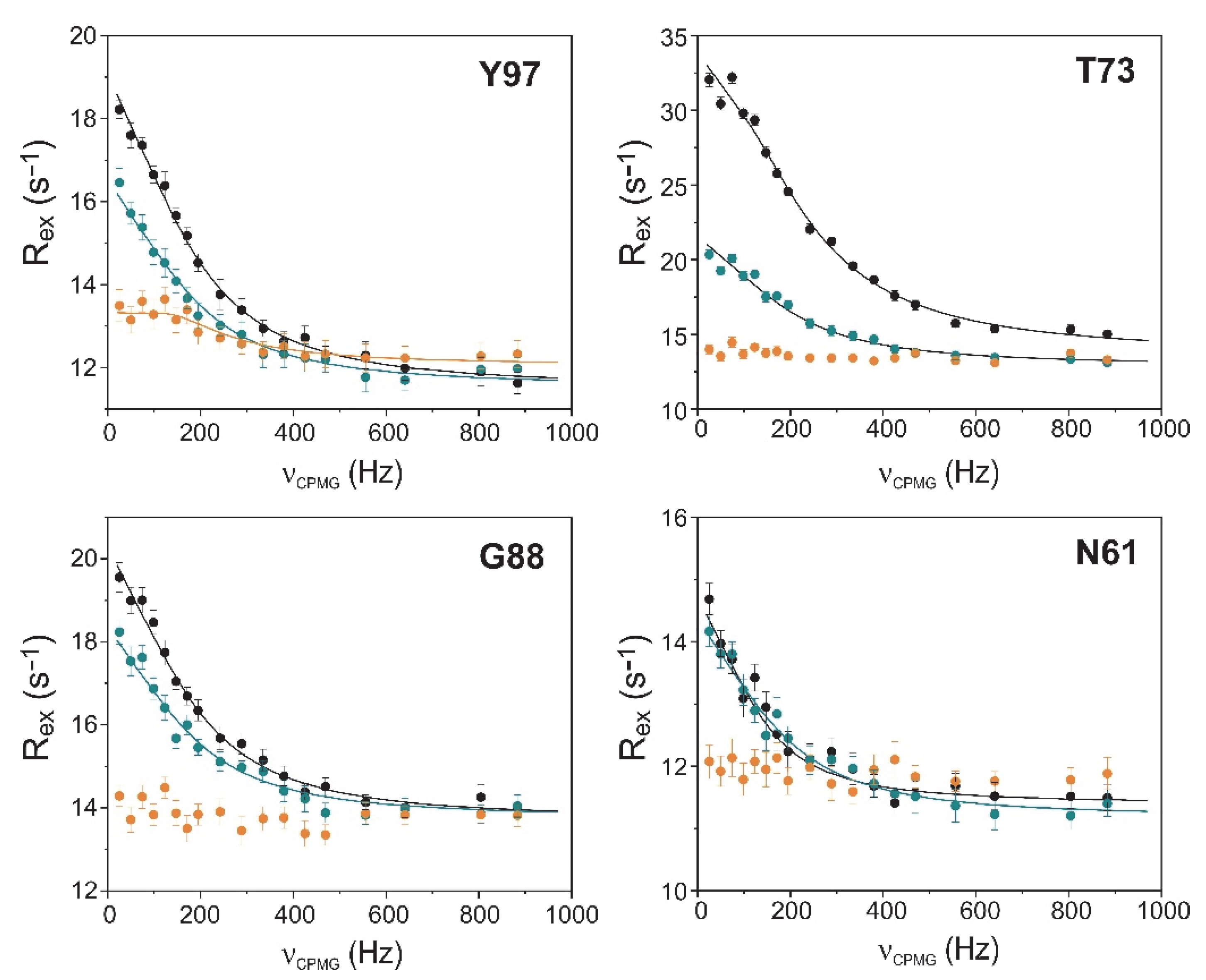
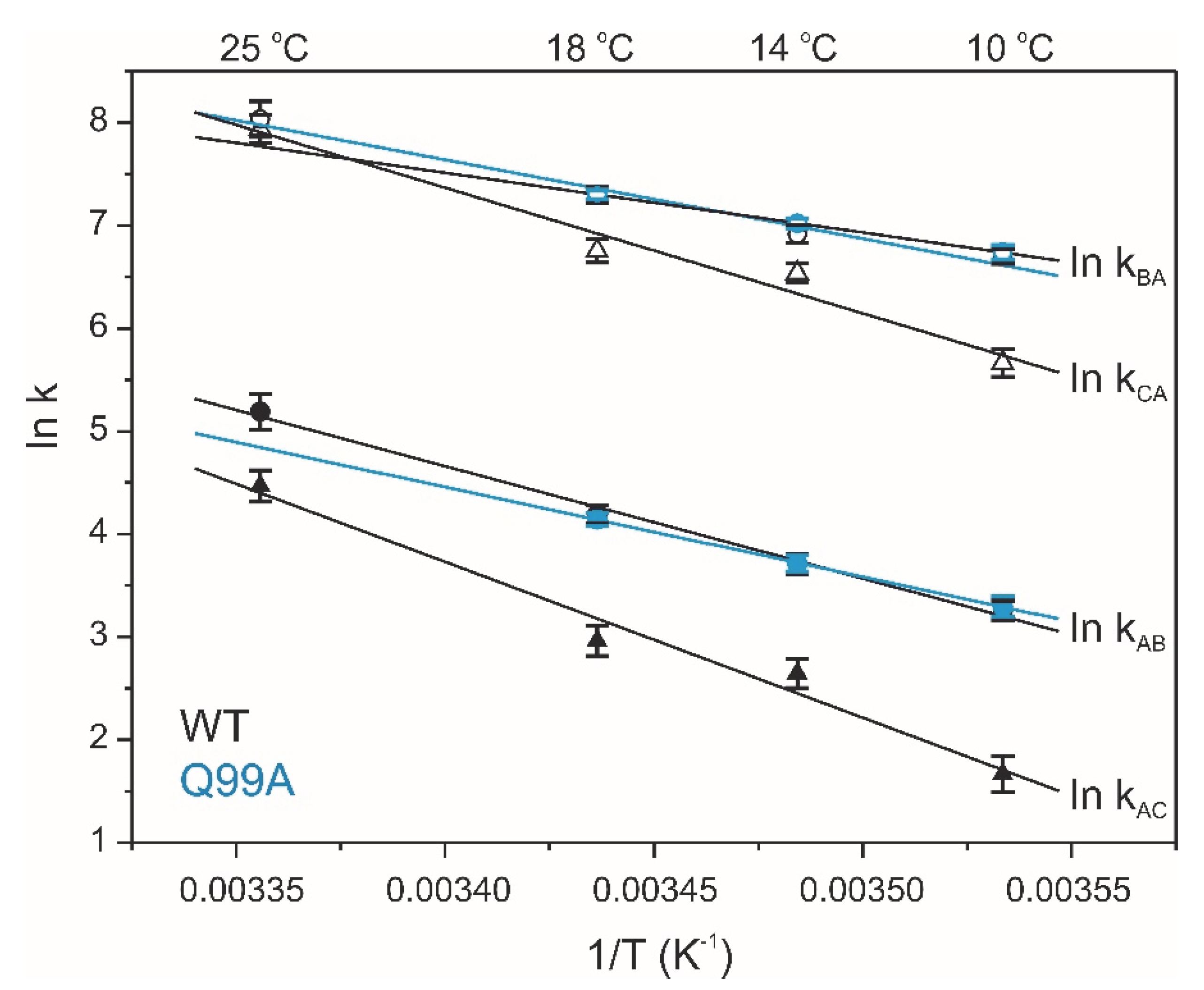
2.4. Relaxation Dispersion Analysis
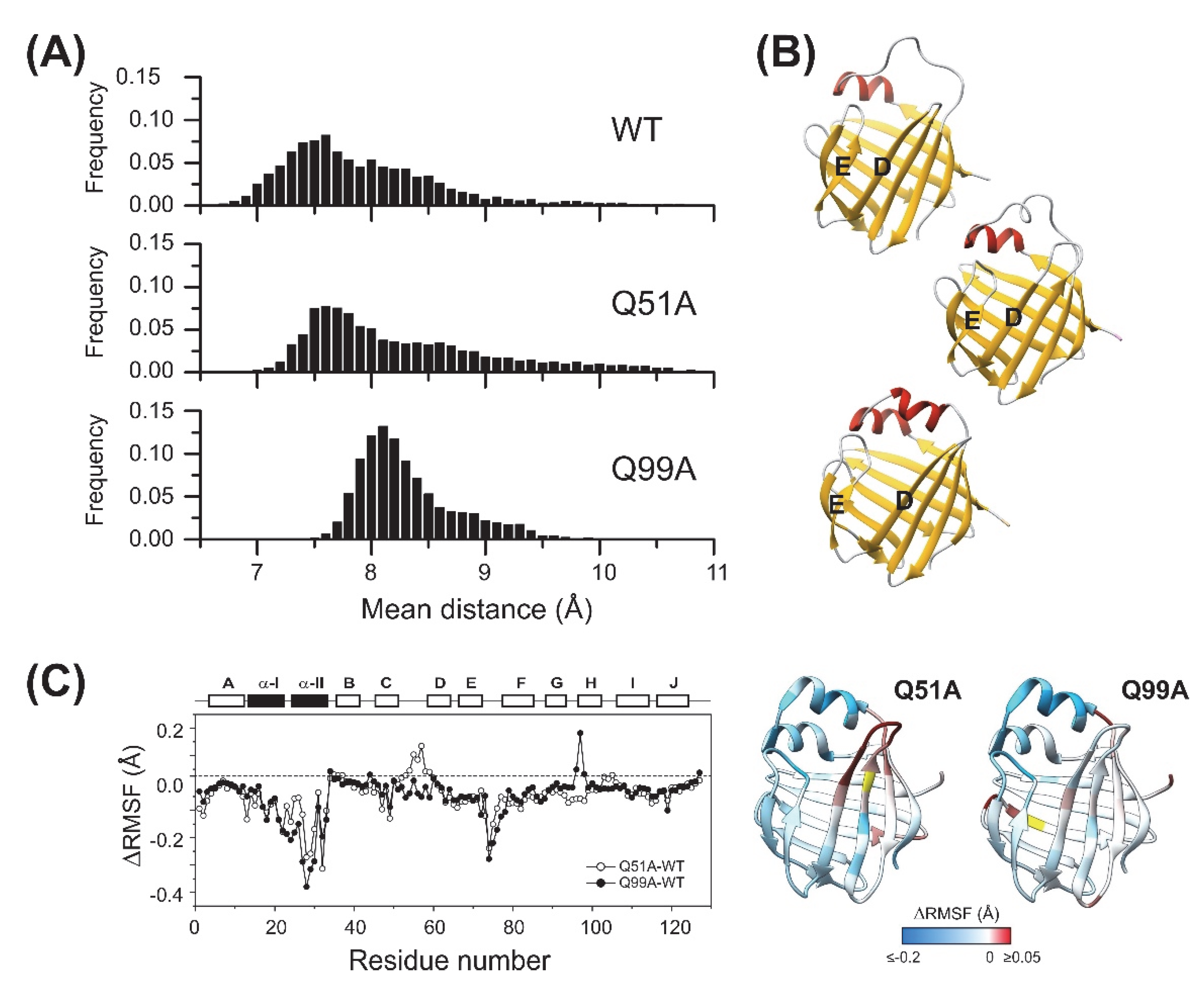
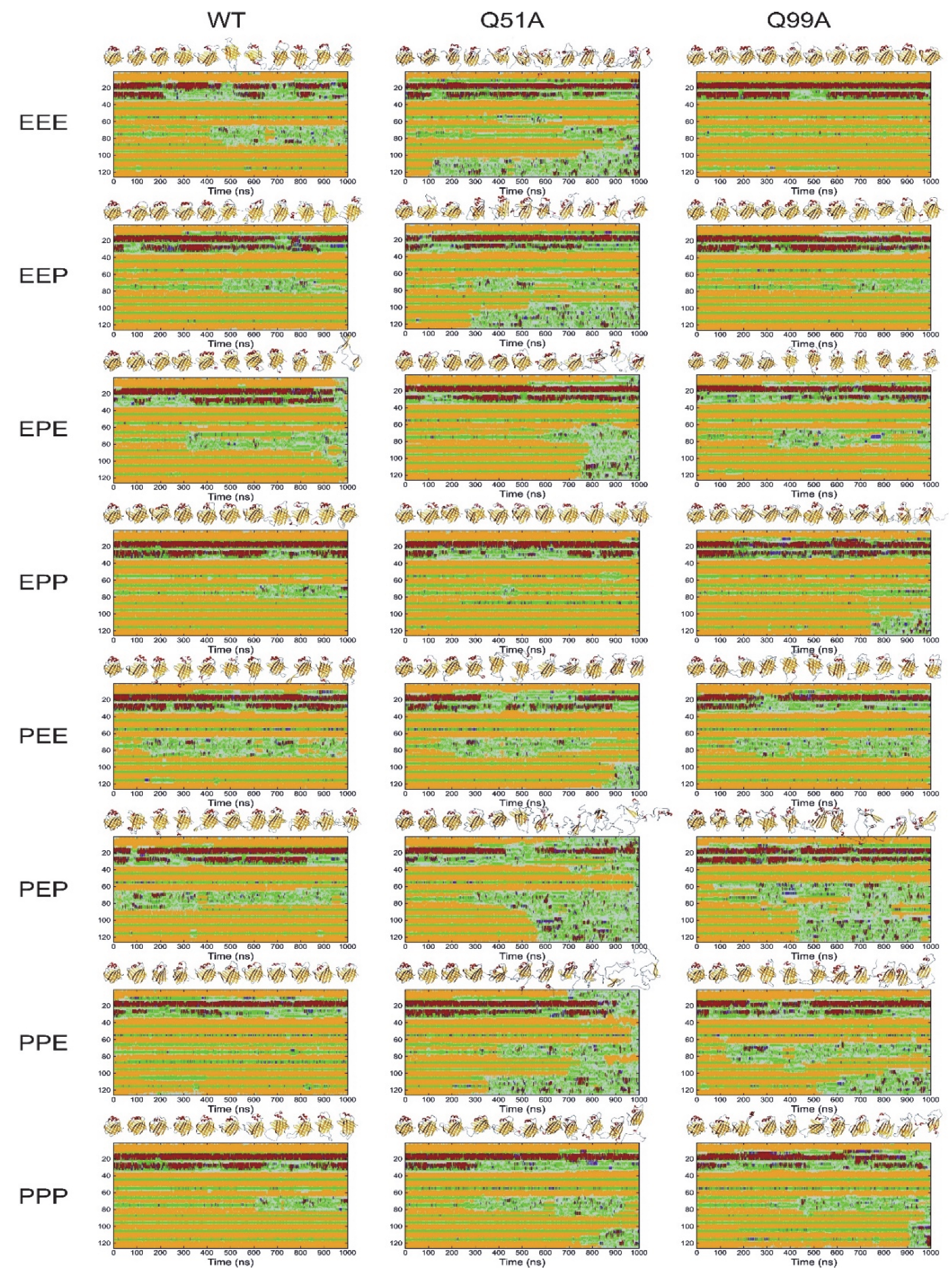
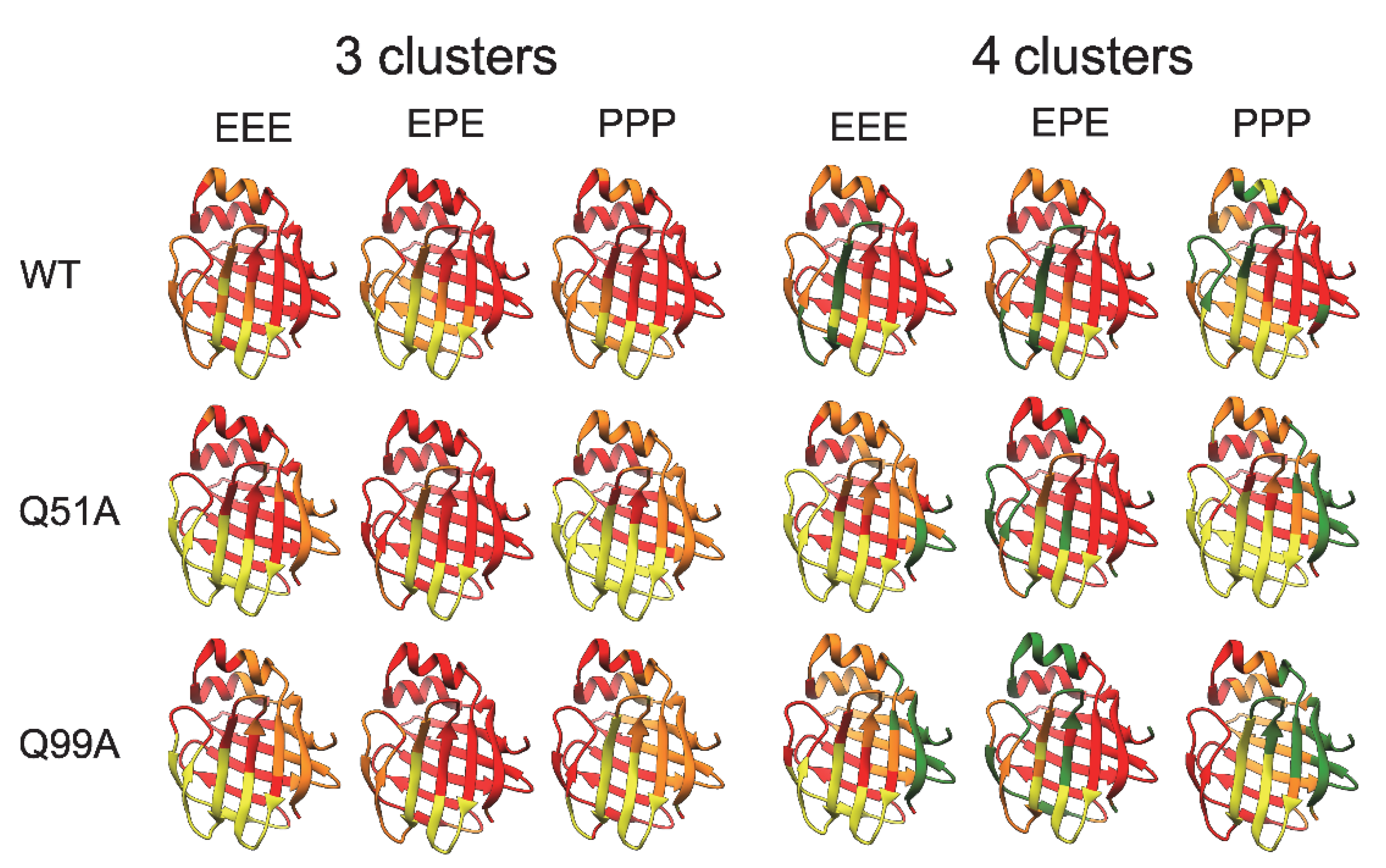
2.5. Molecular Dynamics Simulations
3. Discussion
4. Materials and Methods
4.1. Sample Preparation
4.2. CD Spectroscopy and Thermal Denaturation Experiments
4.3. NMR Data Collection
4.4. NMR Relaxation Analysis
4.5. Molecular Dynamics Simulations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BABP | Bile acid binding protein |
| CD | Circular dichroism |
| cL-BABP | Chicken liver bile acid binding protein |
| CPMG | Carr-Purcell-Meiboom-Gill |
| CRBP | Cellular retinol binding protein |
| FABP | Fatty acid binding protein |
| GCA | Glycocholic acid |
| GCDA | Glycochenodeoxycholic acid |
| hI-BABP | Human ileal bile acid binding protein |
| iLBP | Intracellular lipid binding protein |
| MD | Molecular dynamics |
| NMR | Nuclear magnetic resonance |
| RMSF | Root mean square fluctuation |
| WT | Wild-type |
References
- Small, D.M.; Dowling, R.H.; Redinger, R.N. The enterohepatic circulation of bile salts. Arch. Intern. Med. 1972, 130, 552–573. [Google Scholar] [CrossRef] [PubMed]
- Borgström, B.; Barrowman, J.A.; Lindström, M. Roles of Bile Acids in Intestinal Lipid Digestion and Absorption. In Sterols and Bile Acids; Danielsson, H., Sjövall, J., Eds.; Elsevier: Amsterdam, The Netherlands, 1985; Volume 12, pp. 405–425. [Google Scholar]
- Houten, S.M.; Watanabe, M.; Auwerx, J. Endocrine functions of bile acids. EMBO J. 2006, 25, 1419–1425. [Google Scholar] [CrossRef] [PubMed]
- Makishima, M.; Okamoto, A.Y.; Repa, J.J.; Tu, H.; Learned, R.M.; Luk, A.; Hull, M.V.; Lustig, K.D.; Mangelsdorf, D.J.; Shan, B. Identification of a nuclear receptor for bile acids. Science 1999, 284, 1362–1365. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.J.; Blanchard, S.G.; Bledsoe, R.K.; Chandra, G.; Consler, T.G.; Kliewer, S.A.; Stimmel, J.B.; Willson, T.M.; Zavacki, A.M.; Moore, D.D.; et al. Bile acids: Natural ligands for an orphan nuclear receptor. Science 1999, 284, 1365–1368. [Google Scholar] [CrossRef]
- Maruyama, T.; Miyamoto, Y.; Nakamura, T.; Tamai, Y.; Okada, H.; Sugiyama, E.; Nakamura, T.; Itadani, H.; Tanaka, K. Identification of membrane-type receptor for bile acids (M-BAR). Biochem. Biophys. Res. Commun. 2002, 298, 714–719. [Google Scholar] [CrossRef]
- Kawamata, Y.; Fujii, R.; Hosoya, M.; Harada, M.; Yoshida, H.; Miwa, M.; Fukusumi, S.; Habata, Y.; Itoh, T.; Shintani, Y.; et al. A G protein-coupled receptor responsive to bile acids. J. Biol. Chem. 2003, 278, 9435–9440. [Google Scholar] [CrossRef]
- Gupta, S.; Stravitz, R.T.; Dent, P.; Hylemon, P.B. Down-regulation of cholesterol 7alpha-hydroxylase (CYP7A1) gene expression by bile acids in primary rat hepatocytes is mediated by the c-Jun N-terminal kinase pathway. J. Biol. Chem. 2001, 276, 15816–15822. [Google Scholar] [CrossRef]
- Qiao, L.; Han, S.I.; Fang, Y.; Park, J.S.; Gupta, S.; Gilfor, D.; Amorino, G.; Valerie, K.; Sealy, L.; Engelhardt, J.F.; et al. Bile acid regulation of C/EBPbeta, CREB, and c-Jun function, via the extracellular signal-regulated kinase and c-Jun NH2-terminal kinase pathways, modulates the apoptotic response of hepatocytes. Mol. Cell. Biol. 2003, 23, 3052–3066. [Google Scholar] [CrossRef]
- Lefebvre, P.; Cariou, B.; Lien, F.; Kuipers, F.; Staels, B. Role of bile acids and bile acid receptors in metabolic regulation. Physiol. Rev. 2009, 89, 147–191. [Google Scholar] [CrossRef]
- Ficker, P.; Wagner, M. Biliary bile acids in hepatobiliary injury—What is the link? J. Hepatol. 2017, 67, 619–631. [Google Scholar] [CrossRef] [Green Version]
- Perino, A.; Demagny, H.; Velazquez-Villegas, L.; Schoonjans, K. Moleculer physiology of bile acid signaling in health, disease, and aging. Physiol. Rev. 2021, 101, 683–731. [Google Scholar] [CrossRef]
- Banaszak, L.; Winter, N.; Xu, Z.; Bernlohr, D.A.; Cowan, S.; Jones, T.A. Lipid-binding proteins: A family of fatty acid and retinoid transport proteins. Adv. Protein Chem. 1994, 45, 89–151. [Google Scholar]
- Veerkamp, J.H.; Maatman, R.G. Cytoplasmic fatty acid-binding proteins: Their structure and genes. Prog. Lipid Res. 1995, 34, 17–52. [Google Scholar] [CrossRef]
- Kurz, M.; Brachvogel, V.; Matter, H.; Stengelin, S.; Thüring, H.; Kramer, W. Insights into the bile acid transportation system: The human ileal lipid-binding protein-cholyltaurine complex and its comparison with homologous structures. Proteins 2003, 50, 312–328. [Google Scholar] [CrossRef]
- Nichesola, D.; Perduca, M.; Capaldi, S.; Carrizo, M.E.; Righetti, P.G.; Monaco, H.L. Crystal structure of chicken liver basic fatty acid-binding protein complexed with cholic acid. Biochemistry 2004, 43, 14072–14079. [Google Scholar] [CrossRef]
- Eliseo, T.; Ragona, L.; Catalano, M.; Assfalg, M.; Paci, M.; Zetta, L.; Molinari, H.; Cicero, D.O. Structural and dynamic determinants of ligand binding in the ternary complex of chicken liver bile acid binding protein with two bile salts revealed by NMR. Biochemistry 2007, 46, 12557–12567. [Google Scholar] [CrossRef]
- Capaldi, S.; Saccomani, G.; Fessas, D.; Signorelli, M.; Perduca, M.; Monaco, H.L. The X-ray structure of zebrafish (Danio rerio) ileal bile acid-binding protein reveals the presence of binding sites on the surface of the protein molecule. J. Mol. Biol. 2009, 385, 99–116. [Google Scholar] [CrossRef]
- Horváth, G.; Bencsura, Á.; Simon, Á.; Tochtrop, G.P.; DeKoster, G.T.; Covey, D.F.; Cistola, D.P.; Toke, O. Structural determinants of ligand binding in the ternary complex of human ileal bile acid binding protein with glycocholate and glycochenodeoxycholate obtained from solution NMR. FEBS J. 2016, 283, 541–555. [Google Scholar] [CrossRef]
- Toke, O. Structural and dynamic determinants of molecular recognition in bile acid-binding proteins. Int. J. Mol. Sci. 2022, 23, 505. [Google Scholar] [CrossRef]
- Tochtrop, G.P.; Richter, K.; Tang, C.; Toner, J.J.; Covey, D.F.; Cistola, D.P. Energetics by NMR: Site-specific binding in a positively cooperative system. Proc. Natl. Acad. Sci. USA 2002, 99, 1847–1852. [Google Scholar] [CrossRef]
- Toke, O.; Monsey, J.D.; DeKoster, G.T.; Tochtrop, G.P.; Tang, C.; Cistola, D.P. Determinants of cooperativity and site selectivity in human ileal bile acid binding protein. Biochemistry 2006, 45, 727–737. [Google Scholar] [CrossRef]
- Pedò, M.; D’Onofrio, M.; Ferranti, P.; Molinari, H.; Assfalg, M. Towards the elucidation of molecular determinants of cooperativity in the liver bile acid binding protein. Proteins 2009, 77, 718–731. [Google Scholar] [CrossRef]
- Tochtrop, G.P.; Bruns, J.L.; Tang, C.; Covey, D.F.; Cistola, D.P. Steroid ring hydroxylation patterns govern cooperativity in human bile acid binding protein. Biochemistry 2003, 42, 11561–11567. [Google Scholar] [CrossRef]
- Tochtrop, G.P.; DeKoster, G.T.; Covey, D.F.; Cistola, D.P. A single hydroxyl group governs ligand site selectivity in human ileal bile acid binding protein. J. Am. Chem. Soc. 2004, 126, 11024–11029. [Google Scholar] [CrossRef]
- Ragona, L.; Pagano, K.; Tomaselli, S.; Favretto, F.; Ceccon, A.; Zanzoni, S.; D’Onofrio, M.; Assfalg, M.; Molinari, H. The role of dynamics in modulating ligand exchange in intracellular lipid binding proteins. Biochim. Biophys. Acta Proteins Proteom. 2014, 1844, 1268–1278. [Google Scholar] [CrossRef]
- Hodsdon, M.E.; Cistola, D.P. Discrete backbone disorder in the nuclear magnetic resonance structure of apo intestinal fatty acid-binding protein: Implications for the mechanism of ligand entry. Biochemistry 1997, 36, 1450–1460. [Google Scholar] [CrossRef]
- Lu, J.; Cistola, D.P.; Li, E. Two homologous rat cellular retinol-binding proteins differ in local conformational flexibility. J. Mol. Biol. 2003, 330, 799–812. [Google Scholar] [CrossRef]
- Menozzi, I.; Polverini, E.; Berni, R. Deciphering protein dynamics changes along the pathway of retinol by cellular retinol-binding proteins 1 and 2. Arch. Biochem. Biphys. 2018, 645, 107–116. [Google Scholar] [CrossRef]
- Jenkins, A.E.; Hockenberry, J.A.; Nguyen, T.; Bernlohr, D.A. Testing of the portal hypothesis: Analysis of a V32G, F57G, K85G mutant of the fatty acid binding protein of the murine adipocyte. Biochemistry 2002, 41, 2022–2027. [Google Scholar] [CrossRef]
- Cistola, D.P.; Kim, K.; Rogl, H.; Frieden, C. Fatty acid interactions with a helix-less variant of intestinal fatty acid-binding protein. Biochemistry 1996, 35, 7559–7565. [Google Scholar] [CrossRef]
- Kouvatsos, N.; Meldrum, J.K.; Searle, M.S.; Thomas, N.R. Coupling of ligand recognition to protein folding in an engineered variant of rabbit ileal lipid binding protein. Chem. Commun. 2006, 44, 4623–4625. [Google Scholar] [CrossRef] [PubMed]
- Toke, O.; Monsey, J.D.; Cistola, D.P. Kinetic mechanism of ligand binding in human ileal bile acid binding protein as determined by stopped-flow fluorescence analysis. Biochemistry 2007, 46, 5427–5436. [Google Scholar] [CrossRef] [PubMed]
- Horváth, G.; Király, P.; Tárkányi, G.; Toke, O. Internal motions and exchange processes in human ileal bile acid binding protein as studied by backbone 15N nuclear magnetic resonance spectroscopy. Biochemistry Erratum in Biochemistry, 51, 10119. 2012, 51, 1848–1861. [Google Scholar] [CrossRef] [PubMed]
- Horváth, G.; Egyed, O.; Toke, O. Temperature dependence of backbone dynamics in human ileal bile acid-binding protein: Implications for the mechanism of ligand binding. Biochemistry 2014, 53, 5186–5198. [Google Scholar] [CrossRef] [PubMed]
- Horváth, G.; Biczók, L.; Majer, Z.; Kovács, M.; Micsonai, A.; Kardos, J.; Toke, O. Structural insight into a partially unfolded state preceding aggregation in an intracellular lipid-binding protein. FEBS J. 2017, 284, 3637–3661. [Google Scholar] [CrossRef]
- Ragona, L.; Catalano, M.; Luppi, M.; Cicero, D.; Eliseo, T.; Foote, J.; Fogolari, F.; Zetta, L.; Molinari, H. NMR dynamic studies suggest that allosteric activation regulates ligand binding in chicken liver bile acid-binding protein. J. Biol. Chem. 2006, 281, 9697–9709. [Google Scholar] [CrossRef] [PubMed]
- Cogliati, C.; Ragona, L.; D’Onofrio, M.; Günther, U.; Whittaker, S.; Ludwig, C.; Tomaselli, S.; Assfalg, M.; Molinari, H. Site-specific investigation of the steady-state kinetics and dynamics of the multistep binding of bile acid molecules to a lipid carrier protein. Chem. Eur. J. 2010, 16, 11300–11310. [Google Scholar] [CrossRef]
- Horváth, G.; Egyed, O.; Tang, C.; Kovács, M.; Micsonai, A.; Kardos, J.; Toke, O. Ligand entry in human ileal bile acid-binding protein is mediated by histidine protonation. Sci. Rep. 2019, 9, 4825. [Google Scholar] [CrossRef]
- Micsonai, A.; Wien, F.; Kernya, L.; Lee, Y.-H.; Goto, Y.; Réfrégiers, M.; Kardos, J. Accurate secondary structure prediction and fold recognition for circular dichroism spectroscopy. Proc. Natl. Acad. Sci. USA 2015, 112, E3095–E3103. [Google Scholar] [CrossRef]
- Micsonai, A.; Moussong, É.; Wien, F.; Boros, E.; Vadászi, H.; Murvai, N.; Lee, Y.-H.; Molnár, T.; Réfrégiers, M.; Goto, Y.; et al. BeStSel: Webserver for secondary structure and fold prediction for protein CD spectroscopy. Nucleic Acids Res. 2022, 50, W90–W98. [Google Scholar] [CrossRef]
- Evenäs, J.; Tugarinov, V.; Skrynnikov, N.R.; Goto, N.K.; Muhindaram, K.; Kay, L.E. Ligand-induced structural changes to maltodextrin-binding protein as studied by solution NMR spectroscopy. J. Mol. Biol. 2001, 309, 961–974. [Google Scholar] [CrossRef] [Green Version]
- Forman-Kay, J.D. The ‘dynamics’ in the thermodynamics of binding. Nat. Struct. Biol. 1999, 6, 1086–1087. [Google Scholar] [CrossRef]
- Lipari, G.; Szabo, A. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 1. Theory and range of validity. J. Am. Chem. Soc. 1982, 104, 4546–4559. [Google Scholar] [CrossRef]
- Lipari, G.; Szabo, A. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 2. Analysis of experimental results. J. Am. Chem. Soc. 1982, 104, 4559–4570. [Google Scholar] [CrossRef]
- Clore, G.M.; Driscoll, P.C.; Wingfield, P.T.; Gronenborn, A.M. Analysis of backbone dynamics of interleukin-1β using two-dimensional inverse detected heteronuclear 15N-1H NMR spectroscopy. Biochemistry 1990, 29, 7387–7401. [Google Scholar] [CrossRef]
- Clore, G.M.; Szabo, A.; Bax, A.; Kay, L.E.; Driscoll, P.C.; Gronenborn, A.M. Deviations from the simple two parameter model free approach to the interpretation of 15N nuclear magnetic relaxation of proteins. J. Am. Chem. Soc. 1990, 112, 4989–4991. [Google Scholar] [CrossRef]
- Loria, J.P.; Rance, M.; Palmer, A.G., III. A relaxation-compensated Carr-Purcell-Meiboom-Gill sequence for characterizing chemical exchange by NMR spectroscopy. J. Am. Chem. Soc. 1999, 121, 2331–2332. [Google Scholar] [CrossRef]
- Tollinger, M.; Skrynnikov, N.R.; Mulder, F.A.A.; Forman-Kay, J.D.; Kay, L.E. Slow dynamics in folded and unfolded states of an SH3 domain. J. Am. Chem. Soc. 2001, 123, 11341–11352. [Google Scholar] [CrossRef]
- Basehore, H.K.; Ropson, I.J. Residual interaction in unfolded bile acid-binding protein by 19F NMR. Protein Sci. 2011, 20, 327–335. [Google Scholar] [CrossRef]
- Yeh, S.-R.; Ropson, I.J.; Rousseau, D.L. Hierarchical folding of intestinal fatty acid binding protein. Biochemistry 2001, 40, 4205–4210. [Google Scholar] [CrossRef]
- Turpin, E.R.; Fang, H.-J.; Thomas, N.R.; Hirst, J.D. Cooperativity and site selectivity in the ileal lipid binding protein. Biochemistry 2013, 52, 4723–4733. [Google Scholar] [CrossRef]
- Ayers, S.D.; Nedrow, K.L.; Gillilan, R.E.; Noy, N. Continuous nucleocytoplasmic shuttling underlies transcriptional activation of PPAR by FABP4. Biochemistry 2007, 46, 6744–6752. [Google Scholar] [CrossRef]
- Nakahara, M.; Furuya, N.; Takagaki, K.; Sugaya, T.; Hirota, K.; Fukamizu, A.; Kanda, T.; Fujii, H.; Sato, R. Ileal bile acid-binding protein, functionally associated with the farnesoid X receptor or the ileal bile acid transporter, regulates bile acid activity in the small intestine. J. Biol. Chem. 2005, 280, 42283–42289. [Google Scholar] [CrossRef]
- Cogliati, C.; Tomaselli, S.; Assfalg, M.; Pedò, M.; Ferranti, P.; Zetta, L.; Molinari, H.; Ragona, L. Disulfide bridge regulates ligand-binding site selectivity in liver bile acid-binding proteins. FEBS J. 2009, 276, 6011–6023. [Google Scholar] [CrossRef]
- Pace, C.N.; Vajdos, F.; Fee, L.; Grimsley, G.; Gray, T. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 1995, 4, 2411–2423. [Google Scholar] [CrossRef]
- Shih, P.; Holland, D.R.; Kirsch, J.F. Thermal stability determinats of chicken egg-white lysozyme core mutants: Hyrdophobicity, buried volume, and conserved buried water molecules. Protein Sci. 1995, 4, 2050–2062. [Google Scholar] [CrossRef]
- Marion, D.; Kay, L.E.; Sparks, S.W.; Torchia, D.A.; Bax, A. Three-dimensional heteronuclear NMR of nitrogen-15 labeled proteins. J. Am. Chem. Soc. 1989, 111, 1515–1517. [Google Scholar] [CrossRef]
- Kay, L.E.; Keifer, P.; Saarinen, T. Pure absorption gradient enhanced heteronuclear single quantum correlation spectroscopy with improved sensitivity. J. Am. Chem. Soc. 1992, 114, 10663–10665. [Google Scholar] [CrossRef]
- Kay, L.; Nicholson, L.; Delaglio, F.; Bax, A.; Torchia, D. Pulse sequences for removal of the effects of cross-correlation between dipolar and chemical-shift anisotropy relaxation mechanism on the measurement of heteronuclear T1 and T2 values in proteins. J. Magn. Reson. 1992, 97, 359–375. [Google Scholar] [CrossRef]
- Farrow, N.A.; Muhandiram, R.; Singer, A.U.; Pascal, S.M.; Kay, C.M.; Gish, G.; Shoelson, S.E.; Pawson, T.; Forman-Kay, J.D.; Kay, L.E. Backbone dynamics of a free and phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry 1994, 33, 5984–6003. [Google Scholar] [CrossRef]
- Markley, J.L.; Horsley, W.J.; Klein, M.P. Spin-lattice relaxation measurements in slowly relaxing complex spectra. J. Chem. Phys. 1971, 55, 3604–3605. [Google Scholar] [CrossRef] [Green Version]
- Mulder, F.A.; Skrynnikov, N.R.; Hon, B.; Dahlquist, F.W.; Kay, L.E. Measurement of slow (micros-ms) time scale dynamics in protein side chains by (15) N relaxation dispersion NMR spectroscopy: Application to Asn and Gln residues in a cavity mutant of T4 lysozyme. J. Am. Chem. Soc. 2001, 123, 967–975. [Google Scholar] [CrossRef] [PubMed]
- Abragam, A. Principles of Nuclear Magnetism; Clraendon Press: Oxford, UK, 1961. [Google Scholar]
- Case, D.A. Calculations of NMR dipolar coupling strengths in model peptides. J. Biomol. NMR 1999, 15, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Cole, R.; Loria, J.P. FAST-Modelfree: A program for rapid automated analysis of solution NMR spin-relaxation data. J. Biomol. NMR 2003, 26, 203–213. [Google Scholar] [CrossRef]
- Mandel, A.M.; Akke, M.; Palmer, A.G., III. Backbone dynamics of Escherichia coli ribonuclease HI: Correlations with structure and function in an active enzyme. J. Mol. Biol. 1995, 246, 144–163. [Google Scholar] [CrossRef]
- Palmer, A.G., III; Rance, M.; Wright, P.E. Intramolecular motions of a zinc finger DNA-binding domain from xfin characterized by proton-detected natural abundance 13C heteronuclear NMR spectroscopy. J. Am. Chem. Soc. 1991, 113, 4371–4380. [Google Scholar] [CrossRef]
- Kay, L.E.; Torchia, D.A.; Bax, A. Backbone dynamics of proteins as studied by 15N inverse detected heteronuclear NMR spectroscopy: Application to Staphylococcal nuclease. Biochemistry 1989, 28, 8972–8979. [Google Scholar] [CrossRef]
- Korzhnev, D.M.; Kloiber, K.; Kay, L.E. Multiple-quantum relaxation dispersion NMR spectroscopy probing millisecond time-scale dynamics in proteins: Theory and application. J. Am. Chem. Soc. 2004, 126, 7320–7329. [Google Scholar] [CrossRef]
- Kleckner, I.R.; Foster, M.P. GUARDD:user-friendly MATLAB software for rigorous analysis of CPMG RD NMR data. J. Biomol. NMR. 2012, 52, 11–22. [Google Scholar] [CrossRef]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef]
- Johansson, M.U.; Zoete, V.; Michielin, O.; Guex, N. Defining and searching for structural motifs using DeepView/Swiss-PdbViewer. BMC Bioinform. 2012, 13, 173. [Google Scholar] [CrossRef] [Green Version]
- Aliev, A.E.; Kulke, M.; Khaneja, H.S.; Chudasama, V.; Sheppard, T.D.; Lanigan, R.M. Motional timescale predictions by molecular dynamics simulations: Case study using proline and hydroxyproline sidechain dynamics. Proteins Struct. Funct. Bioinform. 2014, 82, 195–215. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WT | Q51A | Q99A | |
|---|---|---|---|
| 15N R1 (s−1) | 1.4 ± 0.2 | 1.5 ± 0.2 | 1.4 ± 0.1 |
| 15N R2 (s−1) | 14.0 ± 1.3 | 13.3 ± 1.2 | 13.7 ± 1.8 |
| {1H} 15N NOE | 0.80 ± 0.06 | 0.80 ± 0.07 | 0.79 ± 0.06 |
| τm (ns) | 9.6 | 9.3 | 9.4 |
| D||/D⊥ | 1.2 | 1.1 | 1.1 |
| S2 | 0.92 ± 0.07 | 0.91 ± 0.07 | 0.92 ± 0.06 |
| Number of residues | |||
| model 1 (S2) | 78 | 72 | 78 |
| model 2 (τe) | 12 | 18 | 14 |
| model 3 (Rex) | 30 | 9 | 14 |
| model 4 (Rex, τe) | - | 2 | - |
| model 5 (Sf2, Ss2) | 2 | 2 | 3 |
| (A) | Q99A | ||
| global fit analysis | 283 K a | 287 K b | 291 K c |
| kex (s−1) | 871 ± 63 | 1159 ± 57 | 1546 ± 71 |
| pB (%) | 3.1 ± 0.2 | 3.5 ± 0.2 | 4.1 ± 0.2 |
| kAB (s−1) | 27 ± 3 | 41 ± 3 | 63 ± 4 |
| kBA (s−1) | 844 ± 98 | 1118 ± 106 | 1483 ± 123 |
| ΔGAB (kcal/mol) | 2.0 ± 0.1 | 2.0 ± 0.1 | 1.9 ± 0.1 |
| (B) | Q51A | ||
| individual fit analysis (283 K) | kex (s−1) | pE (%) | Rex (Hz) |
| Gly22 | 364 ± 48 | 0.8 ± 0.3 | 2.8 ± 0.2 |
| Val37 | 25 ± 6 | 6 ± 2 | 1.7 ± 0.4 |
| Trp49 | 768 ± 309 | 0.3 ± 0.4 | 2.5 ± 0.6 |
| Gly75 | 35 ± 21 | 5 ± 1 | 1.8 ± 0.3 |
| Gly76 | 112 ± 30 | 2 ± 0.7 | 2.4 ± 0.3 |
| Val91 | 979 ± 57 | 0.9 ± 0.2 | 5.9 ± 0.2 |
| Tyr97 | 27 ± 15 | 3.3 ± 0.9 | 0.9 ± 0.1 |
| Val104 | 1501 ± 304 | 0.2 ± 0.3 | 2.1 ± 0.5 |
| Val109 | 2148 ± 963 | 0.4 ± 0.9 | 2.3 ± 0.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Horváth, G.; Balterer, B.; Micsonai, A.; Kardos, J.; Toke, O. Multiple Timescale Dynamic Analysis of Functionally-Impairing Mutations in Human Ileal Bile Acid-Binding Protein. Int. J. Mol. Sci. 2022, 23, 11346. https://doi.org/10.3390/ijms231911346
Horváth G, Balterer B, Micsonai A, Kardos J, Toke O. Multiple Timescale Dynamic Analysis of Functionally-Impairing Mutations in Human Ileal Bile Acid-Binding Protein. International Journal of Molecular Sciences. 2022; 23(19):11346. https://doi.org/10.3390/ijms231911346
Chicago/Turabian StyleHorváth, Gergő, Bence Balterer, András Micsonai, József Kardos, and Orsolya Toke. 2022. "Multiple Timescale Dynamic Analysis of Functionally-Impairing Mutations in Human Ileal Bile Acid-Binding Protein" International Journal of Molecular Sciences 23, no. 19: 11346. https://doi.org/10.3390/ijms231911346
APA StyleHorváth, G., Balterer, B., Micsonai, A., Kardos, J., & Toke, O. (2022). Multiple Timescale Dynamic Analysis of Functionally-Impairing Mutations in Human Ileal Bile Acid-Binding Protein. International Journal of Molecular Sciences, 23(19), 11346. https://doi.org/10.3390/ijms231911346